

Abstract
Constitutive activation of the transcription factor nuclear factor-κB (NF-κB) plays a major role in inflammatory diseases as well as cancer by inducing the endogenous expression of many proinflammatory proteins such as chemokines, and facilitating escape from apoptosis. The constitutive expression of chemokines such as CXCL1 has been correlated with growth, angiogenesis, and metastasis of cancers such as melanoma. The transcription of CXCL1 is regulated through interactions of NF-κB with other transcriptional regulatory molecules such as poly(ADP-ribose) polymerase-1 (PARP-1) and cAMP response element binding protein (CREB)-binding protein (CBP). It has been proposed that these two proteins interact with NF-κB and other enhancers to form an enhanceosome at the promoter region of CXCL1 and modulate CXCL1 transcription. In addition to these positive cofactors, a negative regulator, CAAT displacement protein (CDP), may also be involved in the transcriptional regulation of CXCL1. It has been postulated that the elevated expression of CXCL1 in melanomas is due to altered interaction between these molecules. CDP interaction with the promoter down-regulates transcription, whereas PARP and/or CBP interactions enhance transcription. Thus, elucidation of the interplay between components of the enhanceosome of this gene is important in finding more efficient and new therapies for conditions such as cancer as well as acute and chronic inflammatory diseases.
I. Chemotactic Cytokines
Chemokines are small, proinflammatory, inducible, secreted cytokines that are involved in trafficking, activation, and proliferation of many cell types such as myeloid, lymphoid, pigment epidermal, and endothelial cells (1). Chemokine proteins are encoded by 70–130 amino acids, which also include a signal peptide sequence of 20–25 amino acids. It is interesting to note that although chemokines share little homology in their primary sequence, their overall tertiary structure is similar (2). Chemokines have been observed to form dimers in concentrated solutions and on crystallization, however, these concentrations are much higher than the biological concentrations. It is now commonly accepted that chemokines act as monomers in biological systems (2–4). To date, over 50 chemokines have been identified and assigned to four classes according to their arrangement of the first two of four conserved cysteine residues: C, CC, CXC, and CX3C chemokines (Table I). The C chemokines such as lymphotactin (XCL1) lack two of the four conserved cysteine residues, whereas CC chemokines have the first two cysteines adjacent to each other; examples are chemoattractant protein-1 (CCL2), macrophage inflammatory protein-1α (CCL3), and regulated upon activation of normal T cells expressed and secreted (CCL5). In CX3C chemokines, three amino acid residues separate the first two cysteines, e.g., fractalkine (CX3CL1), whereas in CXC chemokines such as melanocyte growth stimulatory activity/growth-related oncogene (CXCL1-3), interleukin 8 (CXCL-8), monokine induced by interferon-γ (CXCL9), interferon-γ-inducible protein-10 (CXCL10) and IFN-inducible T cell α-chemoattractant (CXCL11), only one amino acid residue separates the first two cysteines (1, 9). CXC chemokines may contain a Glu-Leu-Arg (ELR) motif at the amino terminus (e.g., CXCL1-3, CXCL8). Chemokines that contain the ELR motif are associated with angiogenesis, whereas chemokines lacking this motif are associated with angiostasis (e.g., CXCL9, CXCL10, and CXCL11) (5, 6).
TABLE I. CXC, C, CX, C, and CC Chemokine and Receptor Famillesa.
| Systematic name | Chromosome | Human ligand | Mouse ligand | Chemokine receptor(s) |
|---|---|---|---|---|
| CXC chemokine/receptor family | ||||
| CXCL1 | 4q21.1 | GROα/MGSAα | GRO/MIP2/KC? | CXCR2>CXCR1 |
| CXCL2 | 4q21.1 | GROβ/MGSAβ | GRO/MIP2/KC? | CXCR2 |
| CXCL3 | 4q21.1 | GROγ/MGSAγ | GRO/MIP2/KC? | CXCR2 |
| CXCL4 | 4q21.1 | PF4 | PF4 | Unknown |
| CXCL5 | 4q21.1 | ENA7B | GCP2/LIX? | CXCR2 |
| CXCL6 | 4q21.1 | GCP2 | GCP2/LIX? | CXCR1, CXCR2 |
| CXCL7 | 4q21.1 | NAP2 | Unknown | CXCR2 |
| CXCL8 | 4q21.1 | IL-8 | Unknown | CXCR1, CXCR2 |
| CXCL9 | 4q21.1 | MIG | MIG | CXCR3 (CD183) |
| CXCL10 | 4q21.1 | IP10 | IP10CRG2 | CXCR3 (CD183) |
| CXCL11 | 4q21.1 | ITAC | ITAC | CXCR3 (CD183) |
| CXCL12 | 10q11.21 | SDF1α/β | SDF1/PBSF | CXCR4 (CD184) |
| CXCL13 | 4q21.1 | BCA1 | BLC | CXCR5 |
| CXCL14 | 5q31.1 | BRAK/Bolckine | BRAK | Unknown |
| (CXCL15) | Unknown | Unknown | Lungkine/WECHE | Unknown |
| CXCL16 | 17p13 | Unknown | Unknown | CXCR6 |
| C chemokine/receptor family | ||||
| XCL1 | 1q24.2 | Lymphotactin/SCM1α/ATAC | Lymphotactin | XCR1 |
| CX3C chemokine/receptor family | ||||
| CX3 CL1 | 16q13 | Fractalkine | Neurotactin/ABCD3 | CX3 CR1 |
| CC chemokine/receptor family | ||||
| CCL1 | 17q11.2 | 1309 | TCA3/P500 | CCR3 |
| CCL2 | 17q11.2 | MCP1/MCAF/TDCF | JE? | CCR2 |
| CCL3 | 17q12 | MIP1α/LD78α | MIP1α | CCR1, CCR5 |
| CCL3L1 | 17q12 | LD78β | Unknown | CCR1, CCR5 |
| CCL4 | 17q12 | MIP1β | MIP1β | CCR5 (CD195) |
| CCL5 | 17q12 | RANTES | RANTES | CCR1, CCR3, CCR5 (CD195) |
| (CCL6) | Unknown | C10/MRP1 | Unknown | |
| CCL7 | 17q11.2 | MCP3 | MARC? | CCR1, CCR2, CCR3 |
| CCL8 | 17q11.2 | MCP2 | MCP2? | CCR3, CCR5 (CD195) |
| (CCL9/10) | Unknown | MRP2/CCF18/MIP1γ | CCR1 | |
| CCL11 | 17q11.2 | Eotaxin | Eotaxin | CCR3 |
| (CCL12) | Unknown | MCP5 | CCR3 | |
| CCL13 | 17q11.2 | MCP4 | Unknown | CCR2, CCR3 |
| CCL14 | 17q12 | HCC1 | Unknown | CCR1, CCR5 |
| CCL15 | 17q12 | HCC/LKN1/MP1δ | Unknown | CCR1, CCR3 |
| CCL16 | 17q12 | HCC4/LEC/LCC1 | Unknown | CCR1, CCR2 |
| CCL17 | 16q13 | TARC | TARC/ABCO2 | CCR4 |
| CCL18 | 17q12 | DC-CK1/PARC/AMAC1 | Unknown | Unknown |
| CCL19 | 9p13.3 | MIP3β/ELC/exodus-3 | MIP3β/ELC/exodus-3 | CCR7 (CD197) |
| CCL20 | 2q36.3 | MIP3α/LARC/exodus-1 | MIP3α/LARC/exodus-1 | CCR6 |
| CCL21 | 9p13.3 | 6Ckine/SLC/exodus-2 | 6Ckine/SLC/exodus-2/TCA4 | CCR7 (CD197) |
| CCL22 | 16q13 | MDC/STCP1 | ABCD1 | CCR4 |
| CCL23 | 17q12 | MPIF1/CKβ8/CKβ8-1 | Unknown | CCR1 |
| CCL24 | 7q11.23 | Eotaxin-2/MPIF2 | MPIF2 | CCR3 |
| CCL25 | 19p13.3 | TECK | TECK | CCR9 |
| CCL26 | 7q11.23 | Eotaxin-3 | Unknown | CCR3 |
| CCL27 | 9p13.3 | CTACK/ILC | ALP/CTACK/ILC/ESkine | CCR10 |
| CCL28 | 5p12 | MEC | CCR3/CCR10 |
Reproduced with permission from International Union of Immunological Societies/World Health Organization Subcommittee on Chemokine Nomenclature. J. Leukoc. Biol. 70, 465–466 (2001). The Society for Leukocyte Biology.
The expression of the chemokine superfamily is regulated through multiple pathways. Cytokines such as interleukin-1 (IL-1)1 and tumor necrosis factor-α (TNF-α) induce their expression through activation of nuclear factor-κB (NF-κB) (7), whereas interferon-γ (IFN-γ) acts through the Janus kinases (JAK)/signal tranducer and activator of transcription proteins (STAT) pathway (8). Transforming growth factor-β (TGF-β) and glucocorticoids negatively regulate chemokine expression (9). The functions of chemokines are mediated through seven transmembrane domain, G protein-coupled cell surface chemokine receptors (9–11). The binding of chemokines to their receptors occurs through interactions of two regions with the receptor. The low-affinity binding of chemokines to the receptors is mediated by an exposed loop of the backbone between the second and third cysteines, whereas high-affinity binding to the receptors requires the N-terminus. The binding of the NH2-terminal to the receptor is required for receptor signaling and its amino acid composition is important in determining the degree of chemokine binding to the receptor (2).
II. Receptors of Chemokines
The functions of chemokines are mediated through seven transmembrane G-protein-coupled cell surface receptors. Chemokine receptors are 340–370 amino acids in length with 25–80% amino acid identity (12). The structures of chemokine receptors have not yet been fully solved; however, they are believed to have common features such as four extracellular domains each with one cysteine residue, a conserved 10 amino acid sequence in the second intracellular loop, an acidic NH2-terminus that contains N-linked glycosylation sites and is involved in the ligand binding, and an intracellular C-terminus containing serine and threonine phosphorylation sites (13, 14).
There is a wide variation in terms of chemokine and chemokine receptor selectivity. Certain chemokines bind only one receptor, whereas others may bind many different receptors. Receptors may also be exclusive for one or more chemokines (15, 16). In general chemokines from the same gene cluster tend to bind similar receptors (17). Once the receptor binds a chemokine, it gets phosphorylated by a G-protein receptor-coupled kinase, an event proposed to stabilize receptor desensitization. Subsequently, the receptor is internalized (18). The receptors may also undergo heterologous desensitization, where its serine residues are phosphorylated without ligand binding (19).
To date 19 human chemokine receptors have been identified among which 6 receptors selectively bind CXC chemokines and thus have been designated CXCR1 through 6, and 10 receptors, CCR1 to CCR10, all bind the CC chemokines. Receptors for fractalkine and lymphotactin were recently identified and named CX3CR1 (20) and XCR1 (21), respectively. The Duffy antigen receptor also includes the chemokine receptor family and binds promiscuously to both CXC and CC chemokines (22).
The biological functions of CXCL1 are known to be mediated through CXCR2. This receptor has been shown to be expressed on all granulocytes, monocytes, and mast cells, and on some CD8+ T cells and CD56+ natural killer cells as well as melanocytes. By binding and activating CXCR2, CXCL1 modulates inflammation, angiogenesis, wound healing, tumorigenesis, and cell motility (23–25).
The signal transduction mechanism of chemokine receptors is dependent on the trimeric G-protein that is coupled to the receptor. G-proteins function as molecular switches that can flip between two states: active when guanosine triphosphate (GTP) is bound, and inactive when guanosine diphosphate (GDP) is in place. When the ligand binds to the receptor, a conformational change in the receptor causes its association with the G-protein to facilitate the exchange of GDP for GTP. In this activated state, the G-protein dissociates into Gα and Gβγ subunits, so that Gβγ is able to activate the membrane bound phospholipase Cβ2 (PLC2). PLC2 in turn catalyzes the synthesis of 1,4,5-trisphosphate (IP3) from phosphatidylinositol 4,5-bisphosphate (PIP2). IP3 mobilizes calcium, leading to activation of calcium-sensitive kinases such as protein kinase C (PKC), which in turn phosphorylate proteins that activate a series of signaling events leading to cellular responses (26, 27). Chemokine receptors can also activate several other intracellular effectors such as Ras and Rho (28), phospholipase A2, phosphatidylinositol-3-kinase (PI-3K) (29), tyrosine kinases (30, 31), and the mitogen-activated protein kinase (MAPK) pathway (32, 33).
Chemokine receptors become desensitized to repeated stimulation with the same or other ligands after activation. Although this process is not fully understood, it is thought to be stabilized by phosphorylation of serine and threonine residues in the C-terminus of the receptor by G-protein-coupled receptor kinases. Receptor phosphorylation in some instances facilitates receptor sequesteration and internalization (34, 35). Desensitization and receptor internalization are thought to be critical for maintaining the cell's capacity to sense a chemoattractant gradient. Recently our laboratory demonstrated that CXCR2 associates with Hsc/Hsp 70-interacting protein (HIP), and this association seems to play an important role in receptor internalization (36). Subsequently, the receptors are targeted for degradation and/or recycling back to the membrane. Thus, desensitization and trafficking of chemokine receptors may also play important roles in the regulation of inflammatory processes and cancer.
III. Chemokines in Wound Healing and Diseases
Chemokines were originally described as mediators of leukocyte recruitment and activation. Chemotaxis of leukocytes is induced by early response mediators and cytokines such as histamine, C5a, TNF-α, and IL-1, which increase the expression of adhesion molecules on the vascular endothelial cells. This allows for localization and adherence of leukocyte populations to areas of inflammation. The early response mediators and cytokines help in slowing the circulatory flow of leukocytes by up-regulating molecules such as selectins on the endothelial surface. Once leukocytes have been localized to the inflamed vascular wall, the presence of adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) allows firm adhesion to the endothelium. After adherence to the vascular endothelium, leukocytes can migrate into the tissue following the chemotactic gradient in the inflamed tissue (37).
Leukocyte migration is one of the important components of wound healing. After acute injury in skin wound models, the destroyed blood vessels release platelets and neutrophils that serve as sources for growth factors and other mediators such as connective tissue-activating peptide III (CTAP-III). CTAPIII is processed to CXCL7 by proteases that are released by neutrophils, which in turn stimulates the migration and extravasation of neutrophils via the receptor CXCR2 (38). The diapedesis of neutrophils is further mediated by CXCL1, which is produced by both endothelial and dermal cells. A third major mediator of the neutrophil migration, CXCL5, is produced by mononuclear cells in the provisional matrix of the wound (39, 40). Furthermore, hypoxia and bacterial products as well as proinflammatory cytokines (TNF-α, IL-1) produced by neutrophils below the wound surface stimulate production of CXCL8, which stimulates the migration and proliferation of keratinocytes in addition to neutrophils.
The keratinocytes in turn express CCL2, which has been considered the main attractant of mast cells, monocytes, and lymphocytes to the wound area (39, 41, 42). Furthermore, CCL2 in addition to other angiogenic chemokines, e.g., CXCL1 and CXCL8, may also stimulate endothelial-cell locomotion during the angiogenesis phase of wound healing (39, 43, 44). CCL2 also may indirectly contribute to fibroblast proliferation by recruiting IL-4-producing mast cells. IL-4 stimulates fibroblast proliferation and helps to limit the inflammatory reaction by down-regulating the expression of chemokines, e.g., CCL2 and CXCL8 (45, 46). Thus, the attraction of resident and inflammatory cells appears to be tightly regulated by the complex and phase-specific expression of chemokines, placing these small molecules at center stage in the process of wound healing.
Almost any stimulus that alters cellular homeostasis may elicit the expression of inducible chemokines leading to an over 300-fold increase in the mRNA level within a few hours of activation (47). However, with a system that is easily inducible, there is also a greater potential for persistent expression, leading to diseases. Chemokines have been associated with various diseases such as the autoimmune disease multiple sclerosis (MS), bacterial and viral infections, atherosclerosis, asthma, graft rejection, as well as neoplasia. MS is a chronic relapsing neuroinflammatory disease in which myelinated nerve fibers are targeted by T lymphocytes and macrophages in the central nervous system. Studies have shown that the appearance of CCL5, CCL3, CCL4, CCL2, and CXCL10 mRNA and protein directly correlates with inflammatory lesions (48–53). Consistent with these data is the presence of the receptors for these chemokines, CCR2, CCR5, and CXCR3 on macrophages, activated microglia and T cell in lesions (54).
Chemokines also play an important role in bacterial and viral infections. Studies have shown increased MIP-2 production during lipopolysaccharide-induced endotoxemia in animal models, which may be responsible for the recruitment of neutrophils (55). Other chemokines such as CCL3, CCL5, and CCL2 have also been implicated in septic responses (56–58). In viral infections however, chemokine receptors seem to play an important role. Studies have shown that some chemokine receptors (CCR3, CCR2b, CCR5, and CXCR4) act as cofactors with CD4 for macrophage cell line-trophic HIV entry into monocytes and T cells (59, 60). Also, other viruses such as cytomegalovirus (CMV) have been shown to encode chemokine receptors (61).
The relevance of chemokines in atherosclerosis was demonstrated in animal models in which chemokines CXCL8, CXCL12, CXCL10, CCL2, and CCL5 were associated with the lesions. A model for involvement of chemokines in progression of atherosclerosis proposes that activated endothelial or arterial smooth muscle cells release chemokines to induce firm adhesion of monocytes to the vascular endothelium. This is followed by diapedesis into the subendothelium, where they take up lipid and become the foam cells within the fatty streak. The smooth muscle migration into the intima and thrombus formation over the plaque may also involve some chemokines (47).
In asthma, a chronic inflammatory disease of small airways, mononuclear, eosinophil, and mast cells infiltrate the submucosa leading to mucous gland hyperplasia and subepithelial fibrosis. Asthma is characterized by airway hyperresponsiveness (smooth muscle contraction) to nonspecific stimuli. This is thought to be due to the involvement of chemokines such as CCL2, CCL12, CCL24, and CCL5. CCL2 was able to induce changes in airway physiology when instilled into the lungs of normal mice, resulting in increased levels of histamine mast cell degranulation (62).
Chemokines may also influence allograft biology through their involvement in immune suppression, inflammatory responses in acute and chronic rejection, as well as recruitment of leukocytes to the allograft leading to ischemia-reperfusion (63). Studies in heart and skin transplants have demonstrated the presence of CXCL1 and CCL2 in the early response and CXCL10, CXCL9, CCL5, and CCL4 at the later stages. The presence of these chemokines may be necessary for the movement of CD4+ and CD8+ T lymphocytes, macrophages, natural killer cells, and antigen-presenting cells that are involved in the acute rejection (64–66).
Interestingly, elevated expression of chemokines has been observed in many tumor cell types, implicating a role for chemokines in neoplasia. The potential of chemokine involvement in tumors has sparked a new interest in the field of cancer biology. Studies of a variety of tumor cell types indicate the role of chemokines as tumor growth factors as well as stimulators of angiogenesis and metastasis. Murine models have shown that chemokine secretion by tumor cells may influence angiogenesis as well as tumor growth. The metastatic and angiogenic abilities of a number of tumors have been attributed to elevated levels of ELR+, angiogenic chemokines (5, 6). Constitutively expressed chemokines have been shown to transform melanocytes and high levels of endogenous CXCL1 and CXCL8 have been detected in melanomas (24, 25, 67–69). The role of these chemokines in tumor growth has also been implicated in many other tumor types such as pancreas, head and neck, and non-small-cell lung (70–72).
The directional metastasis of malignant tumors has also been attributed to the presence of chemokines and their receptors in tumors (73–75). A recent publication on the involvement of CXCR4 and CCR7 in directing breast cancer tumors to their secondary sites further supports this hypothesis (76). The two chemokine receptors CXCR4 and CCR7 were shown to be expressed in high levels in breast tumors compared to the normal breast tissue. Interestingly enough, the ligands for these receptors, CXCL12 and CCL21, respectively, showed high expression in such organs as lung, liver, bone marrow, and regional lymph nodes, which are associated with the distinct breast cancer metastasis pattern. Moreover, the high expression of CXCR4 and CCR7 induced actin polymerization and pseudopodia formation, resulting in enhanced invasiveness of the breast tumors. Thus, chemokines appear to play a significant role in tumor development due to their involvement in tumor growth, angiogenesis, and metastasis.
Thus, with the association of chemokines in a wide variety of diseases, it is imperative to study the mechanisms underlying their regulation in order to develop more efficient therapies. The focus in our laboratory lies on the chemokine CXCL1 in a melanoma model, since CXCL1 appears to contribute to the growth and proliferation of melanoma. Studies in our laboratory have shown that the level of this chemokine is elevated in many types of melanoma cell lines and that this elevation in the expression may be due to deregulation in the transcriptional regulation of the CXCL1 gene. In this review, the possible mechanism of deregulation will be discussed.
IV. Differential Expression of Chemokines
Cytokines are not stored intracellularly and as such their stimulus-dependent secretion heavily relies on de novo protein synthesis. Hence, cytokine expression is regulated primarily at the initial phase of protein synthesis—transcription. This regulation appears to be stimulus and cell type specific and is mediated through inducible transcription factors such as NF-κB and activator protein (AP-1) (77–80). Following a stimulus, early response cytokines such as TNF-α and IL-1 are rapidly produced, which in turn induce macrophages and other cell types to secrete additional cytokines such as CXCL8 and IL-6. The promoters of many of these cytokine genes contain binding sites for NF-κB, which mediates the up-regulation of their transcription in response to the stimulus and is in fact required for their maximal transcription (81–84).
The cause of the differential expression of the NF-κB-dependent cytokines is uncertain, but recent studies support the speculation that involvement of other transcription factors may be important in determining the transcription rates of cytokine genes. Although cytokine promoters have the NF-κB binding site in common, each individual promoter also binds other transactivators as well as repressors that interact with NF-κB to direct transcription. The binding sites for the transcription factors AP1 and nuclear factor activated by interleukin 6 (NF-IL6), for example, are present in the promoters of CXCL8, IL-6, and CCL5 genes, where the positive cooperation between these transcription factors and NF-κB enhances the transcription of these genes (85–87). In addition to these transcription factors, CCL5 gene expression requires IFN-regulatory factor (IRF)-1, 3, and 7 transcription factors as well (88, 89). Other transcription factors common in cytokine gene expression include stimulating protein-1 (Sp1) and high mobility group-I/Y (HMGI/Y) (90, 91). There are additional factors that may lead to differential expression of cytokines. These may include differences in chromatin and transcription factor accessibility, posttranscriptional RNA processing, mRNA stability, as well as differences in translational efficiency (92–94). Thus, the differential activation of the many inducible transcription factors and their accessibility to the binding sites as well as posttranscriptional events may explain the cell type-specific and stimulus-specific expression of cytokines.
V. Transcriptional Regulation of CXC Chemokines
The expression of the CXC chemokines is thought to be NF-κB dependent, thus indicating a disregulation in the activation of the transcription factor NF-κB may be involved in the up-regulation of these chemokines in inflammatory diseases and cancer. However, as mentioned previously, the transcriptional regulation of these chemokines is more complex and involves factors other than NF-κB. In CXCL8 transcription, NF-κB interacts with either AP1, NF-IL6, or CAAT enhancer binding protein (C/EBP) elements (95–97), whereas in CXCL1 transcription, the NF-κB interacting elements are Sp1 (HMG I/Y) and immediate upstream region (IUR) (90) (Fig. 1). It is postulated that the factors binding to these elements may form an enhanceosome-like transcriptional response element in modulating the gene expression (98, 99).
Fig. 1.
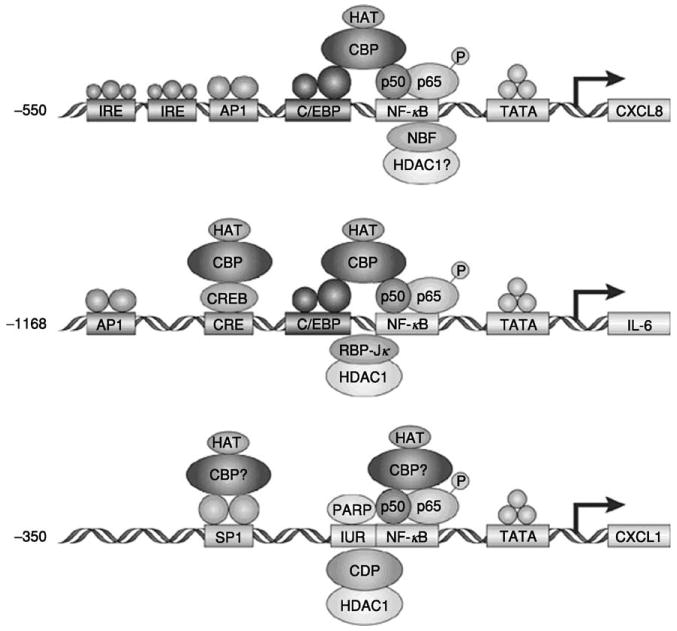
Modulation of transcription of cytokines involves similar regulatory elements. Reprinted by permission from A. Richmond, Nat. Rev. Immunol. 2, 664–674 (2002), ©2002 Macmillan Publishers Ltd.
The transactivation of CXCL8 transcription occurs through the interaction between the NF-κB element of CXCL8 and either the AP1 or the NF-IL6 element in a cell type-specific manner (100–102). Repression of CXCL8 transcription is, on the other hand, regulated by NRF, an NF-κB repressing factor. In the absence of stimulation, NRF inhibits transcription of CXCL8, but after stimulation with IL-1, NRF is necessary for full induction of CXCL8 transcription (103). Upon the binding of NF-κB to its binding site, the p65 subunit activates the promoter via recruitment of CBP to the site. The intrinsic histone acetylase activity (HAT) of CBP/p300 will then stabilize the transcription from the promoter. This stabilization becomes imperiled in the presence of CDP, which has been shown to be capable of recruiting the histone deacetylase activity (HDAC) that would counteract the HAT activity of CBP (104). Thus, it is plausible that similar interactions among NF-κB, CBP, and CDP are involved in the transcriptional regulation of CXCL1.
Within the CXCL1 promoter, the IUR element is thought to regulate the transcription of CXCL1 both positively and negatively. It contains the consensus sequence GGGATCGATC, which binds the negative modulator CDP (105), as well as the TCGATC sequence that binds the positive modulator, PARP-1 (106). Thus, PARP-1 may induce NF-κB-activated transcription of CXCL1, whereas CDP would repress this activity. However, the intricate mechanism of regulation by these molecules is yet to be identified in the context of the CXCL1 promoter. Thus, efforts to shed light on the matter would greatly contribute to the efficacy of therapeutic means used today in inflammatory diseases and cancer.
VI. Regulation of NF-κB Activation
The major known player in the transcription induction of CXCL1 is the transcription factor NF-κB. In the healthy human, NF-κB regulates the expression of genes involved in normal immunological responses (e.g., generation of immunoregulatory molecules such as antibody light chains) in response to proinflammatory cytokines and byproducts of microbial and viral infections (107). However, increased activation of NF-κB results in enhanced expression of proinflammatory mediators, leading to acute inflammatory injury to lungs and other organs, development of multiple organ dysfunction, as well as angiogenesis and tumor growth (107–109).
The NF-κB protein is composed of two subunits, which may vary, affecting the transcriptional activity of the protein. There are five known mammalian NF-κB subunits, each characterized by ankyrin repeat elements: Rel (c-Rel), p65 (RelA), RelB, p50, and p52. The transcription regulatory action of NF-κB depends on the composition of the NF-κB dimers, p50 homodimers lack strong transactivation domains and can actually inhibit gene expression by competing with p65/p50 or other transactivating complexes for the κB sites (107, 108).
In the absence of activation, NF-κB is sequestered in the cytoplasm by being associated with IκB (an inhibitor of κB) protein. The IκB protein binds to the nuclear localization signal (NLS) of NF-κB, inhibiting its translocation into the nucleus (107, 108, 110). When the cell is exposed to activating signals, such as TNF-α, the IκB protein is phosphorylated, ubiquitinated, and then broken down in the 26 S proteasome (111). This frees the NF-κB to translocate into the nucleus where it binds to κB sites in the promoter/enhancer regions of specific genes, including the promoter for IκB, to transactivate transcription. There are five known IκB proteins: IκB-α,β,ε,γ, and Bcl-3. In addition to these five, p105 and p100, the precursors of p50 and p52, respectively, possess domains that act as IκBs. The IκB subtypes show different affinity for the different NF-κB dimers in a cell-specific manner. For example, in endothelial cells, IκB-α has similar inhibitory activity for p50/RelA, p50/RelB, and p50/c-Rel, whereas IκB-β more strongly inhibits p50/RelA than the other two (107, 108).
IκBs are also active in the nucleus and interact with NF-κB dimers as they do in the cytoplasm. IκB-α binds p50/RelA and inhibits its transactivating activity in the nucleus as well as freeing the heterodimer from κB sites on DNA to induce their transport from the nucleus to the cytoplasm. On the contrary, Bcl-3 can act as a transcriptional activator in the nucleus by binding to p50/p50 homodimers, which can act as transcriptional repressors by occupying NF-κB binding sites and preventing the transactivating NF-κB heterodimers such as p50/RelA or p50/c-Rel from binding to these sites (112). The inducible degradation of IκB is controlled by three large multiprotein complexes: IκB kinase (IKK), IκB ubiquitin ligase, and 26 S proteasome. IKK activity is induced upon various stimulation, leading to phosphorylation of two N-terminal serine residues of IκB, Ser-32 and Ser-36 (113, 114). The phosphorylation of IκB targets it for ubiquitination by IκB-ubiquitin ligase and subsequent degradation by the 26 S proteasome (107, 108, 111).
The transactivation function of NF-κB is also regulated in the nucleus through interaction with HDAC corepressor proteins. Ashburner et al. (115) demonstrate in their study a direct interaction between the HDAC1 and NF-κB p65 subunit by which HDAC1 exerts its corepressor function. Overexpression of HDAC1 and HDAC2 was shown to repress TNF-α-induced NF-κB-regulated gene expression. HDAC2 does not interact with NF-κB directly, but can probably regulate NF-κB activity through its association with HDAC1. In accordance with this, chemical inhibitors of HDAC activity such as trichostatin A increased expression of an NF-κB-dependent reporter gene as well as an endogenous IL-8 gene. Thus, the association of NF-κB with the HDAC1 and HDAC2 corepressor proteins functions to repress basal expression of NF-κB-dependent genes as well as to control the induced level of expression of these genes.
Activation of NF-κB may be induced by a variety of pathogenic stimuli, including bacterial products, such as metalloproteases (MMP-3 and 9), viral proteins, cytokines such as IL-1 and TNF-α, growth factors such as PDGF, radiation, ischemia/reperfusion, and oxidative stress through multiple pathways (107, 108). The activation of NF-κB occurs within minutes of stimulation since de novo protein synthesis is not required and the activated NF-κB in turn mediates expression of more than 150 genes involved in inflammatory and immune responses (116). It is important to note that the promoters and enhancers of NF-κB-dependent genes also contain binding sites for other transcription factors and interplay of these factors can potentiate or repress the ability of NF-κB to initiate transcription. However, it is in events of abnormal, constitutive activation of NF-κB that major problems arise resulting in many chronic inflammatory diseases as well as cancer. Persistent activation of NF-κB inhibits apoptosis and promotes proliferation leading to hyperplasia (107–109, 117, 118).
There have been reports on NF-κB activation through IKK-independent pathways as well. Protein kinase A (PKA), casein kinase II (CKII), and p38 MAPK have all been implicated in the phosphorylation of NF-κB Rel A, leading to enhanced interaction of NF-κB with transcriptional coactivators and components of basal transcriptional machinery (119–122). PKA phosphorylation of the NF-κB p65 subunit on serine 276 was shown to weaken the interaction between the N- and C-terminal of p65 and create an additional site for interaction with the coactivator protein CBP (119). Based on these data, it is feasible that there is an interaction between NF-κB and the coactivator protein CBP within the CXCL1 enhanceosome and this interaction among others may be enhanced in melanomas and inflammatory diseases leading to constitutive expression of CXCL1.
A. Signaling Pathways Leading to Activation of NF-κB in Melanoma Cells
The hallmark of cancer cells lies in their ability to escape apoptosis by over-activation of growth and survival pathways. There are two major pathways that are associated with survival and proliferation: PI3K/Akt and Ras/MAPK pathways. Stimulation of any one of these pathways leads to activation of antiapoptotic and prosurvival proteins mediated through NF-κB (Fig. 2).
Fig. 2.
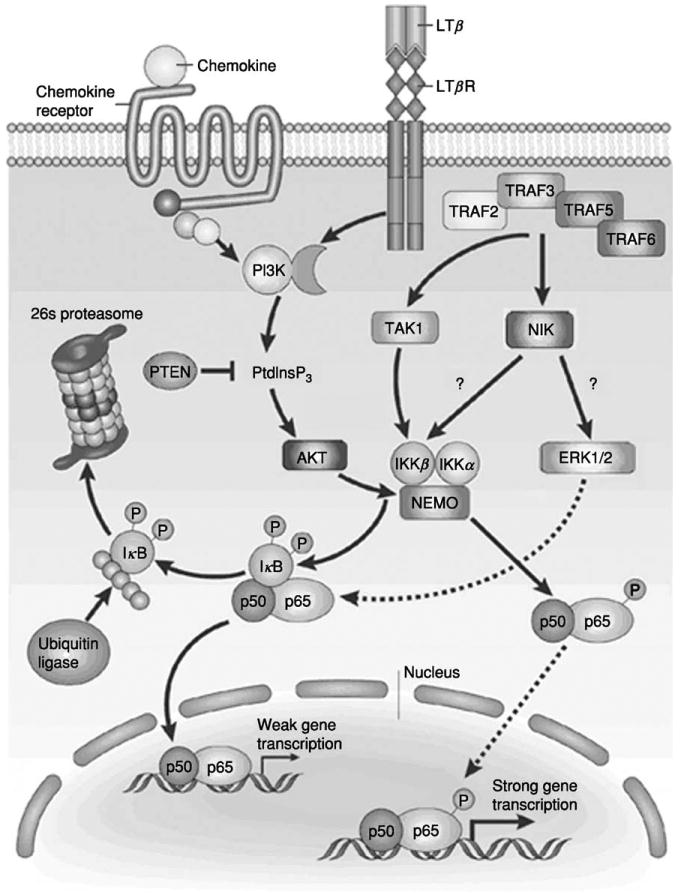
The signal transduction pathways involved in activation of the transcription factor NF-κB. Reprinted by permission from A. Richmond, Nat. Rev. Immunol. 2, 664–674 (2002), ©2002 Macmillan Publishers Ltd.
Akt, originally identified as a homologue of the viral oncogene v-Akt, is closely related to protein kinase A and C and was thus named protein kinase B (PKB). There are three mammalian Akt genes that encode proteins containing a pleckstrin homology (PH) domain in the N-terminus, a central kinase domain, and a regulatory carboxy terminus. There are two regulatory phosphorylation sites within Akt: threonine 308 and serine 473. In unstimulated cells, Akt exists in an unphosphorylated state in the cytoplasm. Upon growth factor stimulation and PI3K activation and subsequent production of PIP3, Akt is recruited to the plasma membrane and is phosphorylated at T308 and S473 by 3-phosphoinositide-dependent protein kinase 1 (PDK1) and PDK2, respectively (123). Fully activated Akt then becomes available to phosphorylate its substrates within the same basic motif, R-X-R-X-X-S/T (124). Thus far, 13 substrates of Akt have been identified, which may be grouped according to their functions in cell survival, cell cycle, glucose metabolism, and protein synthesis. The Akt substrates involved in cell survival regulation include Bad, the forkhead family of transcription factors, FLICE inhibitory protein, and IKK. Thus, activation of Akt may regulate NF-κB activity through IKK. Indeed, NF-κB activation in correlation with Akt activation has been shown in several different carcinomas such as ovarian, breast, pancreatic, and melanoma (125, 126). A model for NF-κB activation through Akt proposed by Madrid et al. (127) suggests Akt utilizes IKK-β in a p38-dependent manner that requires serines 529 and 536 of RelA/p65 to directly stimulate NF-κB activity and Akt signaling in response to IL-1 exposure stimulates NF-κB by activating p38 in a manner dependent on IKK. Whether Akt acts only through IKK to activate NF-κB or directly interacts with NF-κB to activate the transcription factor remains elusive.
Up-regulation of Akt in cancers may be a consequence of mutation or deletion of the phosphatase and tensin homologue deleted on chromosome 10 (PTEN) tumor suppressor gene. PTEN is a lipid phosphatase that plays a crucial role in deactivation of AKT, since one of its primary targets is the direct product of PI3K, PIP3. Loss of PTEN function in cancer cell lines results in accumulation of PIP3 and activation of Akt and escape from apoptosis. Indeed, overexpression of wild-type PTEN sensitizes them to apoptosis probably through inhibition of the PI3K/Akt pathway (128). As in many cancers, PTEN mutations have been reported in melanomas, suggesting a mechanism for overactivation of Akt in this cancer (129).
The second major pathway leading to NF-κB activation involves the Ras family members, which play important roles in cell differentiation, growth, transformation, and apoptosis (130–132). There are four Ras genes in the mammalian genome, expressing the very homologous proteins N-Ras, H-Ras, M-Ras, and K-Ras of 21 kDa. Ras proteins have been shown to activate the PI3K, Rac, RhoA, coupled with activation of the Raf/MAPK pathway to promote oncogenic transformation (133). The ultimate targets of these pathways are transcription factors such as NF-κB (134).
Work in our laboratory has shown that the CXCL1 protein is endogenously expressed in almost 70% of the melanoma cell lines and tumors, but not in normal melanocytes. Overexpression of human CXCL1, 2, or 3 in immortalized murine melanocytes (melan-a cells) enables these cells to form tumors in SCID and nude mice. Differential display examination of the CXCL1 effect on melanocyte transformation revealed overexpression of the Ras genes. One of the mRNAs identified in the screen as overexpressed in CXCL1 transformed melan-a clones was the newly described M-Ras gene. Overexpression of CXCL1 up-regulates M-Ras expression at both the mRNA and protein levels, and this induction requires an intact ELR motif in the CXCL1 protein. K- and N-Ras proteins are also elevated in CXCL1-expressing melan-a clones, leading to an overall increase in the amount of activated Ras. CXCL1-expressing melan-a clones also exhibited enhanced AP-1 activity. Overexpression of wild-type M-Ras or a constitutively activated M-Ras mutant in control melan-a cells as monitored by an AP-1-luciferase reporter also showed enhanced AP-1 activity, whereas expression of a dominant negative M-Ras blocked AP-1-luciferase activity in CXCL1-transformed melan-a clones. In the in vitro transformation assay, overexpression of M-Ras mimicked the effects of CXCL1 by inducing cellular transformation in control melan-a cells, whereas overexpression of dominant negative M-Ras blocked transformation (118). These data suggest that CXCL1-mediated transformation may require Ras activation in melanocytes.
Another NF-κB-regulating kinase that is overexpressed in melanoma is NF-κB-inducing kinase (NIK). NIK was first identified by way of its association with TNF receptor-associated factor 2 (TRAF2) and shares homology with mitogen-activated protein kinase kinase kinases (135). NIK physically associates with and activates both IKK-α and IKK-β (136, 137) and overexpression of NIK has been shown to activate NF-κB (135). Interestingly, our laboratory found that NIK is overexpressed in melanoma cells and the IKK-associated NIK activity is enhanced compared to normal cells. When a catalytically inactive form of NIK was expressed, the constitutive activation of NF-kB and CXCL1 promoter was blocked in Hs294T melanoma cells, but not in normal human epidermal melanocytes. The NIK-induced activation of NF-kB was shown to be through the MAPK signaling kinases extracellular signal-regulated kinase 1 and 2 (ERK1/2), since overexpression of dominant-negative ERK leads to a decrease in NF-kB promoter activity (138). Thus, these data suggest that NIK is involved in up-regulation of NF-kB activity and hence CXCL1 transcription through an NIK/MEKK-IKK-IκB signaling pathway.
VII. Role of CBP in CXCL1 Transcription
Many inducible transcription factors such as NF-κB are activated through interactions with cellular coactivators (139). The transcriptional activity of NF-κB appears to be optimized through interactions with the coactivator CBP as discussed above. CBP is a nuclear protein belonging to a family of highly homologous proteins such as p300 and p270 that are involved in many physiological responses such as proliferation, differentiation, and apoptosis (140). CBP is thought to regulate transcription through its HAT activity, since in vitro transcription experiments have shown that transcription is induced only on the template with chromatin structure, but not on the naked DNA (141, 142). Thus, through its intrinsic HAT activity, CBP activates transcription by transferring an acetyl group to the ε-amino group of a lysine residue of the histone and neutralizes the positive charge of the molecule and thus loosens the interaction between the histone and the negatively charged DNA, leading to chromatin remodeling (143). Furthermore, the HAT activity of CBP is restricted not only to histones, but also to other targets such as transcription factors and the transcription apparatus (144, 145).
The human CBP locus is located in chromosomal region 16p13.3 and codes for a protein containing (1) the bromodomain, which is found in mammalian HATs; (2) an ADA2-homology domain, which is homologous to the yeast transcriptional coactivator, Ada2p; (3) a KIX domain; and (4) three cysteine-histidine (CH)-rich domains referred to as CH1, CH2, and CH3. It has been suggested that the bromodomain recognizes acetylated residues and could function in identification of different acetylated domains. The CH domains and the KIX domain are thought to mediate protein–protein interaction (146). The KIX domain is involved in the interaction between CBP and the p65 (Rel A) subunit of NF-κB, as well as CBP binding to the transcriptionally active serine-133-phosphorylated form of CREB (142, 147).
CBP also appears to be under cell cycle control. Many kinases such as PKA, calcium/calmodulin-dependent kinase, MAP kinase, and cyclin-dependent kinases Cdk2 and Cdc2 have been implicated in the positive and negative phosphorylation control of CBP. The cyclinE–Cdk2 complex has been shown to bind to the C-terminal region of CBP and negatively regulate CBP-mediated coactivation of NF-κB, whereas the other mentioned kinases aid in CBP-mediated transcriptional activation (146). The phosphorylation sites on CBP have not been identified yet, but appear to be critical for its regulation.
There are multiple proposed models for the mode of action of CBP in the regulation of transcription. In the bridging model, CBP acts as a connecting bridge between transcription factors and the transcription apparatus. Data showing the interaction of CBP with a variety of transcription factors as well as components of the basal transcriptional machinery such as TATA box-binding protein (TBP), TFIIB, TFIIE, and TFIIF are supportive of this model (146, 148). In a second model, CBP plays a role in transcriptional activation by assembling a diverse group of cofactor proteins into multi-component coactivator complexes. Thus, CBP serves as a scaffold for the assembly of transcription cofactors, increases the relative concentrations of these factors in the transcription area, and allows for protein–protein and protein–DNA interactions. Yet another model proposes that CBP may take advantage of either its intrinsic HAT activity or other HATs assembled in multi-component complexes to target the chromatin and/or transcription factors for transcription activation. Even though targets of CBP in vivo are yet to be identified, in vitro studies have shown that CBP can acetylate all four core histones (149).
More recently, acetylation of transcription factors such as p53, E2F-1, 2, and 3, MYB, MyoD, GATA-1, CREB, and NF-κB (142, 147, 150, 151) by CBP has been reported and, in almost all cases, the acetylation led to enhanced DNA-binding activity. It is possible that acetylation regulates protein–protein interaction or facilitates protein–DNA binding by changing the conformation of the protein and introducing a DNA-recognition surface (146). These findings also indicate that there may be competitive interactions between the transcription factors for association with CBP, thus providing an additional regulatory event. A good example of this competitive behavior is among CBP, NF-κB, and CREB. CBP recognizes the phosphorylated serine 276 on the p65 subunit of NF-κB through an S domain in the KIX region. The KIX region is also responsible for recognizing the phosphorylated serine 133 form of CREB. Thus phosphorylation of these two proteins promotes their interaction with CBP. Activation of protein kinase A was shown to increase amounts of phosphorylated CREB and decrease NF-κB-mediated transcription (152, 153). Thus, the activity of PKA is a determinant in the binding of CBP to its interacting proteins.
A recent report by Chen et al. (154) demonstrated that the RelA subunit of NF-κB is subject to inducible acetylation by CBP/p300. Acetylation of NF-κB resulted in weak interaction with IκB in the nucleus, which in turn led to decreased IκB-dependent nuclear export of the complex and thus increased NF-κB transactivation. This acetylation was reversed by HDAC3. Thus, CBP/p300 may be involved in the regulation of CXCL1 transcription in more than one way. It could acetylate not only the histones, but also NF-κB itself, and contribute to the prolonged and/or constitutive activation of the transcription factor.
VIII. PARP as a Transcriptional Activator of the CXCL1 Gene
Another putative coactivator of NF-κB in CXCL1 transcription is PARP. The mammalian poly(ADP-ribose) polymerase (PARP)-1, the major isoform of the PARP family, is a nuclear protein that is heavily associated with chromatin. PARP is composed of 1014 amino acids (114 kDa) and is continuously expressed in eukaryotes. It has a 46-kDa DNA-binding domain at the N-terminus that contains two Zn fingers, facilitating the binding of PARP to DNA strand breaks, as well as bipartite nuclear localization signal. The two regions of nuclear localization signals are separated by the DEVD sequence, which is the target of caspase-3 during apoptosis and subsequently gets cleaved, and thus inactivated, resulting in the formation of two proteolytic fragments of PARP, a 29-kDa amino terminus and an 85-kDa carboxyl terminus (155, 156). A 54-kDa domain of PARP located in the carboxyl terminus represents the β-nicotinamide adenine dinucleotide (NAD+)-binding domain containing a highly conserved “PARP signature” sequence comprising the catalytically crucial amino acid residue Glu-988. Between the DNA-binding domain and the NAD+ -binding domain lies a 22-kDa “automodification domain,” which facilitates the homodimerization and/or heterodimerization of PARP with other chromatin proteins (156).
The catalytic activity of PARP is stimulated 500-fold by noncovalent contact of the DNA-binding domain with DNA strand breaks and results in the transfer of successive units of the ADP-ribose moiety from NAD+ to itself and other nuclear protein acceptors such as topoisomerase I and II, histones, HMG proteins, p53, and NF-κB (155, 157–159). It is thought that PARP mediates stress-induced signaling by binding to damaged DNA containing single-strand breaks and nucleotide excisions. Automodification upon DNA binding releases PARP from DNA due to the highly negatively charged poly(ADP-ribose), rendering DNA more accessible to the DNA repair machinery (159). PARP has been shown to associate in vivo with XRCC1, a DNA repair protein involved in base excision repair (BER) of DNA and PARP-1-deficient cells display a severe DNA repair defect that appears to be a primary cause of the observed genomic instability and cytotoxicity of DNA damaging agents that induce BER (160, 161). The catalytic activity of PARP appears to be of importance since in the absence of NAD+, PARP inhibits DNA repair by irreversible binding to damaged DNA (161). When the damage is too extensive, overactivation of PARP leads to depletion of NAD+ and ATP. As apoptosis is ATP dependent, inactivation of PARP by cleavage prevents energy depletion and enables completion of apoptosis. Thus, PARP also serves as a marker for the onset of apoptosis due to its cleavage by caspase-3 into smaller, inactive fragments (162, 163).
Studies with PARP-1-deficient cells and animals have revealed other functions of PARP including roles in genomic recombination and instability, DNA replication, regulation of telomere function, and transcriptional regulation (155, 164). The PARP-1 knockout lines exhibit tetraploidy, increased frequency of sister chromatid exchanges, and micronucleus formation, all markers of genomic instability (164, 165). They also show shorter telomere length, which is associated with aging. Tankyrase1, a telomeric PARP localized at the telomeres, inhibits telomeric-repeat binding factor 1 (TRF1), an inhibitor of telomere elongation, through its ADP-ribosylation activity and thus promotes telomere elongation (158, 166, 167). Microarray analysis of the primary PARP-1 gene, Adprt1, null fibroblasts, revealed down-regulation of several genes involved in the regulation of cell cycle progression, mitosis, DNA replication, chromosomal assembly, or processing. On the other hand, some cytoskeletal and extracellular matrix genes implicated in cancer or in normal or premature aging were up-regulated (158). These data (along with more recent publications) suggest a role for PARP in the regulation of gene expression.
Soldatenkov et al. (168) report a negative role for PARP in transcription regulation, where the direct interaction of the PARP protein with its own gene promoter results in suppression of transcription. However, in response to DNA damage, PARP catalytic activity is stimulated and automodification of the protein will subsequently prevent its interaction with the promoter. This relieves the PARP-mediated block on the promoter and allows for transcription of PARP and other genes suppressed by PARP. Upon DNA damage repair, the DNA binding activity and thus repressor function of PARP are restored. Although in this instance the catalytic activity of PARP relieves suppression, it can in other instances promote suppression. It has been demonstrated that poly(ADP-ribosyl)ation of transcription factors and members of the basal transcriptional machinery prevents binding of these proteins to DNA, thus interrupting formation of new transcription complexes. However, it is important to note that once transcription preinitiation complexes have been formed, the transcription factors are inaccessible to ADP-ribosylation (169). Thus, poly(ADP-ribosyl)ation prevents binding of transcription factors to DNA, whereas binding to DNA prevents their modification.
Conversely, others have reported a positive role for PARP in the formation of the preinitiation complex (PIC). Meisterernst et al. (159) report that the presence of PARP is required during assembly of RNA polymerase II (RNAPII) and other general transcription factors with a preformed complex consisting of TFIID and possibly TFIIA in vitro. They also propose that PARP activation of the PIC is achieved through recruitment of general transcription factors or conformational changes within the components of the basal transcription machinery and that PARP effects on supercoiled templates are DNA concentration dependent and do not require damaged DNA. Furthermore, PARP-1 has been implicated as a transcriptional coactivator in activation of some genes such as B-MYB, reg, and cTNT (170–172).
PARP-1 has also been reported to interact directly with transcription factors such as NF-κB (168, 173, 174). Studies on PARP-1−/− cells revealed that they are defective in NF-κB-dependent transcriptional activation in response to TNF-α and lipopolysaccharide (LPS), indicative of a functional link between PARP-1 and NF-κB. Furthermore, these cells were unable to induce the expression of NF-κB itself after exposure to TNF-α (175, 176). This would suggest a role of PARP-1 as a signaling molecule, controlling the expression of a transcription factor. Moreover, Hassa et al. (177) report that PARP-1 is required for specific NF-κB transcriptional activation, which occurs through interaction of PARP-1 with both subunits of NF-κB, where each subunit binds to a different PARP-1 domain. They also demonstrate that this interaction is independent of PARP-1 activity and/or DNA binding. However, the published literature regarding PARP-1 and NF-κB interaction is somewhat controversial. Inhibition of the enzymatic activity of PARP has been shown not to affect NF-κB DNA binding, which indicates a lack of a direct role for the catalytic activity of PARP-1 in activating NF-κB (173), reinforcing the proposed interaction by Hassa et al. (177). On the other hand Chang and Alvarez-Gonzalez (178) report that direct PARP-1 interaction with NF-κB inhibits NF-κB from binding to its site and this inhibition is relieved by the auto-poly(ADP-ribosyl)ation of PARP-1. Taken together, the data demonstrate that PARP-1 may be a cofactor in the activation of NF-κB through its direct interaction with the transcription factor. However, the nature of this interaction is not yet defined and the question of whether this interaction could be stimulated or inhibited by PARP activity remains to be answered.
IX. CDP as a Transcriptional Repressor of the CXCL1 Gene
Although CBP and PARP are potential positive coactivators in CXCL1 transcription, the negative regulation of CXCL1 transcription is thus far attributed to CDP (10). CDP/Cux/Cut proteins are members of a unique family of homeoproteins that is conserved among higher eukaryotes and contain a Cut homeodomain as well as one or more “Cut repeat” DNA-binding domain(s) (179–181). The CDP, Cux, and Cut proteins contain three Cut repeats and a Cut homeodomain and are referred to as the classic CDP/Cux/Cut proteins. CDP, CCAAT displacement protein, was first identified in the testis of the sea urchin Psammechinus miliaris and later in mammals and was characterized as a transcriptional repressor. There are six evolutionarily conserved domains in mammalian and Drosophila CDP/Cut proteins: a region that forms a coiled-coil structure, three regions of ∼70 amino acids, three Cut repeats (CR1, CR2, and CR3), and a Cut homeodomain (HD) (179, 180).
The cut repeats are believed to function as specific DNA-binding domains, in addition to the HD. These DNA-binding domains enable CDP/Cut to bind to a wide range of DNA sequences including sequences related to CCAAT, ATCGAT, AT-rich matrix attachment regions, and Sp1-sites. Several other types of sequences diverging from the consensus sequences have been isolated as well by way of PCR-mediated site selection using GST-fusion proteins containing various CDP/Cut DNA-binding domains (180). These reports are indicative of the flexibility of the CDP/Cut proteins in choosing their DNA targets.
In vitro studies of CDP/Cut DNA-binding modes have shown several combinations of domains such as CR1CR2, CR1HD, CR2HD, CR3HD, and CR2CR3 implementing this function. However, in vivo experiments in mammalian cells display only two modes of DNA binding. The CCAAT-displacement activity involves primarily CR1 and CR2, whereas binding to ATCGAT involves CR3HD. CR1CR2 exhibits fast “on” and “off” rates, whereas CR3HD displays slow on and off rates. Thus, although CR1CR2 binds to DNA rapidly but only transiently, CR3HD makes a stable interaction with the DNA (179, 180).
The DNA-binding activity of CDP/Cut is regulated in a cell-cycle-dependent manner, where strong binding is observed in S phase as a result of both an increase in CDP/Cut expression and dephosphorylation of CDP/Cut by the Cdc25A phosphatase (180). In G2 phase, however, its DNA-binding activity is inhibited through cyclinA-Cdk1. Santaguida et al. (181) report that the cyclinA–Cdk1 complex activated through the action of the Cdk-activating kinase phosphorylates CDP/Cut on two serine residues around the Cut homeodomain and inhibits its DNA binding. Phosphorylation of conserved sites in the Cut repeats by PKC and CKII or pCAF-mediated acetylation of the Cut homeodomain has also been implicated in the inhibition of CDP/Cut DNA-binding activity (180, 182, 183). These data indicate that down-modulation of CDP/Cut activity may be very important for cell cycle progression in late S and in G2.
The exact mechanism of repression by CDP/Cut is unknown, however, two mechanisms have been proposed: passive repression in which CDP/Cut competes with activators for occupancy of binding sites, hence the name “CCAAT displacement protein,” and active repression involving direct interaction of CDP/Cut with HDAC1 (182). In support of the active repression theory, two active repression domains within the carboxy-terminal domain of CDP/Cut have been identified that are able to interact with HDAC1.
Mammalian CDP/Cut proteins have been shown to repress genes in proliferating precursor cells that are turned on as cells become terminally differentiated and CDP/Cut activity ceases. Thus, CDP/Cut proteins are transcriptional repressors that inhibit terminally differentiated gene expression during early stages of differentiation. CDP/Cut was shown to repress the p21/waf1 gene, a cyclin inhibitor, as well as bind to promoters of various histone genes, which are regulated in a cell-cycle-dependent manner (184). In accordance with this, CDP/Cut DNA-binding activity oscillates during the cell cycle and reaches a maximum at the end of G1 and during the S phase. Interestingly, CDP/Cut regulates histone genes positively, where the peak of expression of these genes coincides with or closely precedes the DNA replication phase (180, 183). Thus, the regulatory effect of CDP/Cut on transcription might vary depending on the proteins with which it interacts and that these CDP/Cut-interacting proteins may be cell and tissue specific.
However, there is limited insight into the function of CDP/Cut in mammals in vivo. To address this question, a few mutational studies have been conducted in which either the CR1 or the Cut HD was mutated. The pheno-types observed in CR1 homozygous mutant mice include curly vibrissae, wavy hair, and high postnatal lethality among the litters, whereas homozygous Cut HD mutants displayed high postnatal lethality, growth retardation, nearly complete hair loss, and severely reduced male fertility (182). Thus absence of fully functional CDP/Cut results only in limited disturbances of normal tissue development.
The repression activity of CDP/Cut has also been observed in non-cell-cycle-related genes such as human cholesterol-7 hydroxylase (CYP7A1), CXCL1, and mouse mammary tumor virus (MMTV) expression. CYP7A1 is expressed only in the liver and is regulated by several liver-enriched transcription factors. CDP/Cut was found to be a major negative regulator of the expression of this gene, where it mediates its repressor activity by displacing two hepatic transcriptional activators, hepatocyte nuclear factor-1 (HNF-1) and C/EBP, from their binding sites within intron 1 of the gene (185). The expression of MMTV in the mammary glands has also been shown to be repressed by CDP/Cut, since mutations in the putative CDP/Cut binding domain elevated reporter gene expression by six-fold, whereas overexpression of CDP/Cut was able to suppress reporter gene expression up to 20-fold (186). The same phenomenon was observed in the regulation of CXCL1 transcription by Nirodi et al. (105). The authors showed that CDP mediates its suppressive function by binding to the GCATCGATC sequence within the IUR element in the promoter of the CXCL1 gene. However, further investigation is needed to establish if the repression activity of CDP/Cut is due to its displacement activity or direct interaction with HDAC1 or both. It is possible that CDP/Cut exerts its transcriptional repression simply by displacing the coactivator PARP-1.
X. Reflection
Thus, based on the above discussion, alterations in the interactions of the CXCL1 enhanceosome proteins may be the cause for overexpression of this chemokine as seen in many malignancies and inflammatory diseases. Based on the available data, these alterations may affect the interactions of CDP with the promoter negatively, but have a positive effect on PARP and NF-κB. Thus, overexpression of CXCL1 may be attributed to a concomitant decrease/loss of CDP binding and enhanced PARP binding to the promoter as well as to NF-κB in melanomas. The question then becomes: How does PARP achieve enhanced interactions with the promoter and NF-κB, whereas CDP interaction is at a loss? To answer this question, it is necessary to explore the explicit role of PARP and CDP in the regulation of CXCL1 transcription in “normal” models that can be compared with melanoma models. The significance of these proteins in the regulation of CXCL1 transcription may be investigated through ablation of their expression as well as mutational studies. It is also important to establish the exact sequences that CDP and PARP bind within the IUR element and to look for any differences in these sequences between normal and melanoma models. The effect of PARP catalytic activity on NF-κB and IUR interaction should also be investigated, as it may be of great importance for therapeutic inventions. Thus, by altering the interaction of these proteins in the CXCL1 enhanceosome complex, we may reduce the inflammatory and tumor growth responses in tissues.
Acknowledgments
We are appreciative of the support for the work described herein from the Department of Veterans Affairs for a SRCS award to Ann Richmond and to the NIH for CA56704 and CA34590.
Footnotes
Abbreviations: IL, interleukin; TNF-α, tumor necrosis factor-α; NF-κB, nuclear factor-κB; IFN-γ, interferon-γ; JAK, Janus kinases; STAT, signal tranducer and activator of transcription proteins; TGF-β, transforming growth factor-β; GTP, guanosine triphosphate; GDP, guanosine diphosphate; PLC2, phospholipase Cβ2; IP3, inositol 1,4,5-triphosphate; PIP2, phosphatidylinositol 4,5-bisphosphate; PKC, protein kinase C; PI3K, phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein kinases; HIP, Hsp70 interacting protein; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1; CTAPIII, connective tissue-activating peptide; MS, multiple sclerosis; AP-1, activator protein-1; NF-IL6, nuclear factor activated by interleukin 6; IRF, IFN-regulatory factor; Sp1, stimulating protein-1; HMGI/Y, high mobility group-I/Y; C/EBP, CAAT enhancer binding protein; IUR, immediate upstream region; NRF, NF-κB repressing factor; CBP, cAMP response element binding protein (CREB)-binding protein; HAT, histone acetyltransferase; HDAC, histone deacetylase; CDP, CAAT displacement protein; PARP, poly(ADP-ribose) polymerase; IκB, inhibitor of κB; NLS, nuclear localization signal; IKK, IκB kinase; PKA, protein kinase A; CKII, casein kinase II; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PDK, 3-phosphoinositide-dependent protein kinase; PTEN, phosphatase and tensin homologue deleted on chromosome 10; NIK, NF-κB-inducing kinase; TRAF2, TNF receptor-associated factor 2; ERK1/2, extracellular signal-regulated kinase 1 and 2; Cdk, cyclin-dependent kinases; NAD+, β-nicotinamide adenine dinucleotide; BER, base excision repair; HD, Cut homeodomain; CYP7A1, human cholesterol-7 hydroxylase; HNF-1, hepatocyte nuclear factor-1.
References
- 1.Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic cytokines—CXC and CC chemokines. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- 2.Clark-Lewis I, Kim KS, Rajarathnam K, Gong JH, Dewald B, Moser B, Baggiolini M, Sykes BD. Structure-activity relationships of chemokines. J Leukoc Biol. 1995;57:703–711. doi: 10.1002/jlb.57.5.703. [DOI] [PubMed] [Google Scholar]
- 3.Clore GM, Appella E, Yamada M, Matsushima K, Gronenborn AM. Three-dimensional structure of interleukin 8 in solution. Biochemistry. 1990;29:1689–1696. doi: 10.1021/bi00459a004. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin ET, Weber IT, St. Charles R, Xuan JC, Appella E, Yamada M, Matsushima K, Edwards BF, Clore GM, Gronenborn AM, et al. Crystal structure of interleukin 8: Symbiosis of NMR and crystallography. Proc Natl Acad Sci USA. 1991;88:502–506. doi: 10.1073/pnas.88.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Marriott D, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem. 1995;270:27348–27357. doi: 10.1074/jbc.270.45.27348. [DOI] [PubMed] [Google Scholar]
- 6.Arenberg DA, Kunkel SL, Polverini PJ, Morris SB, Burdick MD, Glass MC, Taub DT, Iannettoni MD, Whyte RI, Strieter RM. Interferon-gamma-inducible protein 10 (IP-10) is an angiostatic factor that inhibits human non-small cell lung cancer (NSCLC) tumorigenesis and spontaneous metastases. J Exp Med. 1996;184:981–992. doi: 10.1084/jem.184.3.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beg AA, Finco TS, Nantermet PV, Baldwin AS., Jr Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: A mechanism for NF-kappa B activation. Mol Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kotenko SV, Pestka S. Jak-Stat signal transduction pathway through the eyes of cytokine class II receptor complexes. Oncogene. 2000;19:2557–2565. doi: 10.1038/sj.onc.1203524. [DOI] [PubMed] [Google Scholar]
- 9.Richmond A, Luan J, Du J, Haghnegahdar H. The role of ELR+−CXC chemokines in wound healing and melanoma biology. In: Herbert CA, editor. Chemokines in Disease: Biology and Clinical Research. Humana Press Inc; Totowa, NJ: 1999. pp. 191–214. [Google Scholar]
- 10.Ahuja SK, Murphy PM. The CXC chemokines growth-regulated oncogene (GRO) alpha, GRObeta, GROgamma, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the type A, human interleukin-8 receptor. J Biol Chem. 1996;271:20545–20550. doi: 10.1074/jbc.271.34.20545. [DOI] [PubMed] [Google Scholar]
- 11.Combadiere C, Ahuja SK, Tiffany HL, Murphy PM. Cloning and functional expression of CC CKR5, a human monocyte CC chemokine receptor selective for MIP-1 (alpha), MIP-1(beta), and RANTES. J Leukoc Biol. 1996;60:147–152. doi: 10.1002/jlb.60.1.147. [DOI] [PubMed] [Google Scholar]
- 12.Balkwill F. The molecular and cellular biology of the chemokines. J Viral Hepat. 1998;5:1–14. doi: 10.1046/j.1365-2893.1998.00081.x. [DOI] [PubMed] [Google Scholar]
- 13.Murdoch C, Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 2000;95:3032–3043. [PubMed] [Google Scholar]
- 14.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283:R7–28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 15.Neote K, DiGregorio D, Mak JY, Horuk R, Schall TJ. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell. 1993;72:415–425. doi: 10.1016/0092-8674(93)90118-a. [DOI] [PubMed] [Google Scholar]
- 16.Luster AD. Chemokines—chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 17.Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- 18.Ali H, Richardson RM, Haribabu B, Snyderman R. Chemoattractant receptor cross-desensitization. J Biol Chem. 1999;274:6027–6030. doi: 10.1074/jbc.274.10.6027. [DOI] [PubMed] [Google Scholar]
- 19.Le Y, Li B, Gong W, Shen W, Hu J, Dunlop NM, Oppenheim JJ, Wang JM. Novel pathophysiological role of classical chemotactic peptide receptors and their communications with chemokine receptors. Immunol Rev. 2000;177:185–194. doi: 10.1034/j.1600-065x.2000.17704.x. [DOI] [PubMed] [Google Scholar]
- 20.Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida T, Imai T, Kakizaki M, Nishimura M, Takagi S, Yoshie O. Identification of single C motif-1/lymphotactin receptor XCR1. J Biol Chem. 1998;273:16551–16554. doi: 10.1074/jbc.273.26.16551. [DOI] [PubMed] [Google Scholar]
- 22.Szabo MC, Soo KS, Zlotnik A, Schall TJ. Chemokine class differences in binding to the Duffy antigen-erythrocyte chemokine receptor. J Biol Chem. 1995;270:25348–25351. doi: 10.1074/jbc.270.43.25348. [DOI] [PubMed] [Google Scholar]
- 23.Devalaraja RM, Nanney LB, Du J, Qian Q, Yu Y, Devalaraja MN, Richmond A. Delayed wound healing in CXCR2 knockout mice. J Invest Dermatol. 2000;115:234–244. doi: 10.1046/j.1523-1747.2000.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haghnegahdar H, Du J, Wang D, Strieter RM, Burdick MD, Nanney LB, Cardwell N, Luan J, Shattuck-Brandt R, Richmond A. The tumorigenic and angiogenic effects of MGSA/GRO proteins in melanoma. J Leukoc Biol. 2000;67:53–62. doi: 10.1002/jlb.67.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Owen JD, Strieter R, Burdick M, Haghnegahdar H, Nanney L, Shattuck-Brandt R, Richmond A. Enhanced tumor-forming capacity for immortalized melanocytes expressing melanoma growth stimulatory activity / growth-regulated cytokine beta and gamma proteins. Int J Cancer. 1997;73:94–103. doi: 10.1002/(sici)1097-0215(19970926)73:1<94::aid-ijc15>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 26.Bokoch GM. Chemoattractant signaling and leukocyte activation. Blood. 1995;86:1649–1660. [PubMed] [Google Scholar]
- 27.Wu D, LaRosa GJ, Simon MI. G protein-coupled signal transduction pathways for interleukin-8. Science. 1993;261:101–103. doi: 10.1126/science.8316840. [DOI] [PubMed] [Google Scholar]
- 28.Bokoch GM, Vlahos CJ, Wang Y, Knaus UG, Traynor-Kaplan AE. Rac GTPase interacts specifically with phosphatidylinositol 3-kinase. Biochem J. 1996;315:775–779. doi: 10.1042/bj3150775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turner SJ, Domin J, Waterfield MD, Ward SG, Westwick J. The CC chemokine monocyte chemotactic peptide-1 activates both the class I p85/p110 phosphatidylinositol 3-kinase and the class II PI3K-C2alpha. J Biol Chem. 1998;273:25987–25995. doi: 10.1074/jbc.273.40.25987. [DOI] [PubMed] [Google Scholar]
- 30.Huang R, Lian JP, Robinson D, Badwey JA. Neutrophils stimulated with a variety of chemoattractants exhibit rapid activation of p21-activated kinases (Paks): Separate signals are required for activation and inactivation of paks. Mol Cell Biol. 1998;18:7130–7138. doi: 10.1128/mcb.18.12.7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mellado M, Rodriguez-Frade JM, Aragay A, del Real G, Martin AM, Vila-Coro AJ, Serrano A, Mayor F, Jr, Martinez-A C. The chemokine monocyte chemotactic protein 1 triggers Janus kinase 2 activation and tyrosine phosphorylation of the CCR2B receptor. J Immunol. 1998;161:805–813. [PubMed] [Google Scholar]
- 32.Brill A, Hershkoviz R, Vaday GG, Chowers Y, Lider O. Augmentation of RANTES-induced extracellular signal-regulated kinase mediated signaling and T cell adhesion by elastase-treated fibronectin. J Immunol. 2001;166:7121–7127. doi: 10.4049/jimmunol.166.12.7121. [DOI] [PubMed] [Google Scholar]
- 33.Wain JH, Kirby JA, Ali S. Leucocyte chemotaxis: Examination of mitogen-activated protein kinase and phosphoinositide 3-kinase activation by monocyte chemoattractant proteins-1, -2, -3 and -4. Clin Exp Immunol. 2002;127:436–444. doi: 10.1046/j.1365-2249.2002.01764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aramori I, Ferguson SS, Bieniasz PD, Zhang J, Cullen B, Cullen MG. Molecular mechanism of desensitization of the chemokine receptor CCR-5: Receptor signaling and internalization are dissociable from its role as an HIV-1 co-receptor. EMBO J. 1997;16:4606–4616. doi: 10.1093/emboj/16.15.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richmond A, Mueller S, White JR, Schraw W. C-X-C chemokine receptor desensitization mediated through ligand-enhanced receptor phosphorylation on serine residues. Methods Enzymol. 1997;288:3–15. doi: 10.1016/s0076-6879(97)88003-2. [DOI] [PubMed] [Google Scholar]
- 36.Fan GH, Yang W, Sai J, Richmond A. Hsc/Hsp70 interacting protein (hip) associates with CXCR2 and regulates the receptor signaling and trafficking. J Biol Chem. 2002;277:6590–6597. doi: 10.1074/jbc.M110588200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imhof BA. Leukocyte migration and adhesion. Adv Immunol. 1995;58:345–416. doi: 10.1016/s0065-2776(08)60623-9. [DOI] [PubMed] [Google Scholar]
- 38.Brandt E, Petersen F, Ludwig A, Ehlert JE, Bock L, Flad HD. The beta-thromboglobulins and platelet factor 4: Blood platelet-derived CXC chemokines with divergent roles in early neutrophil regulation. J Leukoc Biol. 2000;67:471–478. doi: 10.1002/jlb.67.4.471. [DOI] [PubMed] [Google Scholar]
- 39.Engelhardt E, Toksoy A, Goebeler M, Debus S, Brocker EB, Gillitzer R. Chemokines IL-8, GROalpha, MCP-1, IP-10, and Mig are sequentially and differentially expressed during phase-specific infiltration of leukocyte subsets in human wound healing. Am J Pathol. 1998;153:1849–1860. doi: 10.1016/s0002-9440(10)65699-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gillitzer R, Goebeler M. Chemokines in cutaneous wound healing. J Leukoc Biol. 2001;69:513–521. [PubMed] [Google Scholar]
- 41.Gibran NS, Ferguson M, Heimbach DM, Isik FF. Monocyte chemoattractant protein-1 mRNA expression in the human burn wound. J Surg Res. 1997;70:1–6. doi: 10.1006/jsre.1997.5017. [DOI] [PubMed] [Google Scholar]
- 42.Trautmann A, Toksoy A, Engelhardt E, Brocker EB, Gillitzer R. Mast cell involvement in normal human skin wound healing: Expression of monocyte chemoattractant protein-1 is correlated with recruitment of mast cells which synthesize interleukin-4 in vivo. J Pathol. 2000;190:100–1066. doi: 10.1002/(SICI)1096-9896(200001)190:1<100::AID-PATH496>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 43.Goede V, Brogelli L, Ziche M, Augustin HG. Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int J Cancer. 1999;82:765–770. doi: 10.1002/(sici)1097-0215(19990827)82:5<765::aid-ijc23>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 44.Weber KS, Nelson PJ, Grone HJ, Weber C. Expression of CCR2 by endothelial cells: Implications for MCP-1 mediated wound injury repair and in vivo inflammatory activation of endothelium. Arterioscler Thromb Vasc Biol. 1999;19:2085–2093. doi: 10.1161/01.atv.19.9.2085. [DOI] [PubMed] [Google Scholar]
- 45.Leonard EJ, Skeel A, Yoshimura T, Rankin J. Secretion of monocyte chemoattractant protein-1 (MCP-1) by human mononuclear phagocytes. Adv Exp Med Biol. 1993;351:55–64. doi: 10.1007/978-1-4615-2952-1_7. [DOI] [PubMed] [Google Scholar]
- 46.Trautmann A, Krohne G, Brocker EB, Klein CE. Human mast cells augment fibroblast proliferation by heterotypic cell-cell adhesion and action of IL-4. J Immunol. 1998;160:5053–5057. [PubMed] [Google Scholar]
- 47.Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- 48.Ransohoff RM. Chemokines and chemokine receptors in model neurological pathologies: Molecular and immunocytochemical approaches. Methods Enzymol. 1997;287:319–348. doi: 10.1016/s0076-6879(97)87023-1. [DOI] [PubMed] [Google Scholar]
- 49.Karpus WJ, Kennedy KJ. MIP-1 and MCP-1 differentially regulate acute and relapsing autoimmune encephalomyelitis as well as Th1/Th2 lymphocyte differentiation. J Leukoc Biol. 1997;62:681–687. [PubMed] [Google Scholar]
- 50.Glabinski AR, Tani M, Tuohy VK, Tuthill RJ, Ransohoff RM. Central nervous system chemokine mRNA accumulation follows initial leukocyte entry at the onset of acute murine experimental autoimmune encephalomyelitis. Brain Behav Immunol. 1995;9:315–330. doi: 10.1006/brbi.1995.1030. [DOI] [PubMed] [Google Scholar]
- 51.Karpus WJ, Lukacs NW, McRae BL, Strieter RM, Kunkel SL, Miller SD. An important role for the chemokine macrophage inflammatory protein-1 in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol. 1995;155:5003–5010. [PubMed] [Google Scholar]
- 52.Godiska R, Chantry D, Dietsch GN, Gray PW. Chemokine expression in murine experimental allergic encephalomyelitis. J Neuroimmunol. 1995;58:167–176. doi: 10.1016/0165-5728(95)00008-p. [DOI] [PubMed] [Google Scholar]
- 53.Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, Estes ML, Thomas DM, Tuohy VK. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J. 1993;7:592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]
- 54.Simpson J, Rezaie P, Newcombe J, Cuzner ML, Male D, Woodroofe MN. Expression of the chemokine receptors CCR2, CCR3, and CCR5 in multiple sclerosis central nervous system tissue. J Neuroimmunol. 2000;108:192–200. doi: 10.1016/s0165-5728(00)00274-5. [DOI] [PubMed] [Google Scholar]
- 55.Standiford TJ, Strieter RM, Lukacs NW, Kunkel SL. Neutralization of IL-10 increases lethality in endotoxemia. Cooperative effects of macrophage inflammatory protein-2 and tumor necrosis factor. J Immunol. 1995;155:2222–2229. [PubMed] [Google Scholar]
- 56.Standiford TJ, Kunkel SL, Lukacs NW, Greenberger MJ, Danforth JM, Kunkel RG, Strieter RM. Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak, and early mortality in murine endotoxemia. J Immunol. 1995;155:1515–1524. [PubMed] [Google Scholar]
- 57.VanOtteren GM, Strieter RM, Kunkel SL, Paine R, 3rd, Greenberger MJ, Danforth JM, Burdick MD, Standiford TJ. Compartmentalized expression of RANTES in a murine model of endotoxemia. J Immunol. 1995;154:1900–1908. [PubMed] [Google Scholar]
- 58.Zisman DA, Kunkel SL, Strieter RM, Tsai WC, Bucknell K, Wilkowski J, Standiford TJ. MCP-1 protects mice in lethal endotoxemia. J Clin Invest. 1997;99:2832–2836. doi: 10.1172/JCI119475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 60.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 61.Gao JL, Murphy PM. Human cytomegalovirus open reading frame US28 encodes a functional beta chemokine receptor. J Biol Chem. 1994;269:28539–28542. [PubMed] [Google Scholar]
- 62.Lukacs NW, Kunkel SL. Chemokines and their role in disease. Int J Clin Lab Res. 1998;28:91–95. doi: 10.1007/s005990050025. [DOI] [PubMed] [Google Scholar]
- 63.Melter M, McMahon G, Fang J, Ganz P, Briscoe DM. Current understanding of chemokine involvement in allograft transplantation. Pediatr Transplant. 1999;3:10–21. doi: 10.1034/j.1399-3046.1999.00023.x. [DOI] [PubMed] [Google Scholar]
- 64.Yun JJ, Fischbein MP, Laks H, Fishbein MC, Espejo ML, Ebrahimi K, Irie Y, Berliner J, Ardehali A. Early and late chemokine production correlates with cellular recruitment in cardiac allograft vasculopathy. Transplantation. 2000;69:2515–2524. doi: 10.1097/00007890-200006270-00009. [DOI] [PubMed] [Google Scholar]
- 65.Belperio JA, Burdick MD, Keane MP, Xue YY, Lynch JP, 3rd, Daugherty BL, Kunkel SL, Strieter RM. The role of the CC chemokine, Rantes, in acute lung allograft rejection. J Immunol. 2000;165:461–472. doi: 10.4049/jimmunol.165.1.461. [DOI] [PubMed] [Google Scholar]
- 66.Kapoor A, Morita K, Engeman TM, Vapnek EM, Hobart M, Novick AC, Fairchild RL. Intragraft expression of chemokine gene occurs early during acute rejection of allogeneic cardiac grafts. Transplant Proc. 2000;32:793–795. doi: 10.1016/s0041-1345(00)00985-4. [DOI] [PubMed] [Google Scholar]
- 67.Schadendorf D, Fichtner I, Makki A, Alijagic S, Kupper M, Mrowietz U, Henz BM. Metastatic potential of human melanoma cells in nude mice—characterisation of phenotype, cytokine secretion and tumour-associated antigens. Br J Cancer. 1996;74:194–199. doi: 10.1038/bjc.1996.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh RK, Varney ML, Bucana CD, Johansson SL. Expression of interleukin-8 in primary and metastatic malignant melanoma of the skin. Melanoma Res. 1999;9:383–387. doi: 10.1097/00008390-199908000-00007. [DOI] [PubMed] [Google Scholar]
- 69.Scheibenbogen C, Mohler T, Haefele J, Hunstein W, Keilholz U. Serum interleukin-8 (IL-8) is elevated in patients with metastatic melanoma and correlates with tumour load. Melanoma Res. 1995;5:179–181. doi: 10.1097/00008390-199506000-00006. [DOI] [PubMed] [Google Scholar]
- 70.Takamori H, Oades ZG, Hoch OC, Burger M, Schraufstatter IU. Autocrine growth effect of IL-8 and GROalpha on a human pancreatic cancer cell line, Capan-1. Pancreas. 2000;21:52–56. doi: 10.1097/00006676-200007000-00051. [DOI] [PubMed] [Google Scholar]
- 71.Richards BL, Eisma RJ, Spiro JD, Lindquist RL, Kreutzer DL. Coexpression of interleukin-8 receptors in head and neck squamous cell carcinoma. Am J Surg. 1997;174:507–512. doi: 10.1016/s0002-9610(97)00165-7. [DOI] [PubMed] [Google Scholar]
- 72.Olbina G, Cieslak D, Ruzdijic S, Esler C, An Z, Wang X, Hoffman R, Seifert W, Pietrzkowski Z. Reversible inhibition of IL-8 receptor B mRNA expression and proliferation in non-small cell lung cancer by antisense oligonucleotides. Anticancer Res. 1996;16:3525–3530. [PubMed] [Google Scholar]
- 73.Wakabayashi H, Cavanaugh PG, Nicolson GL. Purification and identification of mouse lung microvessel endothelial cell-derived chemoattractant for lung-metastasizing murine RAW117 large-cell lymphoma cells: Identification as mouse monocyte chemotactic protein 1. Cancer Res. 1995;55:4458–4464. [PubMed] [Google Scholar]
- 74.Youngs SJ, Ali SA, Taub DD, Rees RC. Chemokines induce migrational responses in human breast carcinoma cell lines. Int J Cancer. 1997;71:257–266. doi: 10.1002/(sici)1097-0215(19970410)71:2<257::aid-ijc22>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 75.Wang JM, Deng X, Gong W, Su S. Chemokines and their role in tumor growth and metastasis. J Immunol Methods. 1998;220:1–17. doi: 10.1016/s0022-1759(98)00128-8. [DOI] [PubMed] [Google Scholar]
- 76.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 77.Cogswell JP, Godlevski MM, Wisely GB, Clay WC, Leesnitzer LM, Ways JP, Gray JG. NF-kappa B regulates IL-1 beta transcription through a consensus NF-kappa B binding site and a nonconsensus CRE-like site. J Immunol. 1994;153:712–723. [PubMed] [Google Scholar]
- 78.Roebuck KA, Carpenter LR, Lakshminarayanan V, Page SM, Moy JN, Thomas LL. Stimulus-specific regulation of chemokine expression involves differential activation of the redox-responsive transcription factors AP-1 and NF-kappaB. J Leukoc Biol. 1999;65:291–298. doi: 10.1002/jlb.65.3.291. [DOI] [PubMed] [Google Scholar]
- 79.Stylianou E, Nie M, Ueda A, Zhao L. c-Rel and p65 trans-activate the monocyte chemoattractant protein-1 gene in interleukin-1 stimulated mesangial cells. Kidney Int. 1999;56:873–882. doi: 10.1046/j.1523-1755.1999.00640.x. [DOI] [PubMed] [Google Scholar]
- 80.Shi MM, Chong I, Godleski JJ, Paulauskis JD. Regulation of macrophage inflammatory protein-2 gene expression by oxidative stress in rat alveolar macrophages. Immunology. 1999;97:309–315. doi: 10.1046/j.1365-2567.1999.00798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takaya H, Andoh A, Shimada M, Hata K, Fujiyama Y, Bamba T. The expression of chemokine genes correlates with nuclear factor-kappaB activation in human pancreatic cancer cell lines. Pancreas. 2000;21:32–40. doi: 10.1097/00006676-200007000-00049. [DOI] [PubMed] [Google Scholar]
- 82.Merola M, Blanchard B, Tovey MG. The kappa B enhancer of the human interleukin-6 promoter is necessary and sufficient to confer an IL-1 beta and TNF-alpha response in transfected human cell lines: Requirement for members of the C/EBP family for activity. J Interferon Cytokine Res. 1996;16:783–798. doi: 10.1089/jir.1996.16.783. [DOI] [PubMed] [Google Scholar]
- 83.Garcia GE, Xia Y, Chen S, Wang Y, Ye RD, Harrison JK, Bacon KB, Zerwes HG, Feng L. NF-kappaB-dependent fractalkine induction in rat aortic endothelial cells stimulated by IL-1beta, TNF-alpha, and LPS. J Leukoc Biol. 2000;67:577–584. doi: 10.1002/jlb.67.4.577. [DOI] [PubMed] [Google Scholar]
- 84.Sugita S, Kohno T, Yamamoto K, Imaizumi Y, Nakajima H, Ishimaru T, Matsuyama T. Induction of macrophage-inflammatory protein-3alpha gene expression by TNF-dependent NF-kappaB activation. J Immunol. 2002;168:5621–5628. doi: 10.4049/jimmunol.168.11.5621. [DOI] [PubMed] [Google Scholar]
- 85.Mastronarde JG, He B, Monick MM, Mukaida N, Matsushima K, Hunninghake GW. Induction of interleukin (IL)-8 gene expression by respiratory syncytial virus involves activation of nuclear factor (NF)-kappa B and NF-IL-6. J Infect Dis. 1996;174:262–267. doi: 10.1093/infdis/174.2.262. [DOI] [PubMed] [Google Scholar]
- 86.Akira S, Kishimoto T. IL-6 and NF-IL6 in acute-phase response and viral infection. Immunol Rev. 1992;127:25–50. doi: 10.1111/j.1600-065x.1992.tb01407.x. [DOI] [PubMed] [Google Scholar]
- 87.Casola A, Garofalo RP, Haeberle H, Elliott TF, Lin R, Jamaluddin M, Brasier AR. Multiple cis regulatory elements control RANTES promoter activity in alveolar epithelial cells infected with respiratory syncytial virus. J Virol. 2001;75:6428–6439. doi: 10.1128/JVI.75.14.6428-6439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee AH, Hong JH, Seo YS. Tumour necrosis factor-alpha and interferon-gamma synergistically activate the RANTES promoter through nuclear factor kappaB and interferon regulatory factor 1 (IRF-1) transcription factors. Biochem J. 2000;350:131–138. [PMC free article] [PubMed] [Google Scholar]
- 89.Genin P, Algarte M, Roof P, Lin R, Hiscott J. Regulation of RANTES chemokine gene expression requires cooperativity between NF-kappa B and IFN-regulatory factor transcription factors. J Immunol. 2000;164:5352–5361. doi: 10.4049/jimmunol.164.10.5352. [DOI] [PubMed] [Google Scholar]
- 90.Wood LD, Farmer AA, Richmond A. HMGI(Y) and Sp1 in addition to NF-kappa B regulate transcription of the MGSA/GRO alpha gene. Nucleic Acids Res. 1995;23:4210–4219. doi: 10.1093/nar/23.20.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, Fukushima J, Kawamoto S, Ishigatsubo Y, Okubo T. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol. 1994;153:2052–2063. [PubMed] [Google Scholar]
- 92.Schook LB, Albrecht H, Gallay P, Jongeneel CV. Cytokine regulation of TNF-alpha mRNA and protein production by unprimed macrophages from C57B1/6 and NZW mice. J Leukoc Biol. 1994;56:514–520. doi: 10.1002/jlb.56.4.514. [DOI] [PubMed] [Google Scholar]
- 93.Lin G, Pearson AE, Scamurra RW, Zhou Y, Baarsch MJ, Weiss DJ, Murtaugh MP. Regulation of interleukin-8 expression in porcine alveolar macrophages by bacterial lipopolysaccharide. J Biol Chem. 1994;269:77–85. [PubMed] [Google Scholar]
- 94.Villarete LH, Remick DG. Transcriptional and post-transcriptional regulation of interleukin-8. Am J Pathol. 1996;149:1685–1693. [PMC free article] [PubMed] [Google Scholar]
- 95.Wolf JS, Chen Z, Dong G, Sunwoo JB, Bancroft CC, Capo DE, Yeh NT, Mukaida N, Van Waes C. IL (interleukin)-1alpha promotes nuclear factor-kappaB and AP-1-induced IL-8 expression, cell survival, and proliferation in head and neck squamous cell carcinomas. Clin Cancer Res. 2001;7:1812–1820. [PubMed] [Google Scholar]
- 96.Wu GD, Lai EJ, Huang N, Wen X. Oct-1 and CCAAT/enhancer-binding protein (C/EBP) bind to overlapping elements within the interleukin-8 promoter. The role of Oct-1 as a transcriptional repressor. J Biol Chem. 1997;272:2396–2403. [PubMed] [Google Scholar]
- 97.Roux P, Alfieri C, Hrimech M, Cohen EA, Tanner JE. Activation of transcription factors NF-kappa B and NF-IL-6 by human immunodeficiency virus type 1 protein R (Vpr) induces interleukin-8 expression. J Virol. 2000;74:4658–4665. doi: 10.1128/jvi.74.10.4658-4665.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Luan J, Shattuck-Brandt R, Haghnegahdar H, Owen JD, Strieter R, Burdick M, Nirodi C, Beauchamp D, Johnson KN, Richmond A. Mechanism and biological significance of constitutive expression of MGSA/GRO chemokines in malignant melanoma tumor progression. J Leukoc Biol. 1997;62:588–597. doi: 10.1002/jlb.62.5.588. [DOI] [PubMed] [Google Scholar]
- 99.Wood LD, Richmond A. Constitutive and cytokine-induced expression of the melanoma growth stimulatory activity/GRO gene requires both NF-B and novel constitutive factors. J Biol Chem. 1995;270:30619–30626. doi: 10.1074/jbc.270.51.30619. [DOI] [PubMed] [Google Scholar]
- 100.Mukaida N, Okamoto S, Ishikawa Y, Matsushima K. Molecular mechanism of interleukin-8 gene expression. J Leukoc Biol. 1994;56:554–558. [PubMed] [Google Scholar]
- 101.Roger T, Out T, Mukaida N, Matsushima K, Jansen H, Lutter R. Enhanced AP-1 and NF-kappaB activities and stability of interleukin 8 (IL-8) transcripts are implicated in IL-8 mRNA superinduction in lung epithelial H292 cells. Biochem J. 1998;330:429–435. doi: 10.1042/bj3300429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Matsusaka T, Fujikawa K, Nishio Y, Mukaida N, Matsushima K, Kishimoto T, Akira S. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc Natl Acad Sci USA. 1993;90:10193–10197. doi: 10.1073/pnas.90.21.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nourbakhsh M, Kalble S, Dorrie A, Hauser H, Resch K, Kracht M. The NF-kappa b repressing factor is involved in basal repression and interleukin (IL)-1-induced activation of IL-8 transcription by binding to a conserved NF-kappa b-flanking sequence element. J Biol Chem. 2001;276:4501–4508. doi: 10.1074/jbc.M007532200. [DOI] [PubMed] [Google Scholar]
- 104.Vanden Berghe W, De Bosscher K, Boone E, Plaisance S, Haegeman G. The nuclear factor-kappaB engages CBP/p300 and histone acetyltransferase activity for transcriptional activation of the interleukin-6 gene promoter. J Biol Chem. 1999;274:32091–32098. doi: 10.1074/jbc.274.45.32091. [DOI] [PubMed] [Google Scholar]
- 105.Nirodi C, Hart J, Dhawan P, Nepveu A, Richmond A. The role of CDP in the negative regulation of CXCL1 gene expression. J Biol Chem. 2001;276:26122–26131. doi: 10.1074/jbc.M102872200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nirodi C, NagDas S, Gygi SP, Olson G, Aebersold R, Richmond A. A role for poly(ADP-ribose) polymerase in the transcriptional regulation of the melanoma growth stimulatory activity (CXCL1) gene expression. J Biol Chem. 2001;276:9366–9374. doi: 10.1074/jbc.M009897200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Abraham E. NF-κB activation. Crit Care Med. 2000;28:N100–N104. doi: 10.1097/00003246-200004001-00012. [DOI] [PubMed] [Google Scholar]
- 108.Makarov SS. NF-kappaB as a therapeutic target in chronic inflammation: Recent advances. Mol Med Today. 2000;6:441–448. doi: 10.1016/s1357-4310(00)01814-1. [DOI] [PubMed] [Google Scholar]
- 109.Huang S, DeGuzman A, Bucana CD, Fidler IJ. Nuclear factor-kappaB activity correlates with growth, angiogenesis, and metastasis of human melanoma cells in nude mice. Clin Cancer Res. 2000;6:2573–2581. [PubMed] [Google Scholar]
- 110.Beg AA, Ruben SM, Scheinman RI, Haskill S, Rosen CA, Baldwin AS., Jr I kappa B interacts with the nuclear localization sequences of the subunits of NF-kappa B: A mechanism for cytoplasmic retention. Genes Dev. 1992;6:1899–1913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- 111.DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nolan GP, Fujita T, Bhatia K, Huppi C, Liou HC, Scott ML, Baltimore D. The bcl-3 proto-oncogene encodes a nuclear I kappa B-like molecule that preferentially interacts with NF-kappa B p50 and p52 in a phosphorylation-dependent manner. Mol Cell Biol. 1993;13:3557–3566. doi: 10.1128/mcb.13.6.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 114.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 115.Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 117.Bakker TR, Reed D, Renno T, Jongeneel CV. Efficient adenoviral transfer of NF-kappaB inhibitor sensitizes melanoma to tumor necrosis factor-mediated apoptosis. Int J Cancer. 1999;80:320–323. doi: 10.1002/(sici)1097-0215(19990118)80:2<320::aid-ijc24>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 118.Wang D, Yang W, Du J, Devalaraja MN, Liang P, Matsumoto K, Tsubakimoto K, Endo T, Richmond A. MGSA/GRO-mediated melanocyte transformation involves induction of Ras expression. Oncogene. 2000;19:4647–4659. doi: 10.1038/sj.onc.1203820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 120.Wang D, Westerheide SD, Hanson JL, Baldwin AS., Jr Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem. 2000;275:32592–32597. doi: 10.1074/jbc.M001358200. [DOI] [PubMed] [Google Scholar]
- 121.Berra E, Diaz-Meco MT, Lozano J, Frutos S, Municio MM, Sanchez P, Sanz L, Moscat J. Evidence for a role of MEK and MAPK during signal transduction by protein kinase C zeta. EMBO J. 1995;14:6157–6163. doi: 10.1002/j.1460-2075.1995.tb00306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vanden Berghe W, Plaisance S, Boone E, De Bosscher K, Schmitz ML, Fiers W, Haegeman G. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998;273:3285–3290. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]
- 123.Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90:1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 124.Datta SR, Brunet A, Greenberg ME. Cellular survival: A play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 125.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 126.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 127.Madrid LV, Mayo MW, Reuther JY, Baldwin AS., Jr Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem. 2001;276:18934–18940. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 128.Li J, Simpson L, Takahashi M, Miliaresis C, Myers MP, Tonks N, Parsons R. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res. 1998;58:5667–5672. [PubMed] [Google Scholar]
- 129.Guldberg P, Thor Straten P, Birck A, Ahrenkiel V, Kirkin AF, Zeuthen J. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res. 1997;57:3660–3663. [PubMed] [Google Scholar]
- 130.Gomez J, Martinez-A C, Fernandez B, Garcia A, Rebollo A. Critical role of Ras in the proliferation and prevention of apoptosis mediated by IL-2. J Immunol. 1996;157:2272–2281. [PubMed] [Google Scholar]
- 131.Joneson T, Bar-Sagi D. Suppression of Ras-induced apoptosis by the Rac GTPase. Mol Cell Biol. 1999;19:5892–5901. doi: 10.1128/mcb.19.9.5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Crespo P, Leon J. Ras proteins in the control of the cell cycle and cell differentiation. Cell Mol Life Sci. 2000;57:1613–1636. doi: 10.1007/PL00000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Khosravi-Far R, Solski PA, Clark GJ, Kinch MS, Der CJ. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol Cell Biol. 1995;15:6443–6453. doi: 10.1128/mcb.15.11.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Norris JL, Baldwin AS., Jr Oncogenic Ras enhances NF-kappaB transcriptional activity through Raf-dependent and Raf-independent mitogen-activated protein kinase signaling pathways. J Biol Chem. 1999;274:13841–13846. doi: 10.1074/jbc.274.20.13841. [DOI] [PubMed] [Google Scholar]
- 135.Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 136.Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV. IκB kinase-beta: NF-κB activation and complex formation with IκB kinase-α and NIK. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 137.Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284:309–313. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- 138.Dhawan P, Richmond A. A novel NF-κB-inducing kinase-MAPK signaling pathway up-regulates NF-κB activity in melanoma cells. J Biol Chem. 2002;277:7920–7928. doi: 10.1074/jbc.M112210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–372. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- 141.Michael LF, Asahara H, Shulman AI, Kraus WL, Montminy M. The phosphorylation status of a cyclic AMP-responsive activator is modulated via a chromatin-dependent mechanism. Mol Cell Biol. 2000;20:1596–1603. doi: 10.1128/mcb.20.5.1596-1603.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yuan LW, Gambee JE. Histone acetylation by p300 is involved in CREB-mediated transcription on chromatin. Biochim Biophys Acta. 2001;1541:161–169. doi: 10.1016/s0167-4889(01)00141-0. [DOI] [PubMed] [Google Scholar]
- 143.Lodish H, Berk A, Zipursky LS, Matsudaira P, Baltimore D, Darnell JE. Molecular Cell Biology. 4th. Freeman W. H. & Co.; New York: 1999. Role of deacetylation and hyperacetylation of histone N-terminal tails in yeast transcription control; pp. 384–390. [Google Scholar]
- 144.Nakajima T, Uchida C, Anderson SF, Lee CG, Hurwitz J, Parvin JD, Montminy M. RNA helicase A mediates association of CBP with RNA polymerase II. Cell. 1997;90:1107–1112. doi: 10.1016/s0092-8674(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 145.Bannister AJ, Oehler T, Wilhelm D, Angel P, Kouzarides T. Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene. 1995;11:2509–2514. [PubMed] [Google Scholar]
- 146.Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–2373. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- 147.Shenkar R, Yum HK, Arcaroli J, Kupfner J, Abraham E. Interactions between CBP, NF-kappaB, and CREB in the lungs after hemorrhage and endotoxemia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L418–426. doi: 10.1152/ajplung.2001.281.2.L418. [DOI] [PubMed] [Google Scholar]
- 148.Imhof A, Yang XJ, Ogryzko VV, Nakatani Y, Wolfe AP, Ge H. Acetylation of general transcription factors by histone acetyltransferases. Curr Biol. 1997;7:689–692. doi: 10.1016/s0960-9822(06)00296-x. [DOI] [PubMed] [Google Scholar]
- 149.Bannister A, Konzarides T. The CBP co-activator is a histone acetyl transferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 150.Felzien LK, Farrell S, Betts JC, Mosavin R, Nabel GJ. Specificity of cyclin E-Cdk2, TFIIB, and E1A interactions with a common domain of the p300 coactivator. Mol Cell Biol. 1999;19:4241–4246. doi: 10.1128/mcb.19.6.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Boyes J, Byfield P, Nakatani Y, Ogryzko V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature. 1998;396:594–598. doi: 10.1038/25166. [DOI] [PubMed] [Google Scholar]
- 152.Parry GC, Mackman N. Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J Immunol. 1997;1:5450–5456. [PubMed] [Google Scholar]
- 153.Ollivier V, Parry GC, Cobb RR, de Prost D, Mackman N. Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. J Biol Chem. 1996;271:20828–20835. doi: 10.1074/jbc.271.34.20828. [DOI] [PubMed] [Google Scholar]
- 154.Chen LF, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 155.Smulson ME, Simbulan-Rosenthal CM, Boulares AH, Yakovlev A, Stoica B, Iyer S, Luo R, Haddad B, Wang ZQ, Pang T, Jung M, Dritschilo A, Rosenthal DS. Roles of poly(ADP-ribosyl)ation and PARP in apoptosis, DNA repair, genomic stability and functions of p53 and E2F-1. Adv Enzyme Regul. 2000;40:183–215. doi: 10.1016/s0065-2571(99)00024-2. [DOI] [PubMed] [Google Scholar]
- 156.Alvarez-Gonzalez R, Spring H, Muller M, Burkle A. Selective loss of poly(ADP-ribose) and the 85-kDa fragment of poly(ADP-ribose) polymerase in nucleoli during alkylation-induced apoptosis of HeLa cells. J Biol Chem. 1999;274:32122–32126. doi: 10.1074/jbc.274.45.32122. [DOI] [PubMed] [Google Scholar]
- 157.Hassa PO, Hottiger MO. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol Chem. 1999;380:953–959. doi: 10.1515/BC.1999.118. [DOI] [PubMed] [Google Scholar]
- 158.Burkle A. Physiology and pathophysiology of poly(ADP-ribosyl)ation. BioEssays. 2001;23:795–806. doi: 10.1002/bies.1115. [DOI] [PubMed] [Google Scholar]
- 159.Meisterernst M, Stelzer G, Roeder RG. Poly(ADP-ribose) polymerase enhances activator-dependent transcription in vitro. Proc Natl Acad Sci USA. 1997;94:2261–2265. doi: 10.1073/pnas.94.6.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Conde C, Mark M, Oliver FJ, Huber A, de Murcia G, Menissier-de Murcia J. Loss of poly(ADP-ribose) polymerase-1 causes increased tumour latency in p53-deficient mice. EMBO J. 2001;20:3535–3543. doi: 10.1093/emboj/20.13.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.D'Amours D, Germain M, Orth K, Dixit VM, Poirier GG. Proteolysis of poly(ADP-ribose) polymerase by caspase 3: Kinetics of cleavage of mono(ADP-ribosyl)ated and DNA-bound substrates. Radiat Res. 1998;150:3–10. [PubMed] [Google Scholar]
- 162.Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- 163.Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274:22932–22940. doi: 10.1074/jbc.274.33.22932. [DOI] [PubMed] [Google Scholar]
- 164.Cayuela ML, Carrillo A, Ramirez P, Parrilla P, Yelamos J. Genomic instability in a PARP-1(-/-) cell line expressing PARP-1 DNA-binding domain. Biochem Biophys Res Commun. 2001;285:289–294. doi: 10.1006/bbrc.2001.5178. [DOI] [PubMed] [Google Scholar]
- 165.Simbulan-Rosenthal CM, Ly DH, Rosenthal DS, Konopka G, Luo R, Wang ZQ, Schultz PG, Smulson ME. Misregulation of gene expression in primary fibroblasts lacking poly(ADP-ribose) polymerase. Proc Natl Acad Sci USA. 2000;97:11274–11279. doi: 10.1073/pnas.200285797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Seimiya H, Smith S. The telomeric poly(ADP-ribose) polymerase, tankyrase 1, contains multiple binding sites for telomeric repeat binding factor 1 (TRF1) and a novel acceptor, 182-kDa tankyrase-binding protein (TAB182) J Biol Chem. 2002;277:14116–14126. doi: 10.1074/jbc.M112266200. [DOI] [PubMed] [Google Scholar]
- 167.Ziegler M, Oei SL. A cellular survival switch: Poly(ADP-ribosyl)ation stimulates DNA repair and silences transcription. BioEssays. 2001;23:543–548. doi: 10.1002/bies.1074. [DOI] [PubMed] [Google Scholar]
- 168.Soldatenkov VA, Chasovskikh S, Potaman VN, Trofimova I, Smulson ME, Dritschilo A. Transcriptional repression by binding of poly(ADP-ribose) polymerase to promoter sequences. J Biol Chem. 2002;277:665–670. doi: 10.1074/jbc.M108551200. [DOI] [PubMed] [Google Scholar]
- 169.Oei SL, Griesenbeck J, Schweiger M, Ziegler M. Regulation of RNA polymerase II-dependent transcription by poly(ADP-ribosyl)ation of transcription factors. J Biol Chem. 1998;273:31644–31647. doi: 10.1074/jbc.273.48.31644. [DOI] [PubMed] [Google Scholar]
- 170.Akiyama T, Takasawa S, Nata K, Kobayashi S, Abe M, Shervani NJ, Ikeda T, Nakagawa K, Unno M, Matsuno S, Okamoto H. Activation of Reg gene, a gene for insulin-producing beta-cell regeneration: Poly(ADP-ribose) polymerase binds Reg promoter and regulates the transcription by autopoly(ADP-ribosyl)ation. Proc Natl Acad Sci USA. 2001;98:48–53. doi: 10.1073/pnas.240458597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Butler AJ, Ordahl CP. Poly(ADP-ribose) polymerase binds with transcription enhancer factor 1 to MCAT1 elements to regulate muscle-specific transcription. Mol Cell Biol. 1999;19:296–306. doi: 10.1128/mcb.19.1.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cervellera MN, Sala A. Poly(ADP-ribose) polymerase is a B-MYB coactivator. J Biol Chem. 2000;275:10692–10696. doi: 10.1074/jbc.275.14.10692. [DOI] [PubMed] [Google Scholar]
- 173.Ha HC, Hester LD, Snyder SH. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci USA. 2002;99:3270–3275. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ullrich O, Diestel A, Eyupoglu IY, Nitsch R. Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat Cell Biol. 2001;3:1035–1042. doi: 10.1038/ncb1201-1035. [DOI] [PubMed] [Google Scholar]
- 175.Shall S, de Murcia G. Poly(ADP-ribose) polymerase-1: What have we learned from the deficient mouse model? Mutat Res. 2000;460:1–15. doi: 10.1016/s0921-8777(00)00016-1. [DOI] [PubMed] [Google Scholar]
- 176.Oliver FJ, Menissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la Rubia G, Stoclet JC, de Murcia G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18:4446–4454. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J Biol Chem. 2001;276:45588–45597. doi: 10.1074/jbc.M106528200. [DOI] [PubMed] [Google Scholar]
- 178.Chang WJ, Alvarez-Gonzalez R. The sequence-specific DNA binding of NF-kappa B is reversibly regulated by the automodification reaction of poly (ADP-ribose) polymerase 1. J Biol Chem. 2001;276:47664–47670. doi: 10.1074/jbc.M104666200. [DOI] [PubMed] [Google Scholar]
- 179.Moon NS, Berube G, Nepveu A. CCAAT displacement activity involves CUT repeats 1 and 2, not the CUT homeodomain. J Biol Chem. 2000;275:31325–31334. doi: 10.1074/jbc.M002912200. [DOI] [PubMed] [Google Scholar]
- 180.Nepveu A. Role of the multifunctional CDP/Cut/Cux homeodomain transcription factor in regulating differentiation, cell growth and development. Gene. 2001;270:1–15. doi: 10.1016/s0378-1119(01)00485-1. [DOI] [PubMed] [Google Scholar]
- 181.Santaguida M, Ding Q, Berube G, Truscott M, Whyte P, Nepveu A. Phosphorylation of the CCAAT displacement protein (CDP)/Cux transcription factor by cyclin A-Cdk1 modulates its DNA binding activity in G(2) J Biol Chem. 2001;276:45780–45790. doi: 10.1074/jbc.M107978200. [DOI] [PubMed] [Google Scholar]
- 182.Luong MX, van der Meijden CM, Xing D, Hesselton R, Monuki ES, Jones SN, Lian JB, Stein JL, Stein GS, Neufeld EJ, van Wijnen AJ. Genetic ablation of the CDP/Cux protein C terminus results in hair cycle defects and reduced male fertility. Mol Cell Biol. 2002;22:1424–1437. doi: 10.1128/mcb.22.5.1424-1437.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Coqueret O, Martin N, Berube G, Rabbat M, Litchfield DW, Nepveu A. DNA binding by cut homeodomain proteins is down-modulated by casein kinase II. J Biol Chem. 1998;273:2561–2566. doi: 10.1074/jbc.273.5.2561. [DOI] [PubMed] [Google Scholar]
- 184.Coqueret O, Berube G, Nepveu A. The mammalian Cut homeodomain protein functions as a cell-cycle-dependent transcriptional repressor which downmodulates p21WAF1/CIP1/SDI1 in S phase. EMBO J. 1998;17:4680–4694. doi: 10.1093/emboj/17.16.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Antes TJ, Chen J, Cooper AD, Levy-Wilson B. The nuclear matrix protein CDP represses hepatic transcription of the human cholesterol-7alpha hydroxylase gene. J Biol Chem. 2000;275:26649–26660. doi: 10.1074/jbc.M002852200. [DOI] [PubMed] [Google Scholar]
- 186.Zhu Q, Gregg K, Lozano M, Liu J, Dudley JP. CDP is a repressor of mouse mammary tumor virus expression in the mammary gland. J Virol. 2000;74:6348–6357. doi: 10.1128/jvi.74.14.6348-6357.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]