

Abstract
The cytokine macrophage migration inhibitory factor (MIF) is involved in the pathogenesis of inflammatory and infectious diseases, however its role in HIV-1 infection is unknown. Here we show that HIV-1-infected patients present elevated plasma levels of MIF, that HIV-1-infected peripheral blood mononuclear cells (PBMCs) release a greater amount of MIF, and that the HIV-1 envelope glycoprotein gp120 induces MIF secretion from uninfected PBMCs. The HIV-1 replication in PBMCs declines when these cells were treated with anti-MIF antibodies, or when treated with the ABC-transporter inhibitor probenecid, which also inhibited MIF secretion. The addition of recombinant MIF (rhMIF) to HIV-1-infected PBMCs enhances viral replication of CCR5- or CXCR4-tropic HIV-1 isolates. Using a T CD4+ cell lineage containing an HIV long terminal repeats (LTR)-Luciferase construct, we detected that rhMIF promotes transcription from HIV-1 LTR. Our results show that HIV-1 induces MIF secretion and suggest that MIF influences the HIV-1 biology through activation of HIV-1 LTR.
Keywords: MIF, HIV-1, AIDS, LTR, gp120
INTRODUCTION
Infection by the Human Immunodeficiency Virus type 1 (HIV-1), the etiological agent of the Acquired Immunodeficiency Syndrome (AIDS), is a worldwide spread disease characterized by a persistent viral replication in lymphoid tissues, leading to a profound immunosuppression that can culminate in higher susceptibility to opportunistic infections, tumors and central nervous system degeneration (Stevenson, 2003). HIV-1 infects and replicates in CD4+ lymphocytes and monocyte/macrophages, using the CD4 molecule (Dalgleish et al., 1984) and the chemokine receptors CXCR4 (Feng et al., 1996) or CCR5 (Deng et al., 1996) to penetrate into host cells. A variety of conditions have been associated with increases of viral load and viral persistence in HIV-1-infected individuals, such as coinfections (Orenstein et al., 1997; Toossi et al., 1993; Wahl et al., 1999) and immune activation (Ortigao-de-Sampaio et al., 1998; Stanley et al., 1996). Immune activation in HIV-1-infected patients has been recently related to microbial translocation from the gut to circulation secondary to an increased intestinal permeability associated with destruction of the gut lymphoid tissue (GALT) (Brenchley et al., 2006a; Brenchley et al., 2006b; Lee et al., 2009). Therefore, activation of the immune cells by microbial products (such as LPS) and interaction of HIV-1-infected cells with host molecules can modulate viral replication and influence the disease progression.
One of the most complex aspects of AIDS pathogenesis is the dynamic interplay between HIV-1 and host molecules (Hladik and McElrath, 2008; Neil et al., 2008). In this context, cytokines and other inflammatory mediators play a pivotal role in the viral replicative cycle (Alfano et al., 2008; Kedzierska and Crowe, 2001), and a number of studies support the influence of these molecules over both the course of the disease and HIV-1 biology (Alfano et al., 2008; Copeland, 2005; Kedzierska and Crowe, 2001). Pro-inflammatory cytokines, such as Tumor Necrosis Factor (TNF)-α and Interleukin (IL)-6, are considered to up-regulate HIV-1 replication, whereas certain anti-inflammatory cytokines, such as type 1 Interferon (IFN), IL-10 and IL-27, may diminish viral replication (Alfano et al., 2008; Kedzierska and Crowe, 2001). Some cytokines are also capable of interfering in the HIV-1 replication by means of modulation of cell restriction factors to the virus. For example, production or activation of the proteins of the APOBEC family and recently described Tetherin are regulated by IL-2, IL-7, IL-15 and IFN-α (Neil et al., 2008; Stopak et al., 2007; Varthakavi et al., 2008).
The Macrophage Migration Inhibitory Factor (MIF) is an upstream activator of innate immunity, it is present pre-formed in many cell types and it is endowed with both enzymatic and receptor binding properties (Bernhagen et al., 2007; Calandra and Roger, 2003; Leng et al., 2003). MIF lacks a signal sequence and it is secreted by a specialized pathway that is dependent of ATP-binding cassette (ABC) transporters, specifically from the family ABCA1 (Flieger et al., 2003). Another unique characteristic of MIF is the fact that it binds and activates a multi-component receptor complex comprising CD74, CD44, and the chemokine receptors CXCR2 and CXCR4 (Bernhagen et al., 2007; Calandra and Roger, 2003; Shi et al., 2006). MIF promotes the release of a number of pro-inflammatory cytokines, such as TNF-α, IL-6 and Prostaglandin E2 (PGE2) (Calandra and Roger, 2003). Elevated serum levels of MIF have been detected in many infectious and inflammatory diseases, such as reumathoid arthrits, artherosclerosis (Ayoub et al., 2008), sepsis (Bozza et al., 2004; Bozza et al., 1999; Emonts et al., 2007), West Nile Virus (Arjona et al., 2007) and others (Calandra and Roger, 2003; Hoi et al., 2007). Moreover, MIF has been implicated in the worsening of some pathological conditions, and neutralization of this cytokine ameliorates the disease clinical course (Amano et al., 2007; Bozza et al., 2004; Calandra et al., 2000; Chen et al., 2006; Gando et al., 2007; Ichiyama et al., 2004; Meazza et al., 2002).
Since there are no studies addressing the role of MIF in HIV-1 infection and its ability to modulate HIV-1 replication, we measured the levels of this cytokine in the plasma of HIV-1-infected patients and investigated whether it could influence HIV-1 replication in human primary peripheral blood mononuclear cells (PBMCs). Here, we show for the first time that plasma levels of MIF are elevated in HIV-1-infected individuals, PBMCs release MIF upon HIV-1 infection, and that MIF secretion contributes to viral replication.
MATERIALS AND METHODS
HIV-1 isolates
Assays of PBMC infection were performed with the monocytotropic, CCR5-dependent isolate HIV-1Ba-L and with the CXCR4-dependent isolate HIV-1Tybe, which were donated to us by the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (Bethesda, MD). Both isolates were expanded in phytohemagglutinin-activated PBMCs, as described elsewhere (Lima et al., 2002).
Reagents and ELISA kits
Recombinant human MIF (rhMIF) was obtained as previously described (Bernhagen et al., 1994); anti-MIF polyclonal antibodies and goat IgG were purchased from R&D Systems (Minneapolis, MN); the ABC transporters inhibitor probenecide was obtained from Sigma-Aldrich (MO, EUA). The recombinant HIV-1Ba-L gp120 and the recombinant HIV-1 Tat were donated to us by the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (Bethesda, MD). MIF concentration in cell culture supernatants and in plasma samples was measured by a commercial ELISA kit (R&D Systems), following manufacturer’s instructions.
Acquisition of plasma and assessment of viremia
Plasma of HIV-1 infected and normal individuals were obtained by venopuncture following collection of informed consent and according to FIOCRUZ Institutional Ethics Committee. HIV-1- plasma viral loads were measured using the branched-DNA Versant HIV-1 RNA 3.0 Assay (lower limit of detection 50 copies HIV RNA/ml; Siemens Healthcare Diagnostics Inc., NY), according to the manufacturer’s instructions. We selected 10 patients with undetectable viral load (< 50 copies HIV RNA/ml), 10 with viral load between 1,000 and 10,000 copies HIV RNA/ml, and 10 with viral load between 50,000 and 500,000 HIV RNA/ml. At the moment of plasma collection, HIV-1 patients were not under anti-retroviral treatment and did not present clinical signs of co-infections.
Cells
PBMCs from healthy donors were obtained by density gradient centrifugation (Hystopaque, Sigma) from buffy coat preparations. PBMCs were resuspended in RPMI 1640 (LGC Bio, São Paulo, SP, Brazil) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Hyclone, Logan, UT), penicillin (100 U/mL), streptomycin (100μg/mL), 2mM glutamine and 10mM HEPES, stimulated with 5 μg/mL of phytohemagglutinin (PHA, Sigma) during 2–3 days, and further maintained in culture medium containing 5 U/mL of recombinant human Interleukin-2 (Sigma). Human monocyte-derived macrophages (MDM) were obtained from PBMCs through adherence onto plastic plates. Briefly, 1.5 × 106 PBMCs were plated onto 48-well Plates (Nalge Nunc) in DMEM containing 10% human serum (Sigma) and penicillin- streptomycin (Sigma). Cells were maintained at 37ºC in 5% CO2 for 6–7 days for monocyte differentiation into macrophages. Non-adherent cells were washed out, and macrophages were maintained in DMEM with 10% human serum. Macrophage purity was > 90%, as determined by flow cytometry (FACScan; Becton Dickinson) analysis using anti-CD3 (PharMingen) and anti-CD14 (PharMingen) monoclonal antibodies.
HIV-1 infection
PBMCs were infected with either the HIV-1 R5-isolate Ba-L or the X4-isolate Tybe, using 5 to 10 ng/mL of p24 antigen. After a 2 hours incubation, cells were washed to remove excess virus, and culture medium was added back to infected PBMC. HIV-1 replication was evaluated in cell culture supernatants by a commercial ELISA kit, according to manufacturer’s instructions (ZeptoMetrix Co., NY). Macrophages were infected with HIV-1 R5-isolate Ba-L by exposing them overnight to viral suspensions containing 5 to 10 ng/mL of p24 antigen. Thus, non-internalized viruses were removed by washing, and cell monolayers were replenished with fresh medium. HIV-1 production and MIF secretion were measured in cell culture supernatants as described above.
Effect of MIF on HIV-1 replication
To address the role of MIF in HIV-1 replication, HIV-1-infected PBMCs were distributed in 96-well culture plates (2×105/well/200 μL) and treated either with rhMIF, anti-MIF antibodies (or isotype control; R&D Systems) immediately after cell infection. Cells were maintained in culture for different time-points, and HIV-1 replication was measured as described above.
Inhibition of ABC transporters
To inhibit the functioning of ABC transporters, PBMCs were treated for 45 minutes with probenecide (10 μM) prior to HIV-1 infection. After infection, probenecide was added back to cultures, at the same concentration, for the entire period of the experiment (7 days), when cell culture supernatants were collected for quantification of MIF production and HIV-1 replication.
Treatment of PBMC with recombinant HIV-1 gp120
Uninfected PBMCs were treated with recombinant HIV-1 gp120 derived from the R5-tropic HIV-1 BaL isolate (5 μg/mL) for 16 hours; then, cell culture supernatants were collected for evaluation of MIF release.
Evaluation of transcription from LTR
CD4+ T Jurkat derivative cell line (1G5) containing stably transfection of luciferase under control of HIV-1 LTR was obtained from NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (Bethesda, MD) / Dr Estuardo Aguilar-Cordova and Dr John Belmont. For evaluation of transcription from HIV-1 LTR, 106 1G5 cells were plated in the absence of stimulus or in the presence of MIF, Tat (100 ng/mL) or PHA (2 μg/mL) for 18 hrs at 37ºC/5% CO2. After this time period, cells were collected, washed twice with PBS and processed according Luciferase Detection Kit (Promega, WI). The luminescence was evaluated on a TD-20/20 luminometer (Turner Designs), following instructions provided by the manufacturer of the kit.
Statistical Analysis
All results presented in this study were prepared using Graphpad Prism v4.0 software (San Diego, CA). Statistical analysis calculation was performed using the same software, and the comparisons between values were considered significantly different when the p value was minor than 0.05 using Student’s T test.
RESULTS
HIV-1-infected patients present high plasma levels of MIF
HIV-1 infection increases the secretion of many cytokines in vitro and in vivo (Kedzierska and Crowe, 2001), and enhancement of MIF production has been detected either in the sera of patients or at the site of some inflammatory and infectious diseases (Awandare et al., 2007; He et al., 2006). Also, it is already known that viral infections, such as the dengue and West Nile virus infection, can up-modulate serum levels of MIF (Arjona et al., 2007; Chen et al., 2006). Thus, we hypothesized that a similar phenomenon could be occurring in HIV-1-infected individuals, and we first examined whether these patients would present higher plasma levels of MIF relative to uninfected individuals. We selected a group of HIV-1-infected individuals (n=30) and a control group comprised of healthy volunteers (n=10). Plasma was collected from these individuals and the levels of MIF were investigated by ELISA. It was found that HIV-1-infected individuals presented plasma concentrations of MIF up to 25 times higher than the control group (Figure 1). We found no correlation between plasma viral loads and MIF levels (data not shown).
Figure 1.
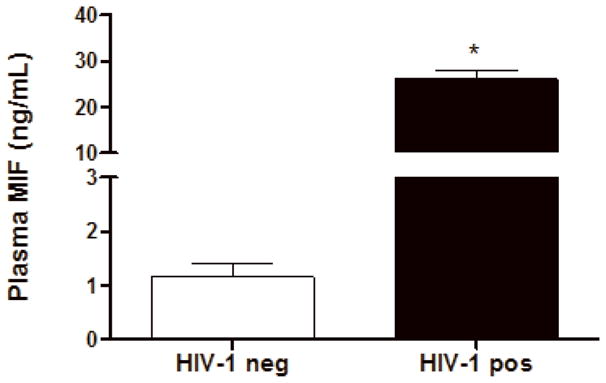
HIV-1-infected patients present higher plasma levels of MIF relative to uninfected individuals. Plasmas from HIV-1-infected individuals (HIV-1 pos), and from healthy volunteers (control plasmas; HIV-1 neg) were assessed for MIF concentrations by ELISA. Bars represent means ± SEM of 10 healthy volunteers (HIV-1 neg) or 30 HIV-1-infected patients (HIV-1 pos). * p<0.0001
MIF is secreted by primary PBMCs infected with HIV-1 in vitro
Based on the findings that some viral infections can up-modulate MIF secretion in vitro, such as West Nile virus, human Cytomegalovirus (CMV) and Influenza A infections (Arjona et al., 2007; Arndt et al., 2002; Bacher et al., 2002), and in order to understand if HIV-1 infection itself could be contributing for these higher levels of MIF in the plasma of infected patients, we infected human primary PBMCs and macrophages and examined whether HIV-1 infection would induce these cells to secrete MIF. PBMCs were infected with either an R5-tropic or an X4-tropic HIV-1 isolate and MIF secretion and viral replication were evaluated 4 and 7 days after infection. We detected that, infection with HIV-1 R5-tropic isolate doubled the release of MIF by these cells relative to MIF secretion by PBMCs (Figure 2, A and B). Of note, there was no significant increase in secreted MIF at 4 days of infection (data not shown), suggesting that MIF release may follow a similar kinetics of virus replication in PBMC, since HIV-1 replication usually peaks about 7 days after infection of these cells. The increment in MIF secretion by HIV-1-infected PBMC is not restricted by the phenotype of the HIV-1 isolate, as evidenced by the fact that the infection by an X4-tropic strain also increased MIF secretion, as measured 7 days after infection (Figure 2-C). We also measured MIF secretion by HIV-1-infected monocyted-derived macrophages and, curiously, HIV-1 infection of macrophages did not alter the secretion of basal levels of MIF (Fig 2-D), as observed in two time points (7 and 14 days after infection), suggesting a phenomenon restricted to PBMCs.
Figure 2.
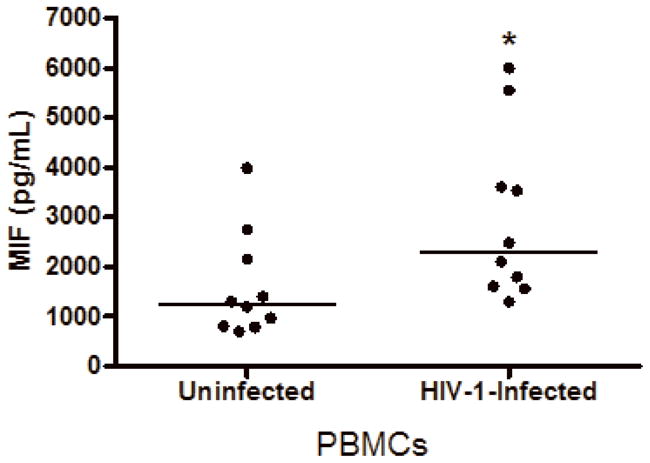
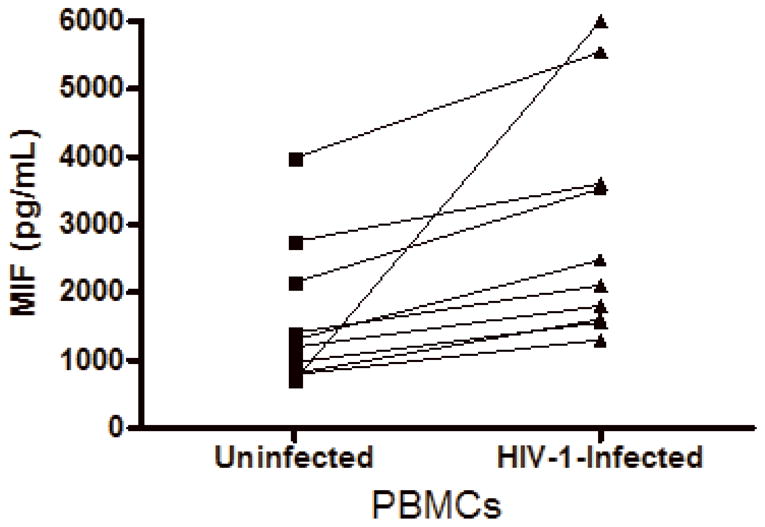
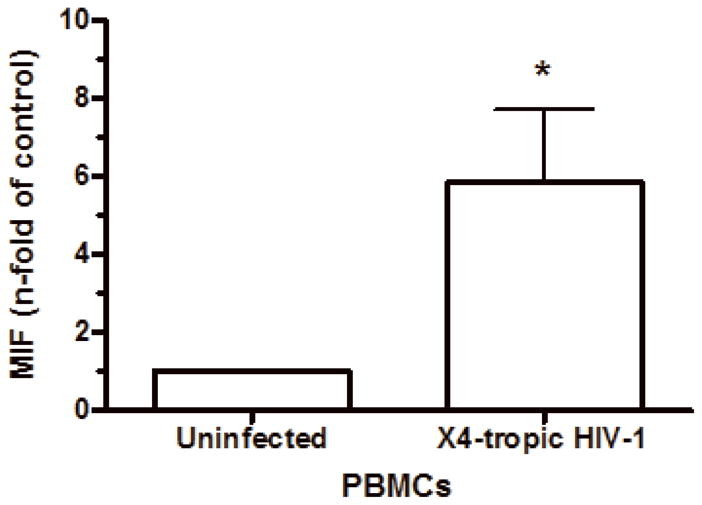
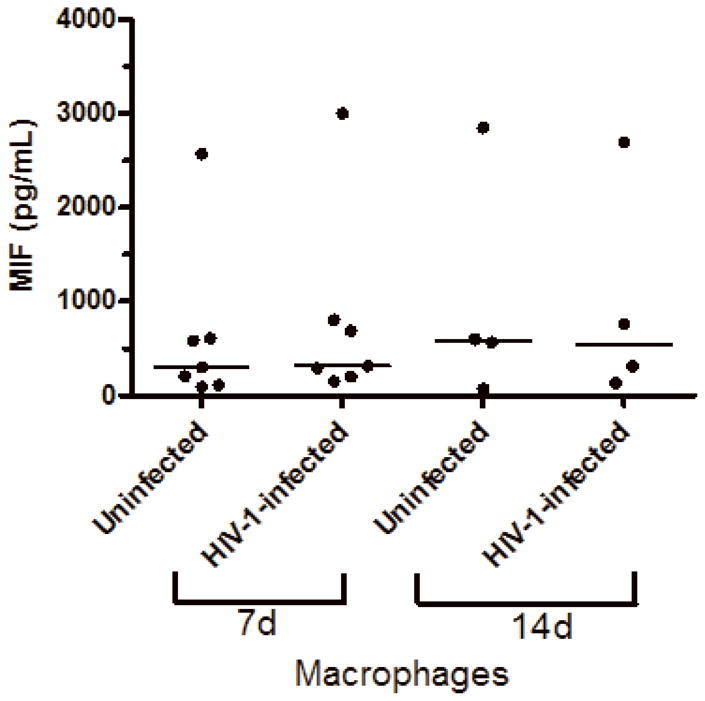
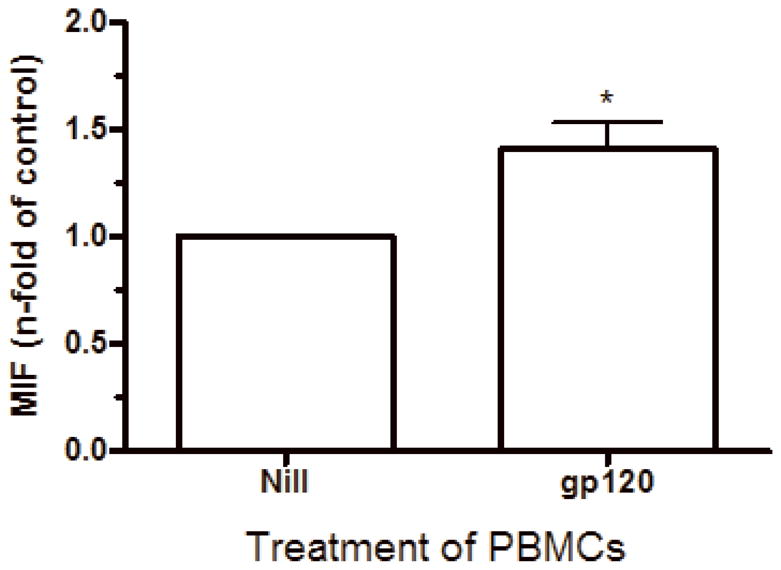
HIV-1 infection and HIV-1 gp120 induce MIF release by PBMCs. (A) PBMCs were infected by an R5-tropic HIV-1 isolate (Ba-L), and after 7 days MIF contents were evaluated in the culture supernatants by ELISA (* p<0.04). Individual values and medians are represented for each group. (B) Lines show individual MIF release in the uninfected and in HIV-1-infected PBMC depicted in figure 2-A. (C) MIF contents in culture supernatants of PBMCs infected by an X4-tropic HIV-1 isolate (Tybe) (* p<0.04). Mean of MIF values by cells cultured only with medium (Nill): 757.8 pg/mL. (D) Macrophages were infected by the HIV-1 isolate Ba-L and MIF release was assessed by ELISA at 7 (7d) and 14 (14d) days after infection. Individual values and medians are represented for each group. (E) Uninfected PBMCs were exposed to recombinant HIV-1 envelope protein gp120 (5 μg/mL) derived from the HIV-1 isolate Ba-L, and MIF secretion was evaluated in culture supernatants by ELISA after 16 hours (* p<0.006). Mean of MIF values by cells cultured only with medium (Nill): 427 pg/mL. In (C) and (E) bars show means ± SEM for 3 and 5 different donors, respectively, and all experiments were done in triplicates per donor.
HIV-1 gp120 also induces MIF secretion by PBMC
Based on our findings that the increment of MIF secretion was significant 7 days after infection, at the same time point when elevated amounts of HIV-1 production are also detected, we hypothesized that the HIV-1 virion per se, or HIV-1 components could induce direct secretion of MIF through interaction with infected or uninfected PBMCs in culture. In fact, it has been described that viral components can accumulate in cultures of HIV-1 infected PBMCs, such as gp120 (Zhang et al., 1997). Rooted on the ability of HIV-1 envelope-derived gp120 protein to trigger the secretion of a variety of cytokines and that gp120 can be detected in serum of HIV-1+ patients (Capobianchi, 1996; Klasse and Moore, 2004), we exposed uninfected PBMCs to a concentration of soluble recombinant BaL gp120 (r gp120) protein previously known to induce cytokine secretion (5 μg/mL) for 16 hours (Schols and De Clercq, 1996). We observed that r gp120 promoted MIF secretion by treated PBMC (Figure 2-E) at approximately 50% more than untreated cells, suggesting that viral components can contribute to the enhanced MIF release by HIV-1-infected PBMC cultures.
HIV-1 infection stimulates MIF release through ABCA1 transporters
The release of MIF upon stimulation by microbial components such as lipopolysaccharide (LPS) has been shown to be dependent on the ABCA1-family of the ATP-binding cassette (ABC) transporters (Flieger et al., 2003). Thus, it would be interesting to examine whether ABC transporters would also participate in the MIF release promoted by an infectious agent such as HIV-1. To proceed with this study, cells were pre-treated with probenecid (10 μM, for 45 minutes), infected with HIV-1, and probenecid re-applied to infected cells prior to measuring MIF release and HIV-1 replication at day 7 post-infection. Probenecid treatment resulted in a reduction of MIF secretion secondary to HIV-1 infection (Figure 3), which is consistent with the notion that the HIV-1-induced MIF release is dependent of ABCA1 transporters.
Figure 3.
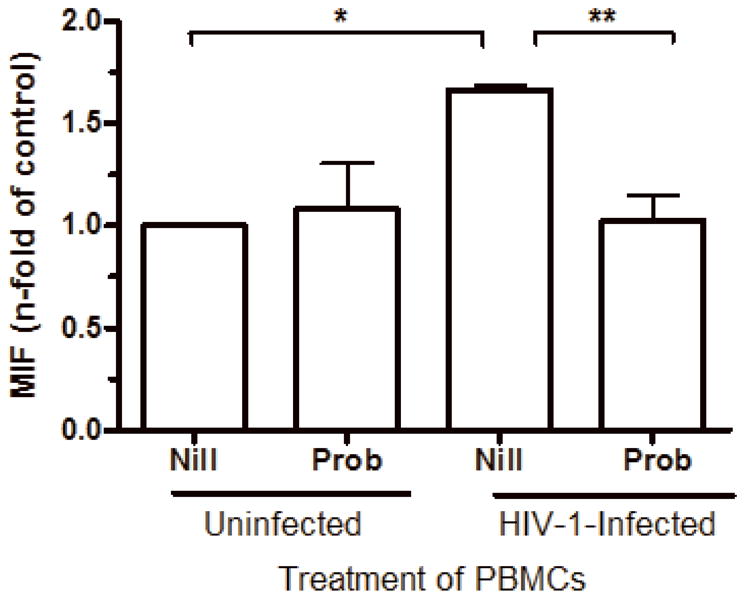
MIF release upon HIV-1 infection is dependent of ABC transporters. PBMCs were treated with probenecid (Prob; 10 μM, 45 minutes), then infected or not by an R5-tropic HIV-1 isolate (BaL), and probenecid was re-applied. MIF contents were measured by ELISA in culture supernatants 7 days after infection (* p<0.0002; ** p<0.004). Mean of MIF values by HIV-1-infected cells cultured only with medium (Nill): 1.6 ng/mL. Data represent means ± SEM for 3 different donors done in triplicates.
Immunoneutralization of secreted MIF diminishes HIV-1 replication
Based on the preceeding result that HIV-1 infection induces MIF secretion and to provide evidence for the role of MIF in HIV-1 replication, HIV-1-infected PBMCs were treated with neutralizing anti-MIF polyclonal antibodies immediately after infection, and viral replication was evaluated 7 days after infection. MIF immunoneutralization reduced HIV-1 replication at approximately 40% (Figure 4), indicating that MIF inhibition down-modulates HIV-1 replication. This result suggests that MIF, similar to other pro-inflammatory cytokines, can promote HIV-1 replication in primary HIV-1-infected cells (Kinter et al., 2000).
Figure 4.
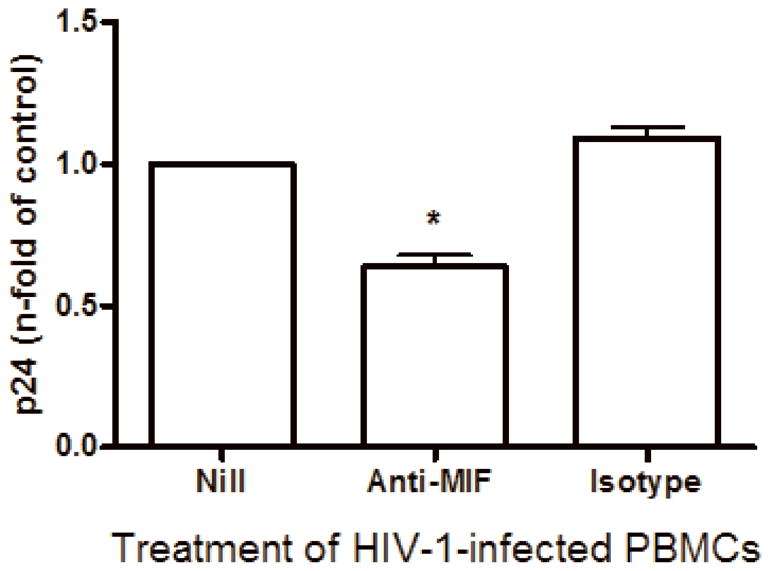
MIF neutralization diminishes HIV-1 replication. PBMCs were infected by an R5-tropic HIV-1 isolate (BaL) and treated with anti- human MIF polyclonal antibodies (20 μg/mL) or isotype control. Viral replication was measured after 7 days in culture supernatants by ELISA (* p<0.0001). Mean of p24 values by HIV-1-infected cells cultured only with medium (Nill): 147 ng/mL. Data represent means ± SEM for 5 different donors, done in triplicates.
Addition of rhMIF to HIV-1-infected PBMCs enhances viral replication
Since immunoneutralization of MIF decreased HIV-1 replication, we next asked whether the addition of rhMIF could augment viral replication. Based on the observation that MIF can be detected at concentrations reaching 25 ng/mL in plasma of HIV-1-infected patients, we added rhMIF (obtained as described on Bernhagen et al., 1994) to HIV-1-infected PBMCs just after the infection, and measured viral replication after 7 days. We found that MIF significantly enhanced HIV-1 replication of a R5-tropic isolate at doses of 12 and 25 ng/mL, when compared to untreated HIV-1 infected cells (Figure 5-A). MIF also exacerbated the viral replication of an X4-tropic isolate (Figure 5-B), indicating that the ability of MIF to enhance viral replication is independent of viral phenotype.
Figure 5.
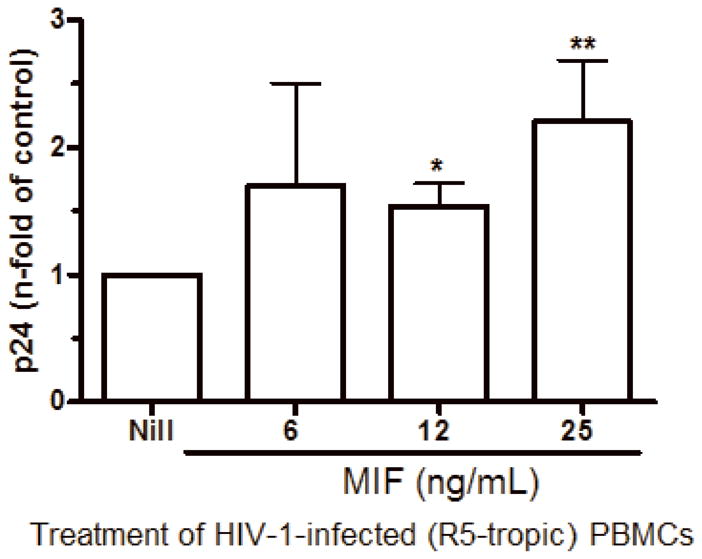
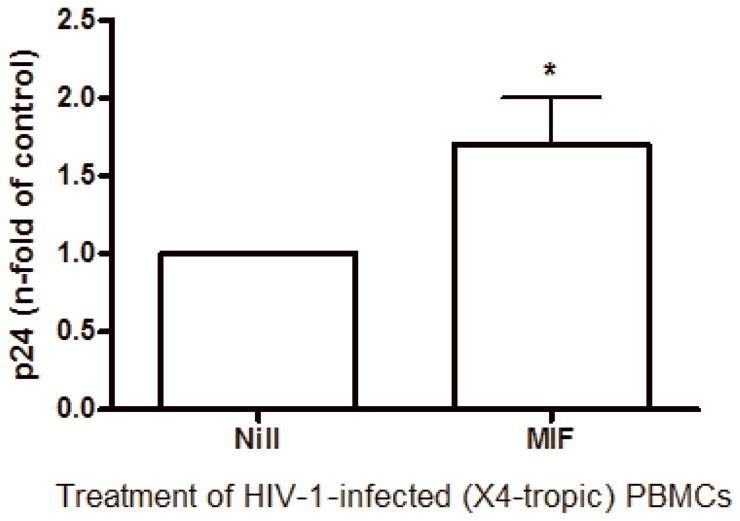
MIF addition exacerbates HIV-1 replication. (A) PBMCs were infected by an R5-tropic HIV-1 isolate (Ba-L), and treated with rhMIF at indicated concentrations. HIV-1 replication was measured after 7 days by a p24 Ag ELISA in culture supernatants (12 ng/mL, * p<0.009; 25 ng/mL, ** p<0.03, both relative to untreated cells). Mean of p24 values by HIV-1-infected cells cultured only with medium (Nill): 123 ng/mL; (B) PBMCs were infected by an X4-tropic HIV-1 (Tybe) and exposed to rhMIF (25 ng/mL). Viral replication was measured 7 days after infection, as described (* p<0.04). Mean of p24 values by HIV-1-infected cells cultured only with medium (Nill): 73 ng/mL. Data represent means ± SEM for 3 (6 ng/ml), 5 (12 ng/mL) and 9 (25 ng/mL) (A), or 5 (B) different donors, done in triplicates.
The rhMIF support HIV-1 transcription from LTR
Next, we addressed whether MIF could be able to stimulate viral replication via induction of HIV-1 transcription from LTR. For this purpose, we added rhMIF to cultures of Jurkat CD4 T derivative cell (1G5) containing a stably integrated HIV-LTR-Luciferase construct. In this system, Luciferase expression is controlled by transcription from LTR, and a quantitative analysis of transcription is possible through luminescence signal emitted by Luciferase (Aguilar-Cordova et al., 1994). As depicted in figure 6, exogenous treatment with 25 ng/mL of rhMIF influenced the transcription from HIV-1 LTR, suggesting that MIF drives a biochemical signaling pathway which culminates on HIV-1 transcription. As positive controls, we used PHA stimulation, as well as HIV1 Tat protein, once this cell lineage was selected for high responsiveness to these molecules. Taken together, these results indicate that rhMIF augments HIV-1 replication via induction of direct HIV-1 transcription.
Figure 6.
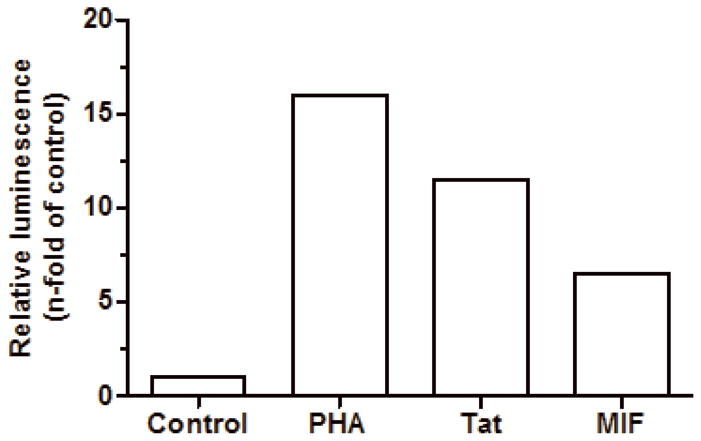
rhMIF promotes HIV-1 transcription from LTR. CD4+ T Jurkat derivative cell lineage (1G5) containing stably transfection of luciferase under control of HIV-1 LTR was left untreated (control) or treated with PHA (2 μg/mL), Tat (100 ng/mL) or MIF (25 ng/mL). After 20hs, cells were washed and lyzed for luminescence evaluation. Bars represent one representative of 3 independent experiments with similar results.
DISCUSSION
The macrophage migration inhibitory factor (MIF) presents a variety of pharmacological activities, including enzymatic activity and the property of being released endocrinally by the pituitary upon stress situations (Bernhagen et al., 1994; Calandra and Roger, 2003; Larson and Horak, 2006). This cytokine has also been implicated in many inflammatory and infectious diseases, such as sepsis and rheumatoid arthritis, to name a few, and in many of these conditions elevated levels of MIF in the circulation were found, and MIF neutralization led to amelioration of the targeted pathology (Javeed and Zhao, 2008).
The HIV-1 infection is known to be profoundly regulated by immune molecules such as cytokines and others soluble factors, that have been described to modulate HIV-1 replication, and which levels are commonly found altered in HIV-1 infection. Therefore, cytokines have been considered pivotal players on the pathogenesis of HIV-1 infection, once they balance the immune response and are able to promote or to inhibit virus replication (Alfano et al., 2008; Kedzierska and Crowe, 2001).
Although the role of cytokines in HIV-1 infection and pathogenesis has largely been explored, there are no reports, so far, about the participation of MIF in this condition. Because MIF can activate several signaling pathways which can ultimately facilitate HIV-1 replication, such as activation of the transcription factor NF-κB, PGE2 induction and TNF-α secretion (Amin et al., 2006; Donnelly et al., 1997; Mitchell et al., 2002), we decided to analyze whether MIF could also be involved in HIV-1 biology, uncovering it as one more factor contributing to the delicate balance of the interaction between host and pathogen in the course of this infection. Our findings constitute the first report that MIF is present in elevated concentrations in the plasma of HIV-1-infected individuals and that MIF is secreted by HIV-1-infected cells or by an HIV-1 component. In addition, our study is the first one to describe secretion of MIF due to a retrovirus infection in humans. Also, we show here that MIF is an up-modulator of HIV-1 replication in vitro, suggesting a novel role for this cytokine in the pathogenesis of HIV-1 infection, and that a continuous MIF release by HIV-1-infected cells can contribute to viral loads and to accelerate progression to AIDS.
First, we investigated if MIF concentrations were elevated in the plasma of individuals infected by HIV-1, and we found that in these patients, levels of MIF were much higher (up to 25 times) than in uninfected individuals. Although there was no positive correlation between plasma viral loads and MIF levels (data not shown), this finding strongly suggests that HIV-1 infection could be responsible for augmenting levels of MIF in vivo. At that point, we could not determine the main source of MIF, whether from HIV-1-infected cells or infected and uninfected cells exposed to HIV-1 antigens (eg. gp120), or even microbial products translocated from gut (Brenchley et al., 2006a). However, we could suggest that the amount of circulating virus is not the only factor triggering MIF release.
To assess if HIV-1-infection could be an event capable of inducing MIF release, we infected PBMC with both an R5 or an X4-tropic strain of HIV-1. We observed that both phenotypes elicited MIF secretion in culture, and that infection with an X4-tropic isolate of HIV-1 resulted in a more pronounced MIF secretion than the one mediated by the R5-virus. One explanation for this difference could be the fact that the expression of CXCR4 on cell membranes is more elevated than CCR5, which, in ultimate analysis could result in a higher number of infected cells in vitro and, thus, more cells releasing MIF. Since it has been reported that in nearly 50% of AIDS patients there is a shift in the viral co-receptor usage from R5 variants to X4-tropic HIV-1 isolates (Connor et al., 1997) our findings suggest that patients harboring both phenotypes of HIV-1 variants may have two simultaneous signals for MIF release during the late phases of HIV-1 infection, and that elevated levels of MIF may be detected in infected patients regardless of the harbored viral phenotype.
The augmentation of MIF release secondary to HIV-1 infection, only 7 days after infection might suggest that induction of MIF secretion requires a peak in the viral replication, or even an accumulation of some viral component in the culture. To address this possibility, we treated uninfected cells with HIV-1 gp120 derived from a R5-tropic strain (Ba-L isolate) and observed a significant, although slight, increase in MIF release after 16 hours of treatment, showing that an HIV-1 molecule could also induce MIF secretion, although it did not appear to be the main factor inducing elevated amounts of MIF release in infected cultures. Certainly, a replicating virus is a more powerful inducer for MIF release than a soluble glycoprotein, through a mechanism not yet described. Taking in account that the gp120 circulates in AIDS patients (Chiodi et al., 1987), this viral protein may contribute to increasing systemic MIF levels as we describe here. Since MIF secretion can be induced by a variety of endogenous/exogenous stimuli (Calandra and Roger, 2003), we understand that it is very difficult to determine the real nature of MIF release in vivo. For an unknown reason, macrophages did not release greater amounts of MIF secondary to HIV-1 infection, despite the fact that macrophages are an important source of MIF (Calandra et al., 1994), suggesting that this phenomen is probably restricted by cell type.
It has already been shown that MIF is released after microbial stimulation, being LPS from gram-negative bacteria one of the most classic MIF inducer (Bernhagen et al., 1994; Calandra and Roger, 2003). MIF release secondary to LPS stimulation is dependent of ABC transporters, as shown by the fact that probenecid, an inhibitor of the ABCA1 family of the ABC transporters, blocks MIF secreted upon LPS stimulation (Flieger et al., 2003). The ABCA1 transporter is considered as a major factor responsible for MIF release, and this cytokine is constitutively expressed and stored in intracellular pools (Calandra and Roger, 2003). To investigate if the MIF released by HIV-1 stimulation was also exported from the cells by means of ABCA1 transporters, we treated HIV-1-infected PBMC with Probenecid and evaluated MIF contents and viral replication. We found that treatment with probenecid decreased MIF release and also blocked HIV-1 growth, indicating that the MIF release upon HIV-1 infection needs the activation of ABCA1 transporters.
The immunoneutralization of MIF in HIV-1-infected cultures revealed the importance of this molecule in viral replication. Diminishing the levels of MIF in the cell cultures resulted in diminished viral replication, implying a direct relation between MIF and HIV-1. The association of MIF with HIV-1 was strengthened by the finding that addition of rhMIF to HIV-1-infected cultures led to an augmentation of viral replication either of R5 or X4-tropic isolates. It is important to mention that rhMIF favored HIV-1 replication when this cytokine was added at the same dose (25 ng/mL) observed in HIV-1 plasma patients, suggesting that this MIF concentration could up-modulate HIV-1 production in vivo.
Based on our results shown in figure 6, we could suggest that exposure to MIF elicits HIV-1 transcription from LTR, thus favoring viral replication. It has been demonstrated that MIF triggers a signaling pathway that culminates in activation of the three transcription factors AP-1, ETS and NF-κB (Amin et al., 2006; Binsky et al., 2007; Calandra and Roger, 2003; Gore et al., 2008; Kleemann et al., 2000; Toh et al., 2006) for which HIV-1 LTR contains binding sites (Copeland, 2005). In this context, cytokines can contribute to HIV-1 production via activation of LTR by positive transcription regulators. For instance, TNF-α is dependent of NF-κB to drive HIV-1 transcription mediated by LTR (Duh et al., 1989; Munoz-Fernandez et al., 1997). In addition, IL-10 cooperates with TNF-α to enhance the binding of AP-1 and NF-κB to the LTR in monocyte/macrophage lineage (Finnegan et al., 1996). Thus, it is plausible to speculate that HIV-1 transcription stimulated by MIF from HIV-1 LTR encompasses activation and binding of AP-1, ETS and NF-κB to HIV-1 LTR.
Taken together, our results suggest a positive, re-entrant pathway for the establishment of HIV-1 in the host whereby viral infection of cells triggers MIF release, which then augments HIV-1 replication.
Because this study was entirely performed in primary cells (except for data presented in figure 6), our results might resemble what is occurring in HIV-1-infected tissues. Thus, MIF may be acting as a detrimental molecule to HIV-1-infected individuals, favoring high viral replication, even supporting viral persistence. Because AIDS patients succumb to opportunistic pathogens and MIF is secreted during the course of a great diversity of infections (Baugh and Bucala, 2002), high levels of MIF induced by co-pathogens could augment HIV-1 replication and contribute to disease progression. Also, it has recently been shown that microbial translocation from the gut to the circulation is enhanced in HIV-1-infected individuals, and plays a critical role on chronic immune activation and HIV-1 pathogenesis (Brenchley et al., 2006b). One of the most common translocated microbial products, the bacterial LPS, is a strong inducer of MIF secretion (Calandra and Roger, 2003), can be found in serum of HIV-1-infected individuals (Brenchley et al., 2006b), and could be contributing to higher levels of MIF in the circulation of HIV-1-infected individuals, thus, favoring viral replication as well. The precise mechanisms whereby MIF promotes HIV-1 replication is unknown, but further studies can enlighten this particular issue and potentially point MIF as a target molecule for anti-HIV-1 therapy.
Acknowledgments
We thank the Hemotherapy Service of the Hospital Clementino Fraga Filho (UFRJ) for providing buffy coats. This work was supported by grants from PAPES/Fiocruz, CNPq, Faperj and CAPES. The HIV-1 isolates Ba-L and Tybe, the recombinant HIV-1 proteins Tat and gp120, and the 1G5 cells were kindly donated by NIH AIDS Research and Reference Reagent Program (Division of AIDS, NIAID, NIH, Bethesda, MD). Eduardo G. Regis is presently a Ph.D. Student of the Cellular and Molecular Biology Post-graduation Program at the Oswaldo Cruz Institute/FIOCRUZ, Rio de Janeiro, RJ, Brazil, and this paper is a partial fulfillment for obtaining his PhD title. Lin Leng and Richard Bucala are supported by NIH grant 2 RO1 AI42310.
References
- Aguilar-Cordova E, Chinen J, Donehower L, Lewis DE, Belmont JW. A sensitive reporter cell line for HIV-1 tat activity, HIV-1 inhibitors, and T cell activation effects. AIDS Res Hum Retroviruses. 1994;10(3):295–301. doi: 10.1089/aid.1994.10.295. [DOI] [PubMed] [Google Scholar]
- Alfano M, Crotti A, Vicenzi E, Poli G. New players in cytokine control of HIV infection. Curr HIV/AIDS Rep. 2008;5(1):27–32. doi: 10.1007/s11904-008-0005-5. [DOI] [PubMed] [Google Scholar]
- Amano T, Nishihira J, Miki I. Blockade of macrophage migration inhibitory factor (MIF) prevents the antigen-induced response in a murine model of allergic airway inflammation. Inflamm Res. 2007;56(1):24–31. doi: 10.1007/s00011-007-5184-9. [DOI] [PubMed] [Google Scholar]
- Amin MA, Haas CS, Zhu K, Mansfield PJ, Kim MJ, Lackowski NP, Koch AE. Migration inhibitory factor up-regulates vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 via Src, PI3 kinase, and NFkappaB. Blood. 2006;107(6):2252–61. doi: 10.1182/blood-2005-05-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjona A, Foellmer HG, Town T, Leng L, McDonald C, Wang T, Wong SJ, Montgomery RR, Fikrig E, Bucala R. Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J Clin Invest. 2007;117(10):3059–66. doi: 10.1172/JCI32218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt U, Wennemuth G, Barth P, Nain M, Al-Abed Y, Meinhardt A, Gemsa D, Bacher M. Release of macrophage migration inhibitory factor and CXCL8/interleukin-8 from lung epithelial cells rendered necrotic by influenza A virus infection. J Virol. 2002;76(18):9298–306. doi: 10.1128/JVI.76.18.9298-9306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awandare GA, Kremsner PG, Hittner JB, Keller CC, Clark IA, Weinberg JB, Perkins DJ. Higher production of peripheral blood macrophage migration inhibitory factor in healthy children with a history of mild malaria relative to children with a history of severe malaria. Am J Trop Med Hyg. 2007;76(6):1033–6. [PubMed] [Google Scholar]
- Ayoub S, Hickey MJ, Morand EF. Mechanisms of disease: macrophage migration inhibitory factor in SLE, RA and atherosclerosis. Nat Clin Pract Rheumatol. 2008;4(2):98–105. doi: 10.1038/ncprheum0701. [DOI] [PubMed] [Google Scholar]
- Bacher M, Eickmann M, Schrader J, Gemsa D, Heiske A. Human cytomegalovirus-mediated induction of MIF in fibroblasts. Virology. 2002;299(1):32–7. doi: 10.1006/viro.2002.1464. [DOI] [PubMed] [Google Scholar]
- Baugh JA, Bucala R. Macrophage migration inhibitory factor. Crit Care Med. 2002;30(1 Supp):S27–S35. [PubMed] [Google Scholar]
- Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13(5):587–96. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF) Biochemistry. 1994;33(47):14144–55. doi: 10.1021/bi00251a025. [DOI] [PubMed] [Google Scholar]
- Binsky I, Haran M, Starlets D, Gore Y, Lantner F, Harpaz N, Leng L, Goldenberg DM, Shvidel L, Berrebi A, Bucala R, Shachar I. IL-8 secreted in a macrophage migration-inhibitory factor- and CD74-dependent manner regulates B cell chronic lymphocytic leukemia survival. Proc Natl Acad Sci U S A. 2007;104(33):13408–13. doi: 10.1073/pnas.0701553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozza FA, Gomes RN, Japiassu AM, Soares M, Castro-Faria-Neto HC, Bozza PT, Bozza MT. Macrophage migration inhibitory factor levels correlate with fatal outcome in sepsis. Shock. 2004;22(4):309–13. doi: 10.1097/01.shk.0000140305.01641.c8. [DOI] [PubMed] [Google Scholar]
- Bozza M, Satoskar AR, Lin G, Lu B, Humbles AA, Gerard C, David JR. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med. 1999;189(2):341–6. doi: 10.1084/jem.189.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. 2006a;7(3):235–9. doi: 10.1038/ni1316. [DOI] [PubMed] [Google Scholar]
- Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006b;12(12):1365–71. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med. 1994;179(6):1895–902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calandra T, Echtenacher B, Roy DL, Pugin J, Metz CN, Hultner L, Heumann D, Mannel D, Bucala R, Glauser MP. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000;6(2):164–70. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3(10):791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capobianchi MR. Induction of lymphomonocyte activation by HIV-1 glycoprotein gp120. Possible role in AIDS pathogenesis. J Biol Regul Homeost Agents. 1996;10(4):83–91. [PubMed] [Google Scholar]
- Chen LC, Lei HY, Liu CC, Shiesh SC, Chen SH, Liu HS, Lin YS, Wang ST, Shyu HW, Yeh TM. Correlation of serum levels of macrophage migration inhibitory factor with disease severity and clinical outcome in dengue patients. Am J Trop Med Hyg. 2006;74(1):142–7. [PubMed] [Google Scholar]
- Chiodi F, Bredberg-Raden U, Biberfeld G, Bottiger B, Albert J, Asjo B, Fenyo EM, Norrby E. Radioimmunoprecipitation and Western blotting with sera of human immunodeficiency virus infected patients: a comparative study. AIDS Res Hum Retroviruses. 1987;3(2):165–76. doi: 10.1089/aid.1987.3.165. [DOI] [PubMed] [Google Scholar]
- Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use correlates with disease progression in HIV-1--infected individuals. J Exp Med. 1997;185(4):621–8. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland KF. Modulation of HIV-1 transcription by cytokines and chemokines. Mini Rev Med Chem. 2005;5(12):1093–101. doi: 10.2174/138955705774933383. [DOI] [PubMed] [Google Scholar]
- Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312(5996):763–7. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381(6584):661–6. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Donnelly SC, Haslett C, Reid PT, Grant IS, Wallace WA, Metz CN, Bruce LJ, Bucala R. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat Med. 1997;3(3):320–3. doi: 10.1038/nm0397-320. [DOI] [PubMed] [Google Scholar]
- Duh EJ, Maury WJ, Folks TM, Fauci AS, Rabson AB. Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc Natl Acad Sci U S A. 1989;86(15):5974–8. doi: 10.1073/pnas.86.15.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emonts M, Sweep FC, Grebenchtchikov N, Geurts-Moespot A, Knaup M, Chanson AL, Erard V, Renner P, Hermans PW, Hazelzet JA, Calandra T. Association between high levels of blood macrophage migration inhibitory factor, inappropriate adrenal response, and early death in patients with severe sepsis. Clin Infect Dis. 2007;44(10):1321–8. doi: 10.1086/514344. [DOI] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272(5263):872–7. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- Finnegan A, Roebuck KA, Nakai BE, Gu DS, Rabbi MF, Song S, Landay AL. IL-10 cooperates with TNF-alpha to activate HIV-1 from latently and acutely infected cells of monocyte/macrophage lineage. J Immunol. 1996;156(2):841–51. [PubMed] [Google Scholar]
- Flieger O, Engling A, Bucala R, Lue H, Nickel W, Bernhagen J. Regulated secretion of macrophage migration inhibitory factor is mediated by a non-classical pathway involving an ABC transporter. FEBS Lett. 2003;551(1–3):78–86. doi: 10.1016/s0014-5793(03)00900-1. [DOI] [PubMed] [Google Scholar]
- Gando S, Sawamura A, Hayakawa M, Hoshino H, Kubota N, Nishihira J. High macrophage migration inhibitory factor levels in disseminated intravascular coagulation patients with systemic inflammation. Inflammation. 2007;30(3–4):118–24. doi: 10.1007/s10753-007-9027-1. [DOI] [PubMed] [Google Scholar]
- Gore Y, Starlets D, Maharshak N, Becker-Herman S, Kaneyuki U, Leng L, Bucala R, Shachar I. Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J Biol Chem. 2008;283(5):2784–92. doi: 10.1074/jbc.M703265200. [DOI] [PubMed] [Google Scholar]
- He XX, Yang J, Ding YW, Liu W, Shen QY, Xia HH. Increased epithelial and serum expression of macrophage migration inhibitory factor (MIF) in gastric cancer: potential role of MIF in gastric carcinogenesis. Gut. 2006;55(6):797–802. doi: 10.1136/gut.2005.078113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hladik F, McElrath MJ. Setting the stage: host invasion by HIV. Nat Rev Immunol. 2008;8(6):447–57. doi: 10.1038/nri2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoi AY, Iskander MN, Morand EF. Macrophage migration inhibitory factor: a therapeutic target across inflammatory diseases. Inflamm Allergy Drug Targets. 2007;6(3):183–90. doi: 10.2174/187152807781696455. [DOI] [PubMed] [Google Scholar]
- Ichiyama H, Onodera S, Nishihira J, Ishibashi T, Nakayama T, Minami A, Yasuda K, Tohyama H. Inhibition of joint inflammation and destruction induced by anti-type II collagen antibody/lipopolysaccharide (LPS)-induced arthritis in mice due to deletion of macrophage migration inhibitory factor (MIF) Cytokine. 2004;26(5):187–94. doi: 10.1016/j.cyto.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Javeed A, Zhao Y. Macrophage-migration inhibitory factor: role in inflammatory diseases and graft rejection. Inflamm Res. 2008;57(2):45–50. doi: 10.1007/s00011-007-7110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzierska K, Crowe SM. Cytokines and HIV-1: interactions and clinical implications. Antivir Chem Chemother. 2001;12(3):133–50. doi: 10.1177/095632020101200301. [DOI] [PubMed] [Google Scholar]
- Kinter A, Arthos J, Cicala C, Fauci AS. Chemokines, cytokines and HIV: a complex network of interactions that influence HIV pathogenesis. Immunol Rev. 2000;177:88–98. doi: 10.1034/j.1600-065x.2000.17708.x. [DOI] [PubMed] [Google Scholar]
- Klasse PJ, Moore JP. Is there enough gp120 in the body fluids of HIV-1-infected individuals to have biologically significant effects? Virology. 2004;323(1):1–8. doi: 10.1016/j.virol.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, Johannes FJ, Roger T, Calandra T, Kapurniotu A, Grell M, Finkelmeier D, Brunner H, Bernhagen J. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature. 2000;408(6809):211–6. doi: 10.1038/35041591. [DOI] [PubMed] [Google Scholar]
- Larson DF, Horak K. Macrophage migration inhibitory factor: controller of systemic inflammation. Crit Care. 2006;10(2):138. doi: 10.1186/cc4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PI, Ciccone EJ, Read SW, Asher A, Pitts R, Douek DC, Brenchley JM, Sereti I. Evidence for Translocation of Microbial Products in Patients with Idiopathic CD4 Lymphocytopenia. J Infect Dis. 2009;199(11):1664–1670. doi: 10.1086/598953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197(11):1467–76. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima RG, Van Weyenbergh J, Saraiva EM, Barral-Netto M, Galvao-Castro B, Bou-Habib DC. The replication of human immunodeficiency virus type 1 in macrophages is enhanced after phagocytosis of apoptotic cells. J Infect Dis. 2002;185(11):1561–6. doi: 10.1086/340412. [DOI] [PubMed] [Google Scholar]
- Lucia MB, Savarino A, Straface E, Golotta C, Rastrelli E, Matarrese P, Rutella S, Malorni W, Cauda R. Role of lymphocyte multidrug resistance protein 1 in HIV infection: expression, function, and consequences of inhibition. J Acquir Immune Defic Syndr. 2005;40(3):257–66. doi: 10.1097/01.qai.0000181280.68046.23. [DOI] [PubMed] [Google Scholar]
- Meazza C, Travaglino P, Pignatti P, Magni-Manzoni S, Ravelli A, Martini A, De Benedetti F. Macrophage migration inhibitory factor in patients with juvenile idiopathic arthritis. Arthritis Rheum. 2002;46(1):232–7. doi: 10.1002/1529-0131(200201)46:1<232::AID-ART10059>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A. 2002;99(1):345–50. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Fernandez MA, Navarro J, Garcia A, Punzon C, Fernandez-Cruz E, Fresno M. Replication of human immunodeficiency virus-1 in primary human T cells is dependent on the autocrine secretion of tumor necrosis factor through the control of nuclear factor-kappa B activation. J Allergy Clin Immunol. 1997;100(6 Pt 1):838–45. doi: 10.1016/s0091-6749(97)70282-3. [DOI] [PubMed] [Google Scholar]
- Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451(7177):425–30. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- Orenstein JM, Fox C, Wahl SM. Macrophages as a source of HIV during opportunistic infections. Science. 1997;276(5320):1857–61. doi: 10.1126/science.276.5320.1857. [DOI] [PubMed] [Google Scholar]
- Ortigao-de-Sampaio MB, Shattock RJ, Hayes P, Griffin GE, Linhares-de-Carvalho MI, Ponce de Leon A, Lewis DJ, Castello-Branco LR. Increase in plasma viral load after oral cholera immunization of HIV-infected subjects. AIDS. 1998;12(14):F145–50. doi: 10.1097/00002030-199814000-00001. [DOI] [PubMed] [Google Scholar]
- Schols D, De Clercq E. Human immunodeficiency virus type 1 gp120 induces anergy in human peripheral blood lymphocytes by inducing interleukin-10 production. J Virol. 1996;70(8):4953–60. doi: 10.1128/jvi.70.8.4953-4960.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Leng L, Wang T, Wang W, Du X, Li J, McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity. 2006;25(4):595–606. doi: 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley SK, Ostrowski MA, Justement JS, Gantt K, Hedayati S, Mannix M, Roche K, Schwartzentruber DJ, Fox CH, Fauci AS. Effect of immunization with a common recall antigen on viral expression in patients infected with human immunodeficiency virus type 1. N Engl J Med. 1996;334(19):1222–30. doi: 10.1056/NEJM199605093341903. [DOI] [PubMed] [Google Scholar]
- Stevenson M. HIV-1 pathogenesis. Nat Med. 2003;9(7):853–60. doi: 10.1038/nm0703-853. [DOI] [PubMed] [Google Scholar]
- Stopak KS, Chiu YL, Kropp J, Grant RM, Greene WC. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J Biol Chem. 2007;282(6):3539–46. doi: 10.1074/jbc.M610138200. [DOI] [PubMed] [Google Scholar]
- Toh ML, Aeberli D, Lacey D, Yang Y, Santos LL, Clarkson M, Sharma L, Clyne C, Morand EF. Regulation of IL-1 and TNF receptor expression and function by endogenous macrophage migration inhibitory factor. J Immunol. 2006;177(7):4818–25. doi: 10.4049/jimmunol.177.7.4818. [DOI] [PubMed] [Google Scholar]
- Toossi Z, Sierra-Madero JG, Blinkhorn RA, Mettler MA, Rich EA. Enhanced susceptibility of blood monocytes from patients with pulmonary tuberculosis to productive infection with human immunodeficiency virus type 1. J Exp Med. 1993;177(5):1511–6. doi: 10.1084/jem.177.5.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varthakavi V, Heimann-Nichols E, Smith RM, Sun Y, Bram RJ, Ali S, Rose J, Ding L, Spearman P. Identification of calcium-modulating cyclophilin ligand as a human host restriction to HIV-1 release overcome by Vpu. Nat Med. 2008;14(6):641–7. doi: 10.1038/nm1778. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wahl SM, Greenwell-Wild T, Peng G, Hale-Donze H, Orenstein JM. Co-infection with opportunistic pathogens promotes human immunodeficiency virus type 1 infection in macrophages. J Infect Dis. 1999;179(Suppl 3):S457–60. doi: 10.1086/314814. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Fadeel B, Hodara V, Fenyo EM. Induction of apoptosis by primary HIV-1 isolates correlates with productive infection in peripheral blood mononuclear cells. AIDS. 1997;11(10):1219–25. doi: 10.1097/00002030-199710000-00004. [DOI] [PubMed] [Google Scholar]