

Abstract
The mechanisms of nitric oxide (NO) signaling include binding to the iron centers in soluble guanylate cyclase and cytochrome c oxidase and posttranslational modification of proteins by S-nitrosation. Low levels of NO control mitochondrial number in cells, but little is known of the impact of chronic exposure to high levels of NO on mitochondrial function in endothelial cells. The focus of this study is the interaction of NO with mitochondrial respiratory complexes in cell culture and the effect this has on iron homeostasis. We demonstrate that chronic exposure of endothelial cells to NO decreased activity and protein levels of complexes I, II, and IV, whereas citrate synthase and ATP synthase were unaffected. Inhibition of these respiratory complexes was accompanied by an increase in cellular S-nitrosothiol levels, modification of cysteines residues, and an increase in the labile iron pool. The NO-dependent increase in the free iron pool and inhibition of complex II was prevented by inhibition of mitochondrial protein synthesis, consistent with a major contribution of the organelle to iron homeostasis. In addition, inhibition of mitochondrial protein synthesis was associated with an increase in heat shock protein 60 levels, which may be an additional mechanism leading to preservation of complex II activity.
Endothelium-derived nitric oxide (NO) is essential for maintenance of vascular function, including control of mitochondrial respiration and apoptosis (1–3). A role for NO in modulation of mitochondrial gene expression and protein synthesis has been demonstrated at several levels in nonvascular cells such as macrophages, where lipopolysaccharide-induced NO production decreased levels of cytochrome b and complex IV subunit I (4, 5). In brown adipose tissue activation of soluble guanylate cyclase has been shown to control mitochondrial number (6). In addition to its effect on protein synthesis, NO can also modulate the activity of a number of respiratory complexes by direct interaction or through the formation of secondary metabolites such as peroxynitrite. The reversible binding of NO to complex IV regulates respiration at low concentrations but at high levels can contribute to cytotoxicity (3). It also has been suggested that fully reduced cytochrome c oxidase can “catalyze” the reduction of NO under anaerobic conditions (7).
The cytotoxic potential of high concentrations of NO is evident only under conditions of an additional stress such as low glucose or compromised bioenergetics (3, 8, 9). In addition, it has been demonstrated that IFNγ/lipopolysaccharide-induced cytotoxicity in fibroblasts requires both NO formation and inhibition of glycolysis (10). Herein, we examine the effects of chronic exposure to NO on mitochondrial function in endothelial cells in the absence of an additional stress.
Several studies using isolated mitochondria have provided important insights into the mechanisms of NO or reactive nitrogen species interaction with mitochondrial proteins (11–13). NO-dependent inhibition at high concentrations has been shown to interact with the complex III and the Q cycle, resulting in the formation of peroxynitrite (14). This formation may be a major source of NO-dependent dysfunction in mitochondria and could underlie the inhibition of respiratory complexes I, II, and V (11). Mitochondria play a central role in cellular iron homeostasis, with synthesis of Fe–S centers and heme occurring in the organelle. It is also well recognized that aconitase is an important target for NO-mediated damage through dissociation of the Fe–S center (15, 16). Indeed, the NO-dependent release of iron has long been recognized as a potential route to cytotoxicity with the formation of low molecular weight carriers of iron such the dinitrosylcysteine complexes (17, 18).
Because the respiratory complexes are critical for maintenance of cellular energy, an interesting possibility is that the interaction of NO with these enzymes can have significant effects on cellular signaling (19, 20). This led us to the hypothesis that the inhibition of mitochondrial respiratory complexes by NO could influence cellular iron homeostasis. To test this hypothesis we examined the inhibition of these enzymes in endothelial cells exposed to rates of formation of NO that could be generated from inducible NO synthase.
Materials and Methods
Chloramphenicol (CAP), dichloroindophenol, ubiquinone, thenoyltrif luoroacetone, cytochrome c, Tris, acetyl-CoA, oxaloacetate, pyruvate, 1,10-phenanthroline, and 5,5′-dithiobis(2,4-nitrobenzoic acid) were obtained from Sigma. (Z)-1-[2-(2-Aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (DETA NONOate) was from Alexis (San Diego). mAbs to respiratory subunits (39-kDa subunit-complex I, 70-kDa subunit-complex II, subunit 1-complex IV, and α subunit-complex V) and Phen green SK were purchased from Molecular Probes. All other reagents used were of analytical grade.
Cell Culture. Bovine aortic endothelial cells (BAECs), passages 5–11, were grown to confluence as described (9) and treated with the NO donor DETA NONOate (100–500 μM) for various times. For inhibition of mitochondrial protein synthesis cells were pretreated with 20 μg/ml CAP (21).
Assay of Respiratory Complex Activities, Citrate Synthase, and ATP Levels. Respiratory enzyme activities were measured spectrophotometrically and corrected by subtraction of the thenoyltrifluoroacetone-, KCN-, and rotenone-insensitive rates for complexes II, IV, and I, respectively (22, 23). Citrate synthase was measured by using the coupled reaction among oxaloacetate, acetyl-CoA, and 5,5′-dithiobis(2,4-nitrobenzoic acid) (24).
Proteomics and Western Blotting. Mitochondria were isolated by differential centrifugation from BAECs and then separated on blue native-PAGE gels as described, and specific respiratory complexes were identified by Western blotting using commercially available mAbs to individual subunits of respiratory proteins (25).
Free sulfhydryl groups on cellular proteins were labeled by using biotin-conjugated iodoacetamide [N-(biotinoyl)-N-(iodoacetyl)ethylenediamine] (BIAM) (26). Cell lysates were labeled with BIAM (100 μM) for 15 min in the dark, followed by isoelectric focusing on an immobilized pH gradient (pH range 3–10). Second-dimension SDS/PAGE was performed on 10–18% gradient acrylamide gels followed by Western blotting and probing with streptavidin-horseradish peroxidase (Amersham Pharmacia). Parallel gels were silver stained to visualize total protein. The Western blots and silver-stained gels were then analyzed by using the PD QUEST software (Bio-Rad).
Measurement of Cellular Labile Iron Pool. The chelatable intracellular iron pool was determined by using the fluorescent probe Phen green SK (27). BAECs were cultured on glass-bottomed dishes and loaded with 20 μM Phen green SK for 30 min in PBS. The cells were then washed with PBS and imaged (Olympus X170) after sequential additions of 1,10-phenanthroline as a cell permeant iron chelator. The intensity of cytosolic fluorescence was then quantitated by using SIMPLEPCI software (Compix, Cranberry Township, PA).
Measurement of S-Nitrosothiols (SNO) by Chemiluminescence. Nitrite and SNO were measured by using a chemiluminescence detector (Antek Instruments, Houston). To detect modifications after treatment with NO, chemiluminescence was measured in samples incubated in 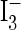 reagent at 37°C after no treatment, treatment with sulfanilamide (0.5%), or treatment with sulfanilamide and HgCl2 (5 mM) to determine total nitrite, SNO, and other adducts, respectively.
reagent at 37°C after no treatment, treatment with sulfanilamide (0.5%), or treatment with sulfanilamide and HgCl2 (5 mM) to determine total nitrite, SNO, and other adducts, respectively.
Western Blotting for Ferritin and Heat Shock Protein (HSP)60 and Immunoprecipitation Experiments. BAECs were lysed and then resolved by SDS/PAGE, followed by Western blotting using anti-ferritin or anti-HSP60 Abs (Sigma). For immunoprecipitation experiments, cell lysates were used for immunoprecipitation with the complex II (70-kDa subunit) Ab, followed by Western blotting and probing for HSP60.
Results
Chronic Exposure to NO Results in Inhibition of Mitochondrial Respiratory Complexes. In the first series of experiments, the effect of NO released from DETA NONOate on the activity and amounts of key mitochondrial proteins was determined in confluent BAECs in complete medium. Under these conditions, no cytotoxicity was associated with NO exposure, in agreement with previous studies (28, 29). After incubation for 2–24 h, the medium was removed and cells were lysed for the measurement of mitochondrial activities. As seen in Fig. 1, NO exposure was associated with a progressive decrease in the activities of complexes I (Fig. 1A) and IV (Fig. 1B), whereas complex II activity (Fig. 1C) was unchanged over the first 7 h, after which a decrease occurred. Complexes IV and II were also inhibited over a range of DETA NONOate concentrations from 100 to 500 μM (data not shown). There was no change in the specific activity of citrate synthase, a mitochondrial matrix enzyme, under these conditions (0.336 ± 0.03 μmol per min per mg of protein with 500 μM DETA NONOate vs. 0.331 ± 0.017 from control, mean ± SEM, n = 3). The activity of complex III was too low to be accurately measured in these cells.
Fig. 1.

NO inhibits mitochondrial respiratory complexes. BAECs were treated with 500 μM DETA NONOate for the indicated time periods, after which the activities of complexes I, II, and IV were measured. Values are mean ± SEM, n = 3.
Recovery of Activity on Inhibition by NO Requires New Protein Synthesis. The mitochondrial genome encodes 13 subunits of the respiratory chain enzymes, including 3 subunits of complex IV and 7 subunits from complex I. However, the 4 subunits of complex II are all encoded by the nuclear genome. To determine whether the inhibition of the respiratory complexes could be reversed and whether this required new protein synthesis, experiments were conducted with the mitochondrial protein synthesis inhibitor CAP. Cells were initially exposed to DETA NONOate for 48 h, after which a subset of cells was used for the measurement of enzyme activities. Cells were also incubated for a further 24–48 h in the absence or continued presence of NO. In a parallel group in which NO had been removed, CAP was added to inhibit mitochondrial protein synthesis. As seen in Fig. 2, the activities of both complex II (Fig. 2A) and complex IV (Fig. 2B) recover on withdrawal of the NO donor. However, in the case of complex IV, recovery of enzyme activity was prevented by CAP, consistent with a requirement for new mitochondrial protein synthesis to repair NO-dependent damage. In contrast, the recovery of complex II after NO withdrawal was not affected. These data indicate that under these conditions significant irreversible damage to the mitochondrial genome and translation machinery has not occurred.
Fig. 2.

Inhibition by NO is reversible and requires new protein synthesis. BAECs were treated with 500 μM DETA NONOate initially for 48 h, and activities of complexes II and IV were measured. The medium was then removed and cells were washed with PBS, followed by addition of fresh medium with or without CAP (20 μg/ml). The cells were lysed and the activities of the complexes were measured again. White bar, NO; black bar, NO withdrawn; gray bar, NO withdrawn + CAP. Values are mean ± SEM, n = 3(*, P > 0.05 vs. control; #, P > 0.05 vs. NO).
NO Treatment Decreased Protein Levels of Mitochondrial Respiratory Complexes. To investigate whether NO treatment resulted in alterations in protein levels of the intact respiratory complexes, blue native-PAGE was used. In this experiment, cells were exposed to DETA NONOate for 24 h before being washed, and a mitochondrially enriched fraction was prepared. These samples were then separated by blue native-PAGE, which was followed by Western blotting using Abs specific to mitochondrial respiratory complexes. As seen in Fig. 3, treatment with NO for 24 h resulted in a decrease in complexes I, II, and IV. However, there was no change in the levels of complex V, indicating that mitochondrial proteins susceptible to NO are not uniquely characterized by being coded for by the mitochondrial genome.
Fig. 3.

NO treatment results in decrease in protein levels of mitochondrial respiratory complexes. BAECs were treated for 24 h with 500 μM DETA NONOate, and the mitochondrial fraction was run on a blue native-PAGE gel and stained with Coomassie blue. The gels were also probed for various respiratory complex subunits. (A) Representative blue native gel and Western blot after probing with the indicated Abs. (B) The intensity of bands in A was determined and plotted as arbitrary units (*, P < 0.05 vs. control).
Chronic Exposure to NO Increases Levels of SNO and Modifies Reactive Cysteines on Cellular Proteins. SNO modification of cysteine residues on proteins has been proposed to be a mechanism for signal transduction by NO in cells and was measured here by chemiluminescence. After 24-h exposure to 500 μM DETA NONOate, each sample was divided into three aliquots, which were left untreated (to determine the total concentration of NO oxidation products formed in the sample), treated with sulfanilamide (to determine the concentration of SNO formed), or treated with sulfanilamide and HgCl2 (resulting in a signal that is indicative of species such as iron-nitrosyl compounds). A significant increase in nitrite levels was detected in the cells treated with NO donor (336.7 ± 49.2 vs. 116 ± 4.97 pmol/mg of protein in control, mean ± SEM, n = 3). The levels of SNO were undetectable in controls (limit of detection 1 pmol/mg of protein), whereas substantial levels were detected in NO-treated samples (11.2 ± 0.07 pmol/mg of protein, mean ± SEM). This value represents a relatively minor level of modification of the total cellular thiol content because the levels of total glutathione are typically 25–40 nmol/mg of protein. The levels of Hg-resistant species (iron nitrosyl compounds and N-nitrosamines) were also undetectable in control cells, whereas a signal was seen after NO treatment (68 ± 7.7 pmol/mg of protein, mean ± SEM, n = 3). It should be noted, however, that any species resistant to mercury oxidation (other than SNO) will result in a signal under these conditions. Taken together, these data demonstrate that endothelial cell proteins are susceptible to modification by chronic exposure to NO, and SNO are formed under these conditions.
In the next series of experiments, we used a proteomics approach to investigate these modifications further. After treatment with the NO donor, cell lysates were prepared, and protein thiols were tagged with biotin by using the reagent BIAM. Under these conditions, NO-dependent modification of the thiol is reflected in decreased BIAM labeling relative to control. The lysates were separated in the first dimension on isoelectric focusing strips, followed by a second-dimension resolution by SDS/PAGE. The proteins were then transferred to nitrocellulose and probed with streptavidin-horseradish peroxidase. Analysis of the silver-stained gels from control cells by using PD QUEST revealed ≈145 spots (Fig. 4A), of which ≈90% matched with the NO-treated samples (data not shown). Treatment with NO resulted in a significant decrease in BIAM labeling (Fig. 4C) compared with control (Fig. 4B). Approximately 135 spots were detected on the control samples, of which only 41 spots were matched to the NO-treated samples. A “master map” was generated from these data (Fig. 4D), which shows spot position and the matching proteins as filled spots. These data indicate that a major proportion of the proteins in endothelial cells separated by this technique contain reactive thiols, and ≈70% of these are modified after chronic exposure to NO. In a separate series of experiments nitration of proteins was also assessed, and no change in nitrotyrosine levels was detected in response to NO exposure (results not shown).
Fig. 4.

NO treatment results in an increase in modifications of reactive cysteines on cellular proteins. BAECs were treated for 16 h with 500 μM DETA NONOate, followed by cell lysis, labeling with BIAM, and 2D separation of proteins. Numbers on right are molecular weight × 10-3. Extent of labeling was determined by Western blotting using streptavidin-horseradish peroxidase. (A) Silver-stained gel from control. (B) BIAM labeling in control. (C) BIAM labeling of NO-treated cells. (D) “Master map” indicating matching of spots between B and C (matched spots in black).
Chronic Exposure to NO Increases the Cellular Labile Iron Pool and Decreases Ferritin Levels. A common factor among the complexes I, II, and IV is that they are metalloproteins with Fe–S centers, copper, or heme, which are all targets for interaction with NO or other reactive nitrogen species such as peroxynitrite (11, 16). NO has been shown to modulate the cellular iron and to test for a mitochondrial contribution to this modulation, cells were exposed to DETA NONOate for 16 h and iron was measured by using Phen green SK (27). BAECs were exposed to 500 μM DETA NONOate for 16 h and then loaded with Phen green SK, which fluoresces when iron is not bound. After incubation of the cells, excess dye was removed and any iron-bound Phen green SK was converted to the free fluorescent form by the addition of the cell-permeant iron chelator 1,10-phenanthroline while monitoring the increase in fluorescence. This fluorescence is shown for control cells by comparison of Fig. 5 A and C. In contrast, cells treated with NO showed an increase in cytosolic fluorescence (Fig. 5 B and D), indicating an increase in the labile iron pool compared with control (Fig. 5E).
Fig. 5.

NO increases the cellular free iron pool and decreases ferritin levels. BAECs were treated for 16 h with 500 μM DETA NONOate, loaded with Phen green SK for 30 min, incubated in 1,10-phenanthroline, and imaged on an inverted fluorescent microscope. (A and B) Baseline fluorescence in control (A) and NO-treated (B) cells. (C and D) Control (C) and NO-treated (D) cells 25 min after 1,10-phenanthroline. (E) Quantitation of the increase in fluorescence in NO-treated cells compared with controls (*, P < 0.05). (F) Western blotting for ferritin after treatment with NO donor (16 h). After NO treatment, cell lysates were probed for ferritin and intensity of bands was plotted as arbitrary units (*, P < 0.05). (Inset) Representative blot showing levels of ferritin in control and NO-treated cells.
It has been reported that increases in the free iron pool are accompanied by an increase in ferritin levels to scavenge the iron and prevent it from participating in redox reactions. To determine the effects of NO on ferritin, cells were again pretreated with DETA NONOate and lysed, and the total cellular ferritin was determined by Western blotting (Fig. 5F). In contrast to other conditions associated with increased cellular iron, NO treatment resulted in a decrease in ferritin level to ≈20% of that in controls.
Interference with Mitochondrial Protein Synthesis Prevents Alteration in the Cellular Labile Iron Pool. To test for a contribution of mitochondrial electron transport proteins to cellular iron, the effect of inhibition of mitochondrial protein synthesis on the NO-dependent iron increase was examined. Treatment of cells with CAP before addition of NO resulted in a significant attenuation of the increase in labile iron (Fig. 6A).
Fig. 6.

(A) Inhibition of mitochondrial protein synthesis prevents increase in cellular labile iron pool. BAECs were treated with CAP for 48 h, followed by 500 μM DETA NONOate for 16 h. The cells were then loaded with Phen green SK and imaged as mentioned earlier (*, P < 0.05 vs. control; #, P < 0.05 vs. NO). (B) Inhibition of mitochondrial protein synthesis induces HSP60 synthesis in BAECs. BAECs were treated with CAP (20 μg/ml for 48 h) or with DETA NONOate (500 μM for 16 h) (parallel to the last 16 h of the CAP treatment). The levels of HSP60 in total cell lysates were then determined by Western blotting, followed by densitometry (*, P < 0.05). (C) Representative blot of total cellular HSP60 after the treatment. (D) HSP60 coimmunoprecipitates with complex II. BAECs were treated with CAP (20 μg/ml) for 48 h and the cell lysates were immunoprecipitated with the complex II (70-kDa subunit) Ab. The immunoprecipitates were then separated by SDS/PAGE and probed with the HSP60 Ab. (E) Decrease in complex II activity by NO is prevented by CAP. BAECs were treated with 500 μM DETA NONOate for 16 h either with or without pretreatment for 48 h with CAP (20 μg/ml), and the activity of complex II was measured. Values are mean ± SEM, n = 3(*, P = 0.0014 vs. control; **, P = 0.02 vs. NO; #, P = 0.02 vs. CAP).
In addition to decreasing levels of mitochondrially encoded proteins, damage of the mitochondrial protein biosynthetic machinery can induce synthesis of heat shock proteins, which may then protect against iron release and oxidative stress (30–32). The effect of inhibition of mitochondrial protein synthesis by CAP on HSP60 levels was then investigated. BAECs were treated with CAP or NO donor, after which the cells were lysed and Western blotting was performed with an anti-HSP60 Ab. As seen in Fig. 6 B and C, treatment with CAP resulted in a significant increase in HSP60, whereas NO had no effect. These data suggested the hypothesis that CAP treatment could protect the NO-induced decrease of respiratory complex activity through an HSP60-dependent mechanism (33). Because complex II is the only respiratory complex that is not affected by CAP treatment per se (9), this enzyme was chosen to examine the potential protective effect of HSP60 induction. Immunoprecipitation of cell lysates from endothelial cells with the complex II (70-kDa subunit) Ab, followed by Western blotting against HSP60, indicates that there is an association between the two proteins (Fig. 6D), which is independent of CAP treatment. To assess the functional significance of HSP60 association with the respiratory complex, enzyme activity was examined after NO treatment in the presence and absence of CAP. As seen in Fig. 6E, pretreatment with CAP resulted in partial protection (≈50%) of the NO-induced decrease in complex II activity.
Discussion
The effect of NO on mitochondrial respiratory complexes has been extensively studied in vitro, and these proteins have been shown to be exquisitely sensitive to modulation by NO or reactive nitrogen species (3). Furthermore, the interaction between NO and the mitochondria in the cell and the effect this has on signal transduction is now emerging as an important area of research (19). NO has been implicated in the etiology of a wide variety of diseases, especially in the cardiovascular system, where endothelial cells can be chronically exposed to high concentrations of this molecule during inflammation (34). It is important to note that at low concentrations NO can be cytoprotective, and recent studies suggest that maintenance of intracellular iron has a critical role to play here (35).
Iron is now known to contribute to signal transduction and the mechanisms of cytotoxicity related to exposure to both high concentrations of NO and reactive oxygen species (36–38). We hypothesized that the inhibition of mitochondrial respiratory complexes by NO could influence cellular iron homeostasis. It has been shown that NO inhibits mitochondrial respiratory complexes in vitro, both by direct interaction as in the case of complex IV, and also through secondary mediators such as peroxynitrite in the case of complex I (3). Endogenous NO can inhibit complex II in rat aortic smooth muscle cells, and one of the mechanisms for this inhibition been suggested to be dissociation of the Fe–S cluster (39, 40).
In the present study, the principal findings were that exposure of NO at high fluxes resulted in the progressive loss of activity of complexes I, II, and IV but not citrate synthase. Inhibition of complex IV was reversible on removal of NO, but new protein synthesis was required to restore activities to control levels. It is known that high concentrations of NO inhibit protein synthesis at the level of ribonucleotide reductase (41), but this inhibition is unlikely to be a major contributory mechanism here because both the levels of complex V and the activity of citrate synthase were unaffected by NO exposure. It is interesting to note that complex II activity decreases at a much slower rate compared with the other complexes after treatment with the NO donor. It has been demonstrated in hepatoma cells that there is a variation in the sensitivity of mitochondrial Fe–S clusters to inactivation by NO, where complex II was more resistant to inhibition, compared with complex I (42).
These responses are similar to those reported for cytochrome b and subunit 1 of complex IV on exposure of macrophages to endogenous NO derived from inducible NO synthase (4, 5). Exposure of hepatocytes to authentic NO led to a concentration-dependent inhibition of mitochondrial aconitase and complexes I and II (43). Treatment of BAECs with the NO donor results in decreased protein levels of complexes I, II, and IV. The Western blotting of the native gels did not reveal complexes after NO treatment at different molecular weights, suggesting that an assembly defect due to the loss of mitochondrial membrane potential is not contributing to NO-dependent damage to the organelle.
S-nitrosation of cysteine thiols may constitute a major route of conveying NO bioactivity through post-translational modifications regulating the activity of proteins involved in cell signaling and metabolism (44). Chronic exposure to NO significantly increased levels of nitrite, SNO, and unidentified nitrosated species (e.g., iron nitrosyl compounds). To determine the effect of these modifications on cellular proteins, we used a proteomics approach coupled to labeling with BIAM, which tags reactive cysteines with a low pKa. Labeling with BIAM has been used to identify proteins containing H2O2-sensitive cysteines (26) and oxidative modifications of red blood cell membrane proteins (45). A significant number of proteins in endothelial cells were labeled with BIAM, and chronic exposure to NO resulted in a substantial decrease in labeling. This observation indicates that there is a population of cellular proteins susceptible to modification by chronic exposure to NO. Previous studies have identified some members of this subproteome. For example, specific S-nitrosation of cysteine residues on caspase enzymes has been identified as a mechanism of NO-induced inhibition of these enzymes (46), and it has also been shown that the majority of mitochondrial, but not cytoplasmic, caspase-3 zymogens contain this inhibitory modification (47). Prolonged exposure to NO in J774 cells results in an inhibition of complex I that appears to result from S-nitrosation of critical thiols in the enzyme complex (48). It has also been shown that S-nitrosation of metallothionein by NO donors is associated with an increase in the labile zinc pool in the pulmonary endothelium (49).
In endothelial cells, the effect of chronic exposure to NO was most prominent on complexes I, II, and IV, and a common link between these proteins is the presence of iron, in heme a in complex IV and in Fe–S centers in complex I and II. NO and peroxynitrite promote complete disruption of the cytosolic isoform of aconitase or iron-regulatory protein 1, resulting in release of iron (16). NO has also been shown to dissociate Fe–S complexes in mitochondrial proteins, and to mobilize iron from these Fe–S clusters (42). The cytosolic pool of labile iron has substantial cytotoxic potential and has been implicated in formation of hydroxyl radicals from hydrogen peroxide (50). It is maintained at minimal levels by modulation of the uptake of iron by the transferrin receptor and sequestration of iron although ferritin, which has been suggested to have a protective role against oxidative stress in a number of different cell types (51), including endothelial cells (52).
We next investigated whether the inhibition of mitochondrial electron transfer proteins by NO had an impact on cellular iron homeostasis. Treatment of BAECs with NO for 16 h resulted in a significant increase in the free iron pool accompanied by a decrease in ferritin levels, so further exacerbating the cytotoxic potential of the chelatable iron pool. A mitochondrial involvement in this process was indicated by the fact that inhibition of mitochondrial protein synthesis abolished the NO-dependent increase in free iron. However, CAP can have a number of effects because it prevents all mitochondrial translation. We have earlier demonstrated that treatment of BAECs with CAP results in mitochondrial respiratory complex assembly defects (25), and it has been shown that accumulation of unfolded protein within the mitochondrial matrix and loss of mitochondrial DNA can result in up-regulation of mitochondrial stress proteins such as HSP60 and HSP10 (30, 31). A 70-kDa member of the HSP family has also been implicated in mitochondrial iron metabolism in yeast (53). In BAECs, the CAP treatment resulted in a significant increase in levels of mitochondrial HSP60 and was associated with prevention of the NO-dependent increase in the cellular labile iron pool. Based on our hypothesis that the increase in cellular free iron was derived from the mitochondrial respiratory complexes, these data implied that the increase in HSP60 levels after CAP treatment should have a protective role against NO-induced inhibition of complex activity. HSP60 overexpression has been shown to protect both nuclear and mitochondrially encoded proteins from ischemic damage (33) and heat stress in the heart (54). Treatment with CAP resulted in a significant protection against the NO-induced decrease in complex II activity, indicating that the HSP60 induction could preserve function of the respiratory complex, probably by maintenance of the Fe–S center. Immunoprecipitation experiments using Abs to the 70-kDa subunit of complex II also indicated that this chaperone protein is associated with the enzyme. HSP60 mediates the folding and assembly of mitochondrial proteins such as complex II, and it has been demonstrated that the chaperone interacts with the flavoprotein subunit in yeast (55). Overexpression of mitochondrial HSP60 in cardiac myocytes has been shown to protect mitochondrial function and prevent apoptotic cell death induced by ischemia–reperfusion (56).
In conclusion, our studies indicate that mitochondria are an important modulator of the effect of NO on cellular iron homeostasis. NO can affect iron-containing proteins beyond the well recognized interaction with aconitase and including complexes I, II, and IV of the mitochondrial respiratory chain. We hypothesize that this effect can lead to an increase in the cellular pools of labile iron, and an important regulatory element is the mitochondrial heat shock proteins.
Acknowledgments
We thank Jack Crawford for help with the chemiluminescence measurements. This study was supported by National Institutes of Health Grant RO1 HL58031. A.R. is supported by a fellowship from the American Heart Association, Southeast Affiliate. E.C. is supported by a predoctoral National Institutes of Health Cardiovascular Fellowship.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: BAEC, bovine aortic endothelial cell; CAP, chloramphenicol; SNO, S-nitrosothiols; HSP, heat shock protein; BIAM, N-(biotinoyl)-N-(iodoacetyl)ethylenediamine; DETA NONOate, (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1, 2-diolate.
References
- 1.Moncada, S. & Erusalimsky, J. D. (2002) Nat. Rev. Mol. Cell. Biol. 3, 214-220. [DOI] [PubMed] [Google Scholar]
- 2.Brown, G. C. & Borutaite, V. (2002) Free Radical Biol. Med. 33, 1440-1450. [DOI] [PubMed] [Google Scholar]
- 3.Ramachandran, A., Levonen, A. L., Brookes, P. S., Ceaser, E., Shiva, S., Barone, M. C. & Darley-Usmar, V. (2002) Free Radical Biol. Med. 33, 1465-1474. [DOI] [PubMed] [Google Scholar]
- 4.Wei, J., Guo, H. & Kuo, P. C. (2002) J. Immunol. 168, 4721-4727. [DOI] [PubMed] [Google Scholar]
- 5.Guo, H., Wei, J. & Kuo, P. C. (2001) Biochem. Biophys. Res. Commun. 289, 993-997. [DOI] [PubMed] [Google Scholar]
- 6.Nisoli, E., Clementi, E., Paolucci, C., Cozzi, V., Tonello, C., Sciorati, C., Bracale, R., Valerio, A., Francolini, M., Moncada, S. & Carruba, M. O. (2003) Science 299, 896-899. [DOI] [PubMed] [Google Scholar]
- 7.Clarkson, R. B., Norby, S. W., Smirnov, A., Boyer, S., Vahidi, N., Nims, R. W. & Wink, D. A. (1995) Biochim. Biophys. Acta 1243, 496-502. [DOI] [PubMed] [Google Scholar]
- 8.Beltran, B., Mathur, A., Duchen, M. R., Erusalimsky, J. D. & Moncada, S. (2000) Proc. Natl. Acad. Sci. USA 97, 14602-14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramachandran, A., Moellering, D. R., Ceaser, E., Shiva, S., Xu, J. & Darley-Usmar, V. (2002) Proc. Natl. Acad. Sci. USA 99, 6643-6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dijkmans, R. & Billiau, A. (1991) Eur. J. Biochem. 202, 151-159. [DOI] [PubMed] [Google Scholar]
- 11.Radi, R., Cassina, A. & Hodara, R. (2002) Biol. Chem. 383, 401-409. [DOI] [PubMed] [Google Scholar]
- 12.Cooper, C. E. (2002) Trends Biochem. Sci. 27, 33-39. [DOI] [PubMed] [Google Scholar]
- 13.Costa, N. J., Dahm, C. C., Hurrell, F., Taylor, E. R. & Murphy, M. P. (2003) Antioxid. Redox. Signal 5, 291-305. [DOI] [PubMed] [Google Scholar]
- 14.Poderoso, J. J., Lisdero, C., Schopfer, F., Riobo, N., Carreras, M. C., Cadenas, E. & Boveris, A. (1999) J. Biol. Chem. 274, 37709-37716. [DOI] [PubMed] [Google Scholar]
- 15.Castro, L. A., Robalinho, R. L., Cayota, A., Meneghini, R. & Radi, R. (1998) Arch. Biochem. Biophys. 359, 215-224. [DOI] [PubMed] [Google Scholar]
- 16.Soum, E. & Drapier, J. C. (2003) J. Biol. Inorg. Chem. 8, 226-232. [DOI] [PubMed] [Google Scholar]
- 17.Yang, W., Rogers, P. A. & Ding, H. (2002) J. Biol. Chem. 277, 12868-12873. [DOI] [PubMed] [Google Scholar]
- 18.Bergamini, S., Rota, C., Canali, R., Staffieri, M., Daneri, F., Bini, A., Giovannini, F., Tomasi, A. & Iannone, A. (2001) Nitric Oxide 5, 349-360. [DOI] [PubMed] [Google Scholar]
- 19.Brookes, P. & Darley-Usmar, V. M. (2002) Free Radical Biol. Med. 32, 370-374. [DOI] [PubMed] [Google Scholar]
- 20.Ranganathan, A. C., Nelson, K. K., Rodriguez, A. M., Kim, K. H., Tower, G. B., Rutter, J. L., Brinckerhoff, C. E., Huang, T. T., Epstein, C. J., Jeffrey, J. J. & Melendez, J. A. (2001) J. Biol. Chem. 276, 14264-14270. [DOI] [PubMed] [Google Scholar]
- 21.Lipton, J. H. & McMurray, W. C. (1977) Biochim. Biophys. Acta 477, 264-272. [DOI] [PubMed] [Google Scholar]
- 22.Ragan, C. I., Wilson, M. T., Darley-Usmar, V. M. & Lowe, P. N. (1987) in Mitochondria: A Practical Approach, eds. Darley-Usmar, V. M., Rickwood, D. & Wilson, M. T. (IRL, Oxford), pp. 79-112.
- 23.Darley-Usmar, V. M., Capaldi, R. A., Takamiya, S., Millett, F., Wilson, M. T., Malatesta, F. & Sarti, P. (1987) in Mitochondria: A Practical Approach, eds. Darley-Usmar, V. M., Rickwood, D. & Wilson, M. T. (IRL, Oxford), pp. 113-152.
- 24.Shepherd, D. & Garland, P. B. (1969) Biochem. J. 114, 597-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brookes, P. S., Pinner, A., Ramachandran, A., Coward, L., Barnes, S., Kim, H. & Darley-Usmar, V. M. (2002) Proteomics 2, 969-977. [DOI] [PubMed] [Google Scholar]
- 26.Kim, J. R., Yoon, H. W., Kwon, K. S., Lee, S. R. & Rhee, S. G. (2000) Anal. Biochem. 283, 214-221. [DOI] [PubMed] [Google Scholar]
- 27.Petrat, F., de Groot, H. & Rauen, U. (2001) Biochem. J. 356, 61-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim, W. K., Chung, J. H., Kim, H. C. & Ko, K. H. (1999) Neurosci. Res. 33, 281-289. [DOI] [PubMed] [Google Scholar]
- 29.Le Goffe, C., Vallette, G., Jarry, A., Bou-Hanna, C. & Laboisse, C. L. (1999) Biochem. J. 344, 643-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao, Q., Wang, J., Levichkin, I. V., Stasinopoulos, S., Ryan, M. T. & Hoogenraad, N. J. (2002) EMBO J. 21, 4411-4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinus, R. D., Garth, G. P., Webster, T. L., Cartwright, P., Naylor, D. J., Hoj, P. B. & Hoogenraad, N. J. (1996) Eur. J. Biochem. 240, 98-103. [DOI] [PubMed] [Google Scholar]
- 32.Cabiscol, E., Belli, G., Tamarit, J., Echave, P., Herrero, E. & Ros, J. (2002) J. Biol. Chem. 277, 44531-44538. [DOI] [PubMed] [Google Scholar]
- 33.Sammut, I. A. & Harrison, J. C. (2003) Clin. Exp. Pharmacol. Physiol. 30, 110-115. [DOI] [PubMed] [Google Scholar]
- 34.Toborek, M. & Kaiser, S. (1999) Basic Res. Cardiol. 94, 295-314. [DOI] [PubMed] [Google Scholar]
- 35.Kotamraju, S., Tampo, Y., Keszler, A., Chitambar, C. R., Joseph, J., Haas, A. L. & Kalyanaraman, B. (2003) Proc. Natl. Acad. Sci. USA 100, 10653-10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tampo, Y., Kotamraju, S., Chitambar, C. R., Kalivendi, S. V., Keszler, A., Joseph, J. & Kalyanaraman, B. (2003) Circ. Res. 92, 56-63. [DOI] [PubMed] [Google Scholar]
- 37.Kalivendi, S. V., Kotamraju, S., Cunningham, S., Shang, T., Hillard, C. J. & Kalyanaraman, B. (2003) Biochem. J. 371, 151-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boullerne, A. I., Nedelkoska, L. & Benjamins, J. A. (1999) J. Neurochem. 72, 1050-1060. [DOI] [PubMed] [Google Scholar]
- 39.Geng, Y., Hansson, G. K. & Holme, E. (1992) Circ. Res. 71, 1268-1276. [DOI] [PubMed] [Google Scholar]
- 40.Welter, R., Yu, L. & Yu, C. A. (1996) Arch. Biochem. Biophys. 331, 9-14. [DOI] [PubMed] [Google Scholar]
- 41.Colasanti, M., Persichini, T., Venturini, G. & Ascenzi, P. (1999) IUBMB Life 48, 25-31. [DOI] [PubMed] [Google Scholar]
- 42.Drapier, J. C. (1997) Methods 11, 319-329. [DOI] [PubMed] [Google Scholar]
- 43.Stadler, J., Billiar, T. R., Curran, R. D., Stuehr, D. J., Ochoa, J. B. & Simmons, R. L. (1991) Am. J. Physiol. 260, C910-C916. [DOI] [PubMed] [Google Scholar]
- 44.Gow, A. J., Chen, Q., Hess, D. T., Day, B. J., Ischiropoulos, H. & Stamler, J. S. (2002) J. Biol. Chem. 277, 9637-9640. [DOI] [PubMed] [Google Scholar]
- 45.Lee, T. H., Kim, S. U., Yu, S. L., Kim, S. H., Park do, S., Moon, H. B., Dho, S. H., Kwon, K. S., Kwon, H. J., Han, Y. H., et al. (2003) Blood 101, 5033-5038. [DOI] [PubMed] [Google Scholar]
- 46.Dimmeler, S., Haendeler, J., Nehls, M. & Zeiher, A. M. (1997) J. Exp. Med. 185, 601-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mannick, J. B., Schonhoff, C., Papeta, N., Ghafourifar, P., Szibor, M., Fang, K. & Gaston, B. (2001) J. Cell Biol. 154, 1111-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clementi, E., Brown, G. C., Feelisch, M. & Moncada, S. (1998) Proc. Natl. Acad. Sci. USA 95, 7631-7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang, Z. L., Wasserloos, K. J., Liu, X., Stitt, M. S., Reynolds, I. J., Pitt, B. R. & St Croix, C. M. (2002) Mol. Cell. Biochem. 234–235, 211-217. [PubMed] [Google Scholar]
- 50.Petrat, F., de Groot, H., Sustmann, R. & Rauen, U. (2002) Biol. Chem. 383, 489-502. [DOI] [PubMed] [Google Scholar]
- 51.Arosio, P. & Levi, S. (2002) Free Radical Biol. Med. 33, 457-463. [DOI] [PubMed] [Google Scholar]
- 52.Balla, G., Jacob, H. S., Balla, J., Rosenberg, M., Nath, K., Apple, F., Eaton, J. W. & Vercellotti, G. M. (1992) J. Biol. Chem. 267, 18148-18153. [PubMed] [Google Scholar]
- 53.Craig, E. A., Voisine, C. & Schilke, B. (1999) Biol. Chem. 380, 1167-1173. [DOI] [PubMed] [Google Scholar]
- 54.Sammut, I. A., Jayakumar, J., Latif, N., Rothery, S., Severs, N. J., Smolenski, R. T., Bates, T. E. & Yacoub, M. H. (2001) Am. J. Pathol. 158, 1821-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robinson, K. M. & Lemire, B. D. (1996) J. Biol. Chem. 271, 4061-4067. [DOI] [PubMed] [Google Scholar]
- 56.Lin, K. M., Lin, B., Lian, I. Y., Mestril, R., Scheffler, I. E. & Dillmann, W. H. (2001) Circulation 103, 1787-1792. [DOI] [PubMed] [Google Scholar]