

Abstract
The availability and interactions of signaling proteins are tightly regulated in time and space to produce specific and localized effects. For calmodulin (CaM), a key transducer of intracellular Ca2+ signaling, binding to its variety of targets initiates signaling cascades and regulates its subcellular localization, thereby making it unavailable for subsequent binding interactions. Among CaM's numerous targets, Ca2+/CaM-dependent protein kinase II is one of the most striking due to its unique ability to increase its affinity for CaM by autophosphorylation and to translocate when bound to Ca2+/CaM. Two-photon fluorescence correlation spectroscopy and cross-correlation spectroscopy were utilized to compare mobility and molecular interactions between CaM and Ca2+/CaM-dependent protein kinase II in solution and in living cells. These techniques revealed that CaM availability in cells could be altered by a change in intracellular conditions. Two-photon fluorescence cross-correlation spectroscopy exemplifies a generally applicable approach for studying protein–protein interactions in living cells that allows access to the behavior of signaling molecules within their native environment to probe for heterogeneities in signaling pathways in different cellular compartments.
Intracellular signal transduction is an elegant process of information transfer through rapid and precise biochemical interactions. The historic concept where receptors and other signaling proteins remain in place while spatial signal transmission is proliferated by the rapid diffusion of second messengers can no longer completely describe all types of signaling (1). Instead, a more dynamic bidirectional scenario emerges where either the protein goes to the signal, or the signal comes to the protein, or both. One such mechanism is the targeted colocalization of interacting signaling proteins for the selective activation of downstream processes (1, 2). These localization mechanisms can be highly regulated and rapidly reversible.
The Ca2+-calmodulin (CaM) pathway is an ideal illustration of this paradigm shift. CaM binds four Ca2+ ions, exposing sites that lead to target binding. An assumption of many models of CaM-dependent regulation has been that a large pool of intracellular CaM is freely diffusible. Based on this assumption, CaM-dependent function would strictly depend on transient Ca2+ signals as the rate-limiting step in enzyme activation. The idea of compartmentalization and limiting concentrations of CaM depicts a different scenario where competition among multiple CaM-binding proteins for limited CaM would ultimately determine the profile of CaM-dependent enzyme activation (3–6). Ca2+/CaM-dependent protein kinase II (CaM-kinase II) is the most concentrated CaM-dependent enzyme in nerve cells. It also has the unique ability to increase its affinity for CaM by several orders of magnitude by autophosphorylation (7, 8). In cells where CaM-kinase II is abundant, such as hippocampal neurons, this characteristic may act as a mechanism to modulate other CaM-dependent processes by sequestering CaM onto CaM-kinase II, making it unavailable for the activation of other enzymes.
Details of earlier models for CaM-dependent regulation were based solely on studies of CaM and its target enzymes in dilute solution. However, the intracellular milieu is considerably more complex where constraints on diffusion may be imposed by factors, such as macromolecular crowding and cytoskeletal elements, that could make some compartments unavailable or even promote intermolecular associations not favored in dilute solutions (9–11). Therefore, CaM availability must be determined empirically in cells and cannot be directly extrapolated from experiments using purified proteins.
Here we use two-photon cross-correlation spectroscopy [TPCCS; (12, 13)] to analyze the dynamics of protein–protein interactions of CaM and CaM-kinase II down to the single-molecule level both in solution and in living cells. The use of these techniques revealed that CaM availability in cells can be altered by a change in intracellular conditions, and that CaM availability can be modulated by protein–protein interactions.
Materials and Methods
Mutagenesis, Expression, and Purification of CaM. The CaM expression plasmid pCR2 was generously provided by Carol Rohl (University of California, Santa Cruz) and Rachel Klevit (University of Washington, Seattle). Introduction of a single Cys residue by conversion of Asp at amino acid 3 to Cys to produce CaM(C3) was accomplished with the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The presence of the desired mutation and the absence of artifacts were verified by DNA sequencing. BL21(DE3)plysS (Novagen) cells expressing CaM were grown and harvested, and recombinant CaM was purified as described (14) with minor modifications, dialyzed into 50 mM Mops, pH 7.0, and stored at –20°C. CaM was quantified by a modified Bradford protein assay (Bio-Rad) and was homogeneous as judged by SDS/PAGE.
Labeling of CaM(C3) with Alexa 633. CaM labeling was performed as described (8) with the following modifications. CaM(C3) was reduced with dithiothreitol; Alexa 633 maleimide (Molecular Probes) was dissolved in DMSO and added in 6.5-fold molar excess over both protein and dithiothreitol. The reaction was allowed to proceed overnight at 4°C and was quenched by using 10 mM 2-mercaptoethanol. The labeled protein was purified by using a 15-cm Bio-Gel P6-DG (Bio-Rad) column and eluted with 50 mM Hepes, pH 7.0. Based on εAlexa633 = 100,000/(M·cm) at 622 nm (Molecular Probes), probe-to-protein ratios were determined to be ≈0.9 for the labeled protein. Full characterization of the fluorescently labeled protein was done to determine its ability to activate CaM-kinase II and its spectral properties over a physiological range of parameters, such as pH, Ca2+ concentration, and ionic strength (unpublished work).
Construction, Expression, and Purification of eGFP-CaM-Kinase II. A eukaryotic expression plasmid for enhanced GFP (eGFP) CaM-kinase II was generated by subcloning a rat CaMKIIα cDNA into the pEGFP-C2 vector (Clontech). The eGFP-CaM-kinase II construct was sequenced to verify that no undesired mutations were present. The eGFP-CaM-kinase II construct created above was subcloned into the pFastBac1 baculovirus expression vector (GIBCO/Invitrogen), and recombinant eGFP-CaM-kinase II was produced by using the baculovirus expression system and purified as described (8) to near homogeneity. A full characterization of eGFP-CaM-kinase II showed that the protein was a holoenzyme and possessed similar specific activity and minimal basal independent activity as wild-type enzyme (S.A.K. and M.N.W., unpublished observation). Purified eGFP-CaM-kinase II preparations were stored at –80°C.
Preparation of Reactions in Solution for Two-Photon Fluorescence Correlation Spectroscopy (TPFCS) and TPCCS Measurements. Purified eGFP-CaM-kinase II samples were spun in a microfuge at 110 × g for 30 min at 4°C to remove particulate material after thawing. All solutions were prepared in a low-fluorescence HPLC grade water (Merck Sharp & Dohme) and were filter sterilized. Twenty-four-microwell chambers (Evotec OAI, Hamburg, Germany) were preincubated with 10% BSA for 10 min followed by two subsequent washes with an enzyme dilution buffer (25 mM Hepes, pH 7.0/150 mM KCl 0.1 mg/ml BSA). Reaction conditions for cross-correlation measurements included: Alexa 633(C3)CaM and eGFP-αCaMKII in 25 mM Hepes, pH 7.0; 100 mM KCl, 10 mM Mg2+ with 0.5 mM CaCl2; and 1 mM MgATP (Amersham Pharmacia) followed by 1 mM EGTA. Each reaction was well mixed prior to each measurement, and measurements were performed at room temperature.
Cell Culture. Human embryonic kidney 293 (HEK293) cells were kindly provided by Ralf Nehring and Andreas Karschin (Max Planck Institute for Biophysical Chemistry) and were originally obtained from American Type Culture Collection (CRL-1573). HEK293 cells were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum and 100 units/ml penicillin and 100 units/ml streptomycin in a humidified incubator containing 5% CO2 at 37°C. Cells were plated at a density of ≈250,000 cells per dish on poly-d-lysine-(0.01 mg/ml) coated MatTek dishes (MatTek, Ashland, MA) and used 24–48 h after plating. To minimize autofluorescence, HEK293 cells were plated in phenol red-free media prior to experiments.
Transfection and Making of Stable Cell Lines. HEK293 cells were transfected with eGFP-CaM-kinase II using Effectene (Qiagen, Hilden, Germany) as described by the manufacturer. Cells were passaged into selective medium containing 1,000 units/ml G418 (GIBCO/BRL/Invitrogen) 3 d posttransfection and maintained under selection for at least 3 wk prior to use. Approximately 50% of the population was positive for eGFP-CaM-kinase II expression in a nanomoler concentration range.
Electroporation. Alexa 633-labeled CaM at amino acid 3 (Cys) [Alexa 633(C3)CaM] was introduced into adherent cells based on a technique developed by Teruel et al. (15). Electroporation was performed at 150–250 V/cm by using three rectangular voltage pulses, each 50 ms long and 1 min apart (BTX, Holliston, MA) in electroporation buffer (125 mM NaCl/5 mM KCl/1.5 mM MgCl2/20 mM Hepes/10 mM glucose, pH 7.4). Prior to experiments, cells were allowed to recover for at least 1 h.
Conditions for Cellular Imaging and Spectroscopy Measurements. During all experiments, cells were incubated at room temperature in a Hepes-buffered saline solution (HBSS) containing: 116 mM NaCl; 5.4 mM KCl; 0.8 mM MgSO4; 1.6 mM CaCl2; 20 mM Hepes; and 10 mM glucose, pH 7.4 (16). For some experiments, cells were incubated in one of the following conditions: HBSS with no added Ca2+, 15 μg/ml α-hemolysin (α-toxin; Sigma), and 200 μM EGTA (Ca2+-free); or HBSS with 15 μg/ml α-hemolysin, 10 mM CaCl2, and 1 mM MgATP (elevated Ca2+). α-Hemolysin permeabilizes the cell membrane, allowing rapid equilibrium of extracellular ions and nucleotides with the intracellular environment. HEK293 cells were prepared in parallel for spectroscopy measurements and Ca2+ imaging. Fluo-4 was used to confirm that Ca2+ was being elevated or decreased uniformly in all cells under the different conditions.
TPCCS Setup. TPFCS and TPCCS measurements were achieved by using a tunable mode-locked titanium:sapphire laser (Tsunami, Spectra-Physics), operating at 80 MHz with 100-fs pulses, coupled to an inverted Olympus (Melville, NY) IX50 microscope (12). A 5× Galilean beam expander (coated for the near-IR spectral range; Thorlabs, Newton, NJ) ensured proper overfilling of the back aperture of the water immersion objective (Uplan-Apo/IR 60×, numerical aperture 1.2, Olympus), creating a diffraction-limited focal spot. Determined from calibration measurements of Rhodamine green (Molecular Probes), a radial dimension (r0) of 0.35 μm and an axial dimension (z0) of 0.95 μm with an effective volume element of ≈0.22 fl were used to describe the two-photon focal volume. The sample was excited with a single laser line at 920 nm. Fluorescence was separated from the excitation light path by the first dichroic mirror (750DCSP, AHF Analysentechnik, Tübingen, Germany) with a bandpass emission filter 600DF200 (AHF) for efficient suppression of IR excitation light. The emitted light was collimated and then split by a second longpass dichroic mirror 615LP (AHF). Additional detection light specificity was obtained by a 525/70M (AHF) bandpass filter in the eGFP and 680/100 (AHF) bandpass filter in the Alexa 633 detection channels. Two optical fiber coupled avalanche photodiodes SPCM-200 (Perkin–Elmer) were used for detection. The optical fibers (Thorlabs) with a diameter of 100 μm were separately adjustable in the x, y, and z directions. The digital pulses of the avalanche photodiodes were amplified, split, and simultaneously correlated online by two personal computer ALV-5000 multiple τ correlator cards (ALV-Laser, Langen, Germany), one operating in a dual autocorrelation, and the other operating in a cross-correlation mode. For single-color measurements, the second dichroic mirror (615LP) was removed. Evaluation of the curves was accomplished by using origin (MicroCal Software, Northampton, MA) with a Levenberg–Marquardt nonlinear least-squares fitting routine.
Spectra for eGFP [Fig. 1b(1) and Alexa 633, Fig. 1b(2)] were taken by using a fiber-coupled spectrometer (Ocean Optics, Dunedin, IL) with fiber diameter of 100 μm. Two-photon excited spectra were measured from micromolar dye solutions within the focal volume element at 920 nm. The transmission characteristics of the dichroic mirror [Fig. 1b(3)] and filters [Fig. 1b(4) and (5)] used for spectral separation of the emission light are also plotted. Optimal measurement conditions for these experiments were determined and will be discussed elsewhere (unpublished observations).
Fig. 1.

Setting up TPCCS. (a) The optical setup is based on an inverted Olympus IX50 microscope. A single Ti:Sa laser line illuminates the sample; the detected light is split by a dichroic mirror behind the collimating lens and focused onto two different optical fiber-coupled detectors [single-photon avalanche diodes (SPADs)]. The signals are correlated online by a personal computer correlator card. (b) Normalized fluorescence emission spectra of the two-photon excited dye system measured within the focal spot by a fiber-coupled spectrometer. Excitation wavelength is 920 nm for both eGFP (trace 1) and Alexa 633 (trace 2). Very little overlap is seen between the two emission spectra, and the dichroic mirror (trace 3, 615LP), coupled to the emission filters (trace 4, eGFP), 525/70M; and (trace 5, Alexa 633, 680/100), cleanly separates the signal for excellent discrimination between the two channels.
Theoretical Concept TPFCS and TPCCS. The technique of fluorescence correlation spectroscopy measures mobility and chemical kinetics of fluorescent molecules (17) by autocorrelation analysis of time-resolved fluorescence intensity fluctuations, F(t), collected from a small group of diffusing molecules (typically nanomolar concentrations). At equilibrium, the number of fluorescent molecules within a small probe volume fluctuates about a mean value due to individual molecules diffusing in and out of the volume. The deviations from the mean in the measured fluorescence are correlated as G(τ), which is generally given by:
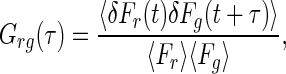 |
[1] |
where the indices refer to one or two measured fluorescence signals, Fr and/or Fg. In the case of one fluorescent species [Alexa 633(C3)CaM or eGFP-CaM-kinase II alone], Eq. 1 (r = g) defines the normalized fluorescence autocorrelation function in a single detection channel.
The autocorrelation function decays with time τ. The shape of the decay and characteristic decay time are related to the mechanisms and rates of the processes that generate the fluorescence fluctuations. The amplitude of the autocorrelation function is inversely proportional to the number of fluorescent particles in the volume element.
For the simplest case (i.e., dye or protein in solution), in a system of one fluorescent species undergoing Brownian motion in 3D, the fluorescence autocorrelation curve can be fit to
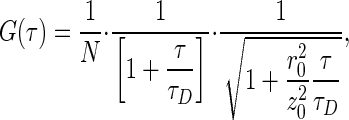 |
[2] |
where r0 and z0 are the half-axes of the detection volume, assumed to have a Gaussian shape; N is the average number of molecules in the detection volume; and τD is average lateral transit time of the molecules through the focus (τD = r20/8D for two-photon excitation, where D is the diffusion coefficient). By calibrating the detection volume with a solution of freely diffusing dye molecules with known diffusion properties, a diffusion coefficient of the probe of interest can be calculated from empirically determined diffusion times, τD. For intracellular measurements, a two-diffusing species model was used by fitting to a weighted sum of two terms in Eq. 2.
Cross-correlation, an extension of FCS using two spectrally distinct fluorophores, analyzes molecular interactions of different molecules (see refs. 18 and 19 for a more detailed discussion of the theory and formalism of cross-correlation analysis). For this type of analysis, fluorescence fluctuations of two different fluorescent molecules [(r ≠ g) in Eq. 1, Alexa 633(C3)CaM and eGFP-CaM-kinase II] are simultaneously recorded and cross-correlated. Grg defines the cross-correlation function, which compares the fluorescence signals of different species for different lag times τ. Under ideal conditions with minimal spectral crosstalk between the dyes and in the absence of fluorescence resonance energy transfer, the amplitude Grg(0) is directly proportional to the relative concentration of bound species. The absolute concentration can be derived from the corresponding autocorrelation curves.
An excess of Alexa 633(C3)CaM was always maintained for in vitro reactions to favor binding. The binding of eGFP-CaM-kinase II molecules that have at least one CaM bound to it is defined as:
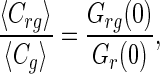 |
[3] |
where 〈Crg〉 = Grg(0)/[Veff·(Gr(0)Gg(0))], 〈Cg〉 = 1/[Veff·Gg (0)], and Veff is the effective detection volume. In the case of no binding, Grg(0) equals zero.
Results
Intracellular Mobility of CaM. Autocorrelation curves of Alexa 633(C3)CaM were measured in HEK293 cells stably transfected with eGFP-CaM-kinase II under Ca2+-free or elevated Ca2+ conditions in the presence of 15 μg of α-hemolysin (used for cell permeabilization to elevate intracellular Ca2+ and MgATP). Qualitatively, the half-maximal value of the curve reflects the approximate diffusion time of the Alexa 633(C3)CaM (molecular mass ≈17 kDa), and a right shift in an FCS curve indicates a slower diffusing species. For changes in diffusion to be detected, CaM must be bound to molecules of significantly larger size, because diffusion time varies with the inverse cube root of the molecular mass.
In the Ca2+-free condition, the majority of Alexa 633(C3)CaM (72.0%) is found to be freely diffusing (τd = 0.73 ms, see Fig. 2 Inset) in the HEK293 cell, with the remainder (28.0%) diffusing very slowly (τd = 15.5 ms, see Fig. 2 Inset). The ≈3-fold slower intracellular diffusion of CaM (τd = 0.73 ms, see Fig. 2 Inset) compared with in-solution measurements (τd = 0.25 ms; see Fig. 3) can be attributed to increased cellular viscosity (20). The slow component is most likely an average of different bound populations of Alexa 633(C3)CaM, because it is poorly resolved under these measurement conditions. Elevating Ca2+ produced a large change in the intracellular diffusion of Alexa 633(C3)CaM (Fig. 2; 2-fold decrease in the rate of diffusion of the faster component in a two-component fit) in the presence of eGFP-CaM-kinase II [12 subunits (21) with a total ≈984 kDa]. Not only was there a decrease in the mobility of Alexa 633(C3)CaM, but also the fractions of the two components shifted to an equal distribution between the two different populations in the elevated Ca2+ condition. As indicated by the mobility, this suggests that a change in intracellular Ca2+ concentration can alter the availability of CaM and perhaps ultimately the profile of CaM-dependent activation.
Fig. 2.

Inducing alterations in intracellular diffusion of Alexa 633(C3)CaM. Autocorrelation curves (normalized) were measured in HEK293 cells stably transfected with eGFP-CaM-kinase II under Ca2+-free (+200 μM EGTA) or elevated Ca2+ (10 mM Ca2+/1 mM MgATP) conditions in the presence of 15 μg of α-hemolysin. A large change in mobility of Alexa 633(C3)CaM is seen in elevated Ca2+. The τd values reported (Inset) are based on a two-component model (see Materials and Methods).
Fig. 3.

Binding reactions of Alexa 633(C3)CaM and eGFP-CaM-kinase II assessed with TPCCS in solution. The central schematic depicts molecules of Alexa 633(C3)CaM and eGFP-CaM-kinase II diffusing through the focal volume, giving rise to the fluorescent signals in the green and red channels (a). Only eGFP-CaM-kinase II having at least one Alexa 633(C3)CaM bound to it appears in both channels simultaneously and contributes to the cross-correlation amplitude (coincident green and red signals in middle of a). Normalized cross-correlation curves show the extent of binding in two different conditions in the presence of 0.5 mM Ca2+/1 mM MgATP (light blue) and 1 mM EGTA (dark blue) (b). The corresponding autocorrelation [green, eGFP-CaM-kinase II; red, Alexa 633(C3)CaM] and cross-correlation (Lower, broken red–green curve) curves are also provided for the two conditions in c and d.
Assessment of Binding Between CaM and CaM-Kinase II in Solution by Using TPCCS. Although a change in diffusion of CaM, in the presence of added binding sites (CaM-kinase II, Fig. 2), indicates an alteration of the availability of CaM, it does not directly specify the cause for the change. Therefore, we utilized cross-correlation to investigate binding interactions between CaM and CaM-kinase II. An extension of the standard FCS scheme, TPCCS is a sensitive tool to follow the association or dissociation of different molecules.
In the central schematic of Fig. 3, both bound and unbound Alexa 633(C3)CaM and eGFP-CaM-kinase II (shown here as a monomer for clarity) are depicted diffusing through the focal volume, giving rise to the fluorescent signals in the green and red channels (Fig. 3a, seen as schematic fluorescence fluctuations). Red CaMs, both free and bound to CaM-kinase II, contribute to the red CaM autocorrelation curve (red curves in Fig. 3 c and d), whereas green CaM-kinase IIs with and without red CaM bound contribute to the green autocorrelation curve (green curves in Fig. 3 c and d). However, only green CaM-kinase IIs having at least one red CaM bound to it appear in both channels simultaneously (coincident green and red signals in middle of Fig. 3a) and contribute to the cross-correlation amplitude (broken red–green line in Fig. 3d).
The cross-correlation amplitude (bound CaM-kinase II and CaM) relates to the red CaM autocorrelation amplitude as the number of bound molecules to the total number of green CaM-kinase II in the system (Eq. 3; Fig. 3 c Lower and d Lower; red curves indicate Alexa 633(C3)CaM autocorrelation, and broken red–green lines show cross-correlation). The greater the fraction of bound Ca2+-CaM–CaM-kinase II complexes, the higher the cross-correlation amplitude relative to the red autocorrelation amplitude (Fig. 3d Lower). Because the nonnormalized cross-correlation amplitude is the same height as the red autocorrelation curve for CaM, almost all green CaM-kinase II molecules observed must have at least one bound red CaM.
Several types of information (Table 1) can be culled from these data, including: concentrations of each of the interacting molecules, absolute ratios of the average molecules, extent of binding, and the diffusion of each of the molecules involved. These data is tabulated in Table 1 for the experiment described in Fig. 3. In this particular reaction, increased binding (95%) was seen in the presence of 0.5 mM Ca2+/1 mM MgATP; subsequently, 1 mM EGTA (causing only minimal dilution) was added to the same reaction to show the reversal of the binding (Fig. 3b). This resulted in reduced cross correlation (12%) due to the dissociation of CaM from CaM-kinase II. Negative control reaction of Alexa 633 hydrazide and CaM-kinase II (data not shown) under all conditions showed 8–12% cross correlation, indicating that the 11.5% of binding is nonspecific.
Table 1. Summary of parameters in reaction.
| +EGTA | +Ca2+/MgATP | |
|---|---|---|
| eGFP-αCaMKII | 0.7 molecules | 1.3 molecules |
| (5 nM) | (10 nM) | |
| Alexa 633(C3)CaM | 29.7 molecules | 44.6 molecules |
| (224 nM) | (337 nM) | |
| Ratio of molecules of αCaMKII to total CaM | 1:42 | 1:36 |
| Binding efficiency | 12 | 95 |
Summary of the calculated values from the analysis of autocorrelation [Fig. 3 c and d; green, eGFP-CaM-kinase II; red, Alexa 633(C3)CaM] and cross-correlation (Fig. 3 c Lower and d Lower, broken red-green curve) curves including the observed ratio between the two molecules, extent of binding, concentrations, and diffusion of each of the interacting molecules in the presence of 1 mM EGTA (Fig. 3c, dark blue) and 0.5 mM Ca2+/1 mM MgATP (Fig. 3d, light blue).
Monitoring Intracellular CaM and CaM-Kinase II Binding Dynamics. Historically, colocalization of two molecules was used as a method for estimating possible interactions between proteins. Confocal laser scanning images of stably transfected eGFP-CaM-kinase II HEK293 cells electroporated with Alexa 633(C3)CaM show little noticeable difference in their distribution in the presence of elevated Ca2+ (Fig. 4a) or Ca2+-free (Fig. 4b) conditions. In both images, Alexa 633(C3)CaM was localized to the cytosol with a higher concentration in the nucleus, whereas eGFP-CaM-kinase II was generally cytoplasmic with very little signal in the nucleus. Although the molecules were colocalized in the cytoplasm in both conditions, analysis using TPCCS shows quite different binding dynamics.
Fig. 4.

Intracellular changes of binding of Alexa 633(C3)CaM and eGFP-CaM-kinase II. Confocal images of stably transfected eGFP-CaM-kinase II HEK293 cell line electroporated with Alexa633(C3)CaM show little change in appearance under 10 mM Ca2+/1 mM MgATP (a) followed by +200 μM EGTA (b) conditions in the presence of 15 μg/ml α-hemolysin in the same dish. Intracellular cross-correlation curves (normalized) were measured under each of the conditions. The amplitude of the cross-correlation, corresponding to the binding efficiency, increased with elevated Ca2+/MgATP (light blue) and decreased with no Ca2+ (dark blue). (Bar = 10 μm.)
Cross-correlation curves were measured in the living cell under elevated Ca2+ followed by Ca2+-free conditions in the same dish, and curves were calibrated to the autocorrelation of Alexa 633(C3)CaM (Fig. 2b) that were collected simultaneously. Increases of intracellular [Ca2+] and [ATP] (with α-hemolysin to provide optimal conditions for maximal intracellular binding) increased the binding interaction in a predictable manner. The amplitude of the cross-correlation, corresponding to the binding efficiency, increased with elevated Ca2+ (light blue) and decreased with Ca2+-free conditions (dark blue; Fig. 4c). Experimentally, we find no evidence of binding in cells after they were bathed in Ca2+-free conditions.
Discussion
We successfully show the proof of the principle utilizing TPCCS to study the reversible binding dynamics of Alexa 633(C3)CaM and eGFP-CaM-kinase II both in solution and in the living cell. TPCCS provides a sensitive method for measuring protein–protein interactions and offers a complementary approach to the popular technique of fluorescence resonance energy transfer. TPCCS as applied in the present study exploits the technical and biological advantages of two-photon excitation to optimize cross correlation. Two-photon excitation confers the ability to simultaneously excite multiple dyes with a single laser line and requires no pinholes for detection, minimizing optical alignment issues and guaranteeing overlap of the excitation volumes. In addition to the general advantages for cellular applications of inherent optical sectioning, two-photon excitation reduces cumulative photobleaching, which can artificially decrease the cross-correlation amplitude. Thus, TPCCS allows access to studying protein–protein interactions of signaling molecules within their native environment and was specifically used here for the assessment of CaM-binding to CaM-kinase II inside living cells.
CaM-kinase II is a large dodecameric molecule with 12 potential binding sites for CaM that has the ability to modulate its own CaM-binding affinity when activated by Ca2+-saturated CaM (7, 8). Coupled with its particularly high concentrations in certain cells (22), these observations led to the proposal that a potential role for CaM-kinase II may be to modulate CaM availability for other CaM-dependent processes (7, 8, 23). The autocorrelation curves presented in Fig. 2 provide experimental in situ evidence for this hypothesis. In HEK-293 cells transfected with CaM-kinase II, conditions of elevated Ca2+ and Mg/ATP produced significant (2.2-fold) slowing of CaM diffusion. However, standard one-color FCS suffers from the ambiguity that a slowed CaM diffusion can be attributed to environmental effects, such as different local viscosities or nonspecific interactions, rather than specific binding to CaM-kinase II. We therefore applied TPCCS to directly assess CaM and CaM-kinase II interactions in vitro and subsequently in situ. The optimal conditions for measuring TPCCS data were established in vitro by using purified eGFP-tagged CaM-kinase II and Alexa 633-labeled CaM. As anticipated, these data revealed that in the presence of Ca2+ and Mg/ATP, the binding efficiency between CaM and CaM-kinase II increased dramatically from 12% (same as negative control) to 95%. By applying TPCCS to the HEK cell model, it was possible to establish that one cause of the slowed CaM diffusion was directly due to its interactions with CaM-kinase II under elevated Ca2+ and Mg/ATP conditions. In total, our data experimentally support the hypothesis that intracellular CaM availability, and therefore activation of the entire family of CaM-dependent enzymes, can be regulated by the presence of CaM-kinase II. We generally conclude that the combination of TPFCS and TPCCS is feasible, and both, when combined, are powerful techniques to assess the interactions of signaling molecules in vitro and inside living cells.
Acknowledgments
We thank Drs. Warren Zipfel and Rebecca Williams for help with the design of the custom-built electroporation system and Dr. Gary St. John Bird for advice regarding Ca2+ imaging in HEK293 cells. We also thank Kirsten Bacia, Elke Haustein, and Drs. Andy Hudmon and John Putkey for helpful scientific discussions and constructive criticism on the manuscript, and Karin Birkenfeld for technical assistance with cell culture. Fig. 1a of the cross-correlation spectroscopy setup was kindly provided by Elke Haustein (Max Planck Institute for Biophysical Chemistry). This work was supported by grants from the National Institute of Mental Health (to S.A.K.), the Fulbright Kommission (to S.A.K.), the University of Texas Graduate School of Biomedical Sciences (to S.A.K.), the National Institute of Neurological Disease and Stroke (to M.N.W.), the Human Frontiers Science Program (to M.N.W. and P.S.), and the German Ministry for Education and Research (to P.S.).
Abbreviations: CaM, calmodulin; CaM-kinase II, Ca2+/CaM-dependent protein kinase II; TPFCS, two-photon fluorescence correlation spectroscopy; TPCCS, two-photon cross-correlation spectroscopy; Alexa 633(C3)CaM, Alexa 633-labeled CaM at amino acid 3 (Cys); eGFP, enhanced GFP; HEK293, human embryonic kidney 293 cells.
References
- 1.Teruel, M. N. & Meyer, T. (2000) Cell 103, 181–184. [DOI] [PubMed] [Google Scholar]
- 2.Pawson, T. & Scott, J. D. (1997) Science 278, 2075–2080. [DOI] [PubMed] [Google Scholar]
- 3.Persechini, A. & Cronk, B. (1999) J. Biol. Chem. 274, 6827–6830. [DOI] [PubMed] [Google Scholar]
- 4.Luby-Phelps, K., Hori, M., Phelps, J. M. & Won, D. (1995) J. Biol. Chem. 270, 21532–21538. [DOI] [PubMed] [Google Scholar]
- 5.Bayley, P. M., Findlay, W. A. & Martin, S. R. (1996) Protein Sci. 5, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Persechini, A. & Stemmer, P. M. (2002) Trends Cardiovasc. Med. 12, 32–37. [DOI] [PubMed] [Google Scholar]
- 7.Meyer, T., Hanson, P. I., Stryer, L. & Schulman, H. (1992) Science 256, 1199–1202. [DOI] [PubMed] [Google Scholar]
- 8.Putkey, J. A. & Waxham, M. N. (1996) J. Biol. Chem. 271, 29619–29623. [DOI] [PubMed] [Google Scholar]
- 9.Minton, A. P. (2001) J. Biol. Chem. 276, 10577–10580. [DOI] [PubMed] [Google Scholar]
- 10.Luby-Phelps, K. (2000) Int. Rev. Cytol. 192, 189–221. [DOI] [PubMed] [Google Scholar]
- 11.Seksek, O., Biwersi, J. & Verkman, A. S. (1997) J. Cell Biol. 138, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinze, K. G., Koltermann, A. & Schwille, P. (2000) Proc. Natl. Acad. Sci. USA 97, 10377–10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohl, T., Heinze, K. G., Kuhlemann, R., Koltermann, A. & Schwille, P. (2002) Proc. Natl. Acad. Sci. USA 99, 12161–12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singla, S. I., Hudmon, A., Goldberg, J. M., Smith, J. L. & Schulman, H. (2001) J. Biol. Chem. 276, 29353–29360. [DOI] [PubMed] [Google Scholar]
- 15.Teruel, M. N., Blanpied, T. A., Shen, K., Augustine, G. J. & Meyer, T. (1999) J. Neurosci. Methods 93, 37–48. [DOI] [PubMed] [Google Scholar]
- 16.Pedrosa Ribeiro, C. M., McKay, R. R., Hosoki, E., Bird, G. S. & Putney, J. W., Jr. (2000) Cell Calcium 27, 175–185. [DOI] [PubMed] [Google Scholar]
- 17.Magde, D., Elson, E. L. & Webb, W. W. (1972) Phys. Rev. Lett. 29, 705–708. [Google Scholar]
- 18.Schwille, P., Meyer-Almes, F. J. & Rigler, R. (1997) Biophys. J. 72, 1878–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwille, P. (2001) in Fluorescence Correlation Spectroscopy: Theory and Applications, eds. Elson, E. & Rigler, R. (Springer, Berlin), pp. 383–408.
- 20.Schwille, P., Haupts, U., Maiti, S. & Webb, W. W. (1999) Biophys. J. 77, 2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolodziej, S. J., Hudmon, A., Waxham, M. N. & Stoops, J. K. (2000) J. Biol. Chem. 275, 14354–14359. [DOI] [PubMed] [Google Scholar]
- 22.Erondu, N. E. & Kennedy, M. B. (1985) J. Neurosci. 5, 3270–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanson, P. I., Meyer, T., Stryer, L. & Schulman, H. (1994) Neuron 12, 943–956. [DOI] [PubMed] [Google Scholar]