

Abstract
Cells lacking the GTPase Ypt6p have defects in intracellular traffic and are temperature sensitive. Their growth is severely impaired by additional mutation of IMH1, which encodes a non-essential Golgi-associated coiled-coil protein. A screen for mutants that, like ypt6, specifically impair the growth of imh1 cells led to the identification of RIC1. Ric1p forms a tight complex with a previously uncharacterized protein, Rgp1p. The Ric1p–Rgp1p complex binds Ypt6p in a nucleotide-dependent manner, and purified Ric1p–Rgp1 stimulates guanine nucleotide exchange on Ypt6p in vitro. Deletion of RIC1 or RGP1, like that of YPT6, blocks the recycling of the exocytic SNARE Snc1p from early endosomes to the Golgi and causes temperature-sensitive growth, but this defect can be relieved by overexpression of YPT6. Ric1p largely colocalizes with the late Golgi marker Sec7p. Ypt6p shows a similar distribution, but this is altered when RIC1 or RGP1 is mutated. We infer that the Ric1p–Rgp1p complex serves to activate Ypt6p on Golgi membranes by nucleotide exchange, and that this is required for efficient fusion of endosome-derived vesicles with the Golgi.
Keywords: Golgi/Rab GTPase/Saccharomyces cerevisiae/vesicle fusion/Ypt6p
Introduction
A central feature of intracellular membrane traffic is the specific fusion of transport vesicles with their target organelles. Key components of this process are the SNAREs, membrane proteins found on both vesicles and target whose interactions are intimately involved in initiating bilayer fusion. Families of SNARE proteins exist, each with a characteristic distribution, and it is thought that the preferential formation of complexes comprising specific sets of SNAREs contributes to the specificity of membrane fusion (Rothman, 1994; Nichols and Pelham, 1998; Pelham, 1999). Further specificity is provided by additional proteins that aid the initial interaction of vesicles with their targets. These include both tethering factors with coiled-coil motifs and large protein complexes (Pfeffer, 1999; Waters and Pfeffer, 1999). Another set of proteins that may control the initial tethering or docking event are the small GTPases termed rabs (in animal cells) or Ypts (in yeast). Representatives of this family are found at each step of transport (Lazar et al., 1997; Chavrier and Goud, 1999; Gotte et al., 2000). Examples in yeast are Sec4p, required for transport and fusion of exocytic vesicles with the plasma membrane, and Ypt1p, which plays a crucial role in the docking of endoplasmic reticulum (ER)-derived vesicles with early Golgi compartments containing the SNARE Sed5p.
Another GTPase associated with the Golgi is Ypt6p, the yeast homologue of mammalian rab6. Unlike Ypt1p, Ypt6p is not required (in most strains) for secretion or for viability, but null mutants have a number of phenotypes including temperature-sensitive growth, partial mis- sorting of the vacuolar protease carboxypeptidase Y and mis-localization of the late Golgi protein Kex2p to the vacuole (Li and Warner, 1996; Tsukada et al., 1999). These phenotypes are quite similar to those of mutants lacking the SNARE Tlg1p, which has been shown to be involved in the recycling of proteins from early endosomes to the Golgi apparatus (Holthuis et al., 1998a,b; Lewis et al., 2000). Hence, a possible role for Ypt6p would be to assist the fusion of endosome-derived vesicles with the late Golgi. A role for Ypt6p in the Golgi is also consistent with some genetic evidence, which suggests a link between the functions of Ypt6p and Ypt1p (Li and Warner, 1998).
Despite these clues, rather little is known about the molecules with which Ypt6p might interact. Genetic studies have identified several genes that interact with ypt6, by suppressing its temperature-sensitive growth defect when they are overexpressed (Li and Warner, 1996; Tsukada and Gallwitz, 1996). One such gene was identified as a low-copy suppressor of a ypt6 allele and named IMH1 (Li and Warner, 1996). The same gene was isolated as a high-copy suppressor of a deletion allele of ypt6, and named SYS3 (Tsukada and Gallwitz, 1996); for simplicity we use the name IMH1 here. Since Imh1p has effects even in the absence of Ypt6p, it must be able to act independently of it. However, its function is clearly related, since loss of Imh1p, which normally has little effect on cells, severely reduces the growth of a ypt6 mutant (Tsukada et al., 1999).
In this paper we describe a search for proteins interacting with Ypt6p using two independent approaches. In the first, mutations were sought that, like ypt6, show synthetic interactions with imh1. In the second, proteins binding to Ypt6p were identified by protein purification and mass spectrometry. Both approaches led to the identification of a complex consisting of the products of the RIC1 and RGP1 genes. These are peripheral membrane proteins that colocalize with the Golgi marker Sec7p, and we show that they act in vitro as a nucleotide exchange factor for Ypt6p. Their activity does not seem to influence the ability of Ypt6p to bind to membranes, but in their absence the distribution of Ypt6p is distinct from that of the Golgi. We suggest that Ypt6p is able to bind to endosome-derived vesicles, but becomes activated to the GTP-bound form by the Ric1p–Rgp1p complex only at Golgi membranes; Ypt6p–GTP then stimulates the fusion of endosome-derived vesicles with the Golgi. Proteins related to the C-terminal domain of Ric1p are found in a variety of species, including humans.
Results
Genetic screen for mutants showing synthetic lethality with imh1
Null mutants of YPT6 are viable but temperature sensitive for growth. Though deletion of the IMH1 gene alone has little or no effect on growth, imh1Δ ypt6Δ double mutants show a very severe growth defect (Tsukada et al., 1999). We reasoned that mutations in genes with functions related to that of ypt6 might also show synthetic lethality with imh1, and therefore undertook a screen for such mutants using a colony sectoring assay as described previously (Kranz and Holm, 1990).
An imh1Δ yeast strain that carried IMH1 on a plasmid was mutagenized and some 13 000 colonies screened (see Materials and methods). Six mutants were identified that were unable to grow without the plasmid-borne IMH1 gene. Of these, one could be complemented by YPT6, confirming the interaction between imh1 and ypt6. From a yeast genomic library we identified a plasmid that was able to complement four of the remaining mutants; the fifth has proven, so far, refractory to cloning. We restricted the complementary activity of the recovered plasmid to a single intact open reading frame, which corresponded to the RIC1 gene (YLR039c). Figure 1A shows typical data for one of the mutants, sl233. The mutant strain, which lacked the chromosomal copy of IMH1 but carried a URA3 plasmid with the IMH1 gene, was transformed with LEU2 plasmids carrying either RIC1, RIC1–GFP, IMH1 or no inserted gene and the transformants plated on medium containing 5-fluoro-orotic acid (5-FOA) to select cells that had lost the URA3–IMH1 plasmid. As shown in Figure 1, the strain was able to grow only if it contained a plasmid expressing either Imh1p or Ric1p, or the green fluorescent protein (GFP)-tagged version of Ric1p.
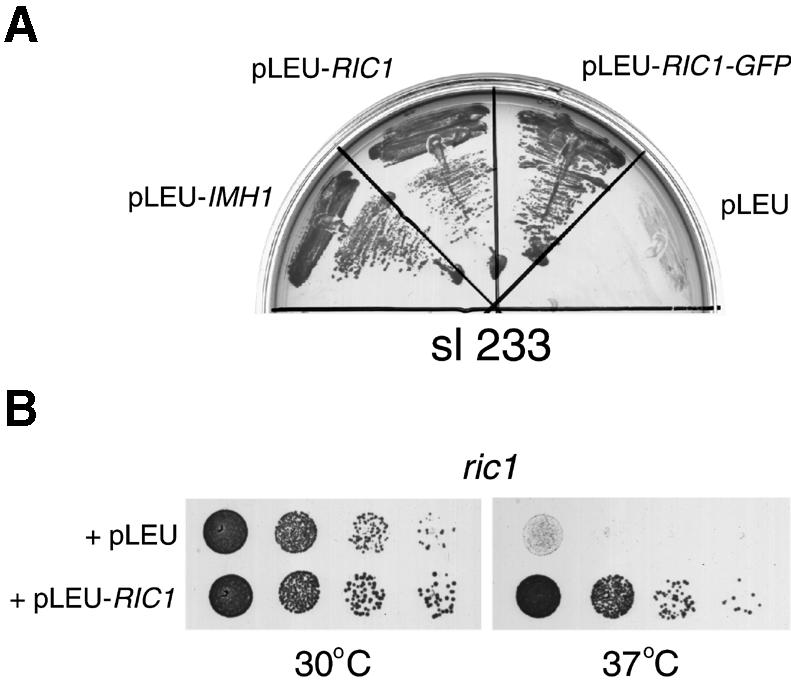
Fig. 1. Functional interaction between IMH1 and RIC1. (A) Synthetic lethal interaction between imh1Δ and ric1 mutants. The sl233 mutant was transformed with the indicated Ycplac111 plasmid (pLEU), either with no insert, or carrying IMH1, RIC1 or a RIC1–GFP fusion. Transformants were grown on plates containing 5-FOA for 3 days at 30°C. (B) Growth properties of the ric1Δ deletion mutant (ric1). Cells from a ric1Δ strain transformed either with an empty LEU2 plasmid or with the same plasmid carrying the wild-type RIC1 gene were diluted in YEPD, spotted on to selective (–Leu) plates and grown at 30 and 37°C for 2 days.
RIC1 is a non-essential gene originally isolated in a screen for mutants with altered regulation of ribosome synthesis (Mizuta et al., 1997). This phenotype is common for secretory mutants (Mizuta and Warner, 1994), and indeed the same screen also yielded ypt6 (Li and Warner, 1996). Recently, RIC1 was also identified in a screen for mutants that are synthetically lethal with a temperature-sensitive clathrin heavy chain (Bensen et al., 2000). A role for Ric1p in membrane traffic thus seemed likely, so we characterized it further. Deletion of the RIC1 gene resulted in temperature-sensitive growth, which could be overcome by provision of RIC1 on a plasmid (Figure 1B). We confirmed that a ric1Δ imh1Δ strain was unviable, but found that a ric1Δ ypt6Δ strain was no more defective in terms of growth than a ypt6Δ single mutant (data not shown). The fact that removal of Ric1p had no additional effect once Ypt6p was removed suggests that Ric1p performs a function that is not only related to that of Ypt6p, but is dependent on the presence of Ypt6p.
Ric1p binds to Rgp1p
To identify proteins that might bind to Ric1p, we expressed a version of the protein tagged at its C-terminus with two IgG-binding domains of protein A (Ric1p–PtA). This was expressed from the RIC1 promoter, on a centromere-containing vector, in ric1Δ cells; it fully complemented the temperature sensitivity of this strain (data not shown). Purification of the fusion protein on IgG–Sepharose revealed a second protein in roughly equimolar amounts (Figure 2, left panel, lane 2). Tryptic digestion of this protein and mass spectrometry of the products (see Materials and methods) showed that it corresponded to Rgp1p, the product of a largely uncharacterized gene reported to be required for efficient growth (Aguilera et al., 1990). Protein A tagging and purification of Rgp1p, performed exactly as for Ric1p, resulted in the copurification of a single protein, which was identified by mass spectrometry as Ric1p (Figure 2, left panel, lane 1). Cleavage of either fusion protein with tobacco etch virus (TEV) protease at a site introduced between the proteins and the protein A tag yielded ∼1:1 molar ratios of Ric1p to Rgp1p, regardless of which was originally tagged (Figure 2, right panel). This indicates that essentially all of the Ric1p and Rgp1p in the cell is in the form of a complex between the two proteins.

Fig. 2. Ric1p forms a complex with Rgp1p in vivo. (Left) Affinity purification of Rgp1p–PtA and Ric1p–PtA from rgp1Δ and ric1Δ strains, respectively. The purified proteins eluted from the IgG–Sepharose column with low pH were analysed by SDS–PAGE and Coomassie staining. The position of the protein A fusions and the copurifying proteins, identified as Ric1p and Rgp1p by mass spectrometry, are indicated. Molecular mass standards are indicated on the left side of the gel. (Right) The IgG–Sepharose beads carrying the Ric1p–PtA and Rgp1p–PtA fusions were digested by TEV protease and the released fractions containing the cleaved Ric1p and Rgp1p were analysed by SDS–PAGE and Coomassie staining. Molecular mass standards were resolved on the left side of the gel. The position of TEV protease is indicated; the asterisk indicates immunoglobulin heavy chains derived from the affinity column.
Deletion of Rgp1p in our standard wild-type strain resulted in a temperature-sensitive growth phenotype very similar to that of ric1Δ cells (data not shown, see also Figure 6). The similar properties of the deletion mutants makes it likely that both proteins are required for the activity of the Ric1p–Rgp1p complex.
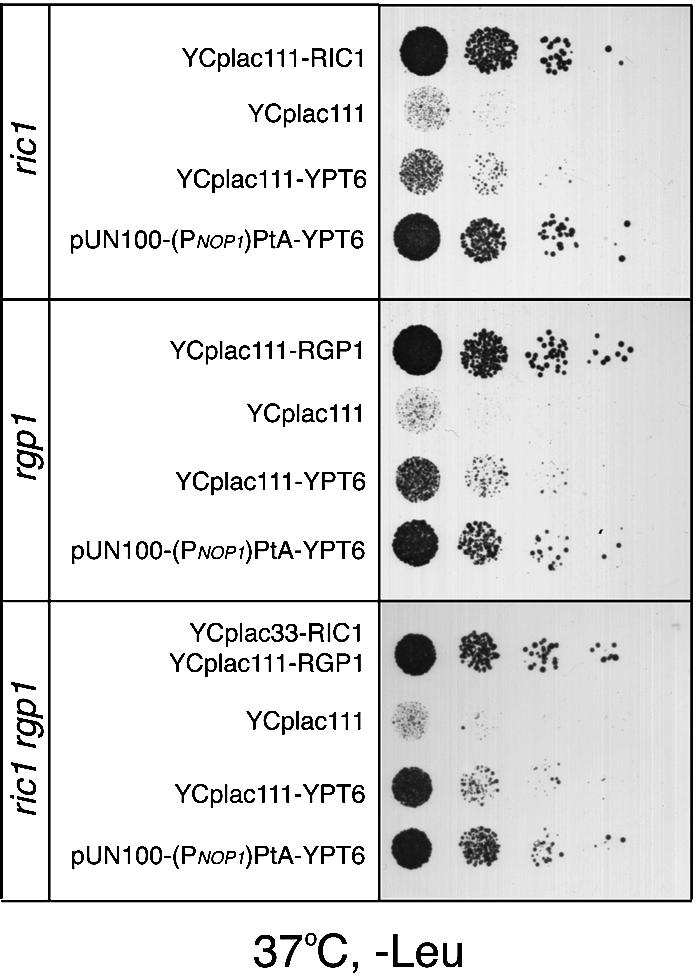
Fig. 6. YPT6 suppresses the temperature-sensitive growth defect of the ric1, rgp1 and ric1 rgp1 deletion mutants. The ric1, rgp1 and ric1 rgp1 deletion mutants were transformed with an empty centromeric plasmid (YCplac111), YCp plasmid(s) carrying the complementing gene(s) as indicated, or with a centromeric plasmid over-expressing PtA–YPT6 [pUN100-(PNOP1)PtA-YPT6]. Transformants were spotted onto selective (–Leu) plates and incubated for 2 days at 37°C.
Ric1p and Rgp1p bind to Ypt6p and act as a nucleotide exchange factor
In an independent series of experiments, we expressed a protein A–Ypt6p (PtA–Ypt6p) chimera in yeast cells, under the control of the NOP1 promoter. This fusion protein was functional since it could complement the ypt6 deletion mutant (data not shown). We then isolated PtA–Ypt6p by affinity chromatography on IgG–Sepharose and looked for associated proteins. Since the interactions of the rab/Ypt GTPases are controlled by the state of the bound nucleotide, we incubated a portion of the cell extract with the non-hydrolysable GTP analogue, GTP-γS, prior to purification and performed all subsequent steps in the presence of it. As shown in Figure 3A, two proteins that were present in very low amounts relative to Ypt6p were selectively lost when the extract was incubated with GTP-γS. Mass spectrometry of tryptic digests of these proteins identified them as Ric1p and Rgp1p. To confirm this we co-expressed GFP-tagged Ric1p with PtA–Ypt6p, isolated the Ypt6p and detected Ric1p–GFP by immunoblotting. Figure 3B shows that Ric1p again copurified with Ypt6p, and that it could be released by addition of GTP-γS in the lysis buffer. GDP also released Ric1p, but less efficiently. Thus, we conclude that the Ric1p–Rgp1p complex can bind to Ypt6p, and that it interacts preferentially with the nucleotide-free form and, to a lesser extent, with the GDP form, but not with the GTP form. These binding characteristics are typical for guanine nucleotide exchange factors (GEFs).

Fig. 3. The Ric1p–Rgp1p complex binds to Ypt6p in vivo in a nucleotide-dependent manner. (A) Affinity purification of a PtA–Ypt6p fusion and identification of proteins that bind to it. A detergent-solubilized supernatant from a ypt6Δ strain expressing a PtA–YPT6 fusion, in the absence (–) or presence of GTP-γS, was loaded onto an IgG–Sepharose column and the purified fusion protein eluted at low pH. Samples from the supernatant loaded on to the column (S) and the eluted PtA–Ypt6p (E), (3700-fold concentrated as compared with the supernatant), were analysed by SDS–PAGE followed by Coomassie staining. The position of PtA–Ypt6p and the copurifying Ric1p and Rgp1p, identified by mass spectrometry, are indicated by asterisks. (B) PtA–Ypt6p was affinity purified from a ypt6Δ ric1Δ double deletion strain co-expressing a Ric1p–GFP fusion from a Ycplac33-URA3 plasmid, in the absence of nucleotide, or in the presence of GDP or GTP-γS. The eluates from the IgG–Sepharose column were analysed by SDS–PAGE followed by western blotting using anti-protein A or anti-GFP antibodies.
As a direct test of GEF activity, we purified Ypt6p from a yeast strain lacking both Rgp1p and Ric1p (see Materials and methods), loaded it with [3H]GDP, incubated it with excess unlabelled GDP and monitored the release of labelled GDP from the protein. As shown in Figure 4A, the spontaneous rate of release was low but addition of the Ric1p–Rgp1p complex, purified by protein A affinity chromatography and TEV protease cleavage as in Figure 2, accelerated release in a dose-dependent manner. Identical results were obtained when unlabelled GTP was used as competitor (data not shown). This effect was clearly catalytic, since complete release of [3H]GDP from 90 pmol Ypt6p could be induced by only 8 pmol of the complex; only 20% of the [3H]GDP was released without the complex within the same time period.

Fig. 4. The Ric1p–Rgp1p complex stimulates GDP exchange on Ypt6p. (A) Ypt6p and the Ric1p–Rgp1p complex were purified from yeast as described in Materials and methods. Ninety picomoles of Ypt6p pre-incubated with [3H]GDP were incubated with the indicated amounts of Ric1p–Rgp1p at 24°C, and protein-bound radioactivity was determined at intervals by filtration of aliquots through nitrocellulose. Background binding (with bovine serum albumin in place of Ypt6p) was equivalent to ∼10% of the total and has not been subtracted. (B) Ypt1p preloaded with [3H]GDP was incubated with the Ric1p–Rgp1p complex at 24°C and nucleotide exchange was determined as in (A). (C) Ypt6p preloaded with [3H]GDP was incubated independently with either Ric1p or Rgp1p or the Ric1p–Rgp1p complex at 24°C. Nucleotide exchange was determined as in (A).
The exchange activity was specific, since no effect of Ric1p–Rgp1p was seen when Ypt1p was used in place of Ypt6p (Figure 4B). We also tested Ric1p and Rgp1p separately, expressing each from a multicopy vector in the appropriate deletion strain. This was necessary because Rgp1p, in particular, appeared unstable in the absence of its partner. However, neither protein alone provided detectable exchange activity (Figure 4C).
Taken together these data show that the Ric1p–Rgp1p complex can bind to Ypt6p and function as an exchange factor. Like other exchange factors, it probably acts primarily by destabilizing the Ypt6p–GDP complex.
Role of Ric1p–Rgp1p in Ypt6p-mediated traffic
If Ric1p and Rgp1p are important for the formation of Ypt6p–GTP, the presumed active form of the protein, then one would expect cells lacking one or both to have phenotypes similar to those of ypt6 mutants (Li and Warner, 1996; Tsukada et al., 1999). Indeed, like ypt6 cells, a ric1 mutant showed little defect in secretion of invertase or transport to the plasma membrane of an endocytosis-defective version of the exocytic SNARE Snc1p (data not shown), and only a mild mis-sorting of the vacuolar protease carboxypeptidase Y (Bensen et al., 2000). ypt6, ric1 and rgp1 cells all showed a similar characteristic fragmentation of vacuoles, as visualized by uptake of the dye FM4-64 (Figure 5A). Since many of the phenotypes described for ypt6 are similar to those of tlg1, a gene required for recycling of proteins such as Snc1p through early endosomes to the Golgi, we also examined the distribution of GFP–Snc1p in the various mutants. In wild-type cells this protein travels from Golgi to plasma membrane, is endocytosed and returns to the Golgi for re-use (Lewis et al., 2000); in the steady state it is visible on the plasma membrane and in internal dots corresponding to endosomes and/or Golgi cisternae. In ypt6, ric1 and rgp1 cells it was entirely intracellular, in an indistinct hazy or dotty pattern that probably corresponds at least in part to transport vesicles that are unable to fuse with their target (Figure 5B). Hence, as previously proposed for Ypt6p (Tsukada et al., 1999), all three proteins are likely to be involved in endosome–Golgi recycling.
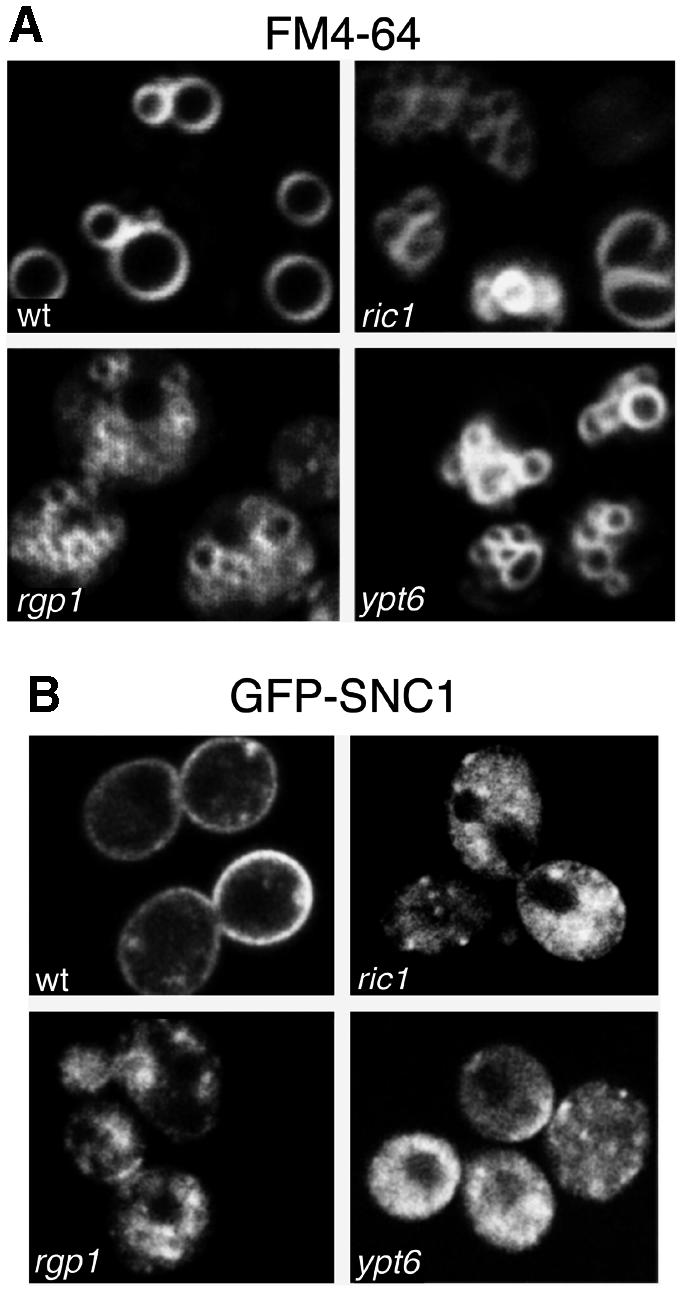
Fig. 5. Defects in vacuolar morphology and GFP–Snc1p recycling in ric1Δ, rgp1Δ and ypt6Δ mutants. (A) The ric1Δ, rgp1Δ and ypt6Δ deletion mutants and a wild-type strain were grown at 30°C to exponential phase, labelled with the endocytic tracer dye FM4-64 for 15 min and chased for 1 h. Note that only vacuoles are visible under these conditions. (B) The deletion mutants were transformed with a plasmid expressing GFP–Snc1p. Transformants were grown to exponential phase at 30°C and inspected by confocal microscopy.
Despite these similarities, we had observed a more severe growth defect for ypt6Δ cells than for ric1Δ, rgp1Δ or a ric1Δ rgp1Δ double mutant, especially on selective medium at elevated temperatures. This implied that Ypt6p was capable of some function even without the GEF activity provided by Ric1p and Rgp1p. In confirmation of this, we found that even very mild overexpression of Ypt6p (from a low-copy plasmid and the YPT6 promoter) was sufficient to improve growth of ric1Δ, rgp1Δ and the double mutant at 37°C, and when a stronger promoter was used to drive YPT6 expression, growth was restored to wild-type levels (Figure 6). This could be accounted for by a low level of Ypt6p–GTP being generated spontaneously, or by the action of some other GEF. It strongly suggests that the major role of Ric1p and Rgp1p in vivo is to activate Ypt6p. Conversely, we can conclude that once activated by nucleotide exchange, Ypt6p does not require Ric1p or Rgp1p to stimulate membrane traffic.
Location of Ypt6p and Ric1p–Rgp1p in cells
We used functional GFP chimeras, expressed at endogenous levels in the absence of the corresponding untagged protein, to localize Ypt6p, Ric1p and Rgp1p. All of these fusion proteins retained their function (Figure 1A and data not shown). None are integral membrane proteins, but confocal microscopy showed that each was present in a dotted pattern typical of the yeast Golgi (Figure 7A). To identify the dots, we colocalized Ric1p–GFP with a YFP-tagged version of Sec7p (Figure 7; see Materials and methods for details). Analysis of fields of cells showed that 75–80% of the Ric1p dots also contained Sec7p. The dots containing the two proteins sometimes showed slight differences in shape or position, suggesting segregation of the proteins within a single structure. Since Sec7p is a late Golgi marker (Rossanese et al., 1999; Lewis et al., 2000), the colocalization confirms that the Ric1p–Rgp1p complex is present on Golgi membranes. A similar analysis showed that Ypt6p also overlapped with Sec7p (Figure 7), but interestingly, the degree of overlap appeared lower: only 50–60% of the Ypt6p dots contained Sec7p. Hence, Ypt6p may not be restricted to late Golgi membranes alone.

Fig. 7. Localization of Ric1p–GFP, Rgp1p–GFP and GFP–Ypt6p fusions. (A) RIC1–GFP, RGP1–GFP and GFP–YPT6 were expressed from centromeric plasmids under the control of their own promoters, in the corresponding deletion mutants. (B) The Ric1p–GFP and GFP–Ypt6p strains were mated with a strain bearing a chromosomal copy of YFP–SEC7, under the control of the TPI1 promoter. YFP and GFP images of diploid cells were obtained as described in Materials and methods. Arrows indicate identical positions in the two images.
Ric1p–Rgp1p affects the localization of Ypt6p
We next examined Ric1p–GFP and GFP–Ypt6p individually in a ypt6Δ ric1Δ strain; since the GFP-tagged proteins are functional, this allowed the effects of removal of one on the distribution of the other to be tested. As might be expected, since the Golgi complex remains functional, the dotty pattern of Ric1p–GFP was unchanged in ypt6 cells (Figure 8A). This shows that binding of the Ric1p–Rgp1p complex to membranes does not require Ypt6p. However, removal of Ric1p had a dramatic effect on the distribution of GFP–Ypt6p: the pattern changed from dots (when RIC1 was provided on a plasmid) to an even haze (Figure 8B). Similar results were obtained with an rgp1 deletion mutant (data not shown). Since removal of Ric1p is not likely to have any greater effect on Golgi structure than removal of Ypt6p, the absence of a dotty pattern for GFP–Ypt6p suggests that this protein is absent from Golgi membranes in a ric1 mutant.
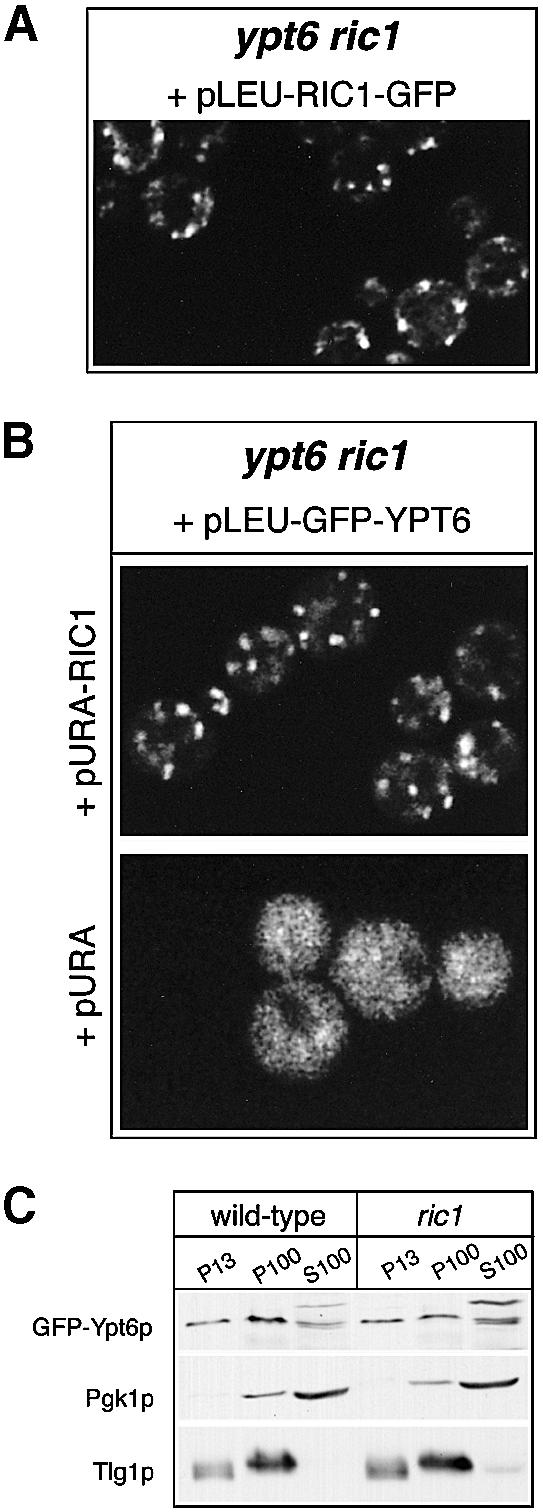
Fig. 8. Subcellular distribution of GFP–Ypt6p is drastically altered in ric1 deletion mutants. (A) The ypt6 ric1 double deletion mutant was transformed with a centromeric plasmid expressing Ric1p–GFP and examined under the confocal microscope. (B) A ypt6 ric1 double deletion mutant transformed with the indicated plasmids was examined under the MRC-600 confocal microscope. (C) The GFP–YPT6 plasmid was transformed into ypt6 cells, thus complementing them (‘wild-type’) or into the double ypt6 ric1 deletion mutant (‘ric1’) and fractionated into 13 000 and 100 000 g pellets (P13 and P100, respectively) and 100 000 g supernatant (S100). Equivalent amounts of each fraction were analysed by SDS–PAGE followed by western blotting with anti-GFP, anti-Pgk1p and anti-Tlg1p antibodies. The upper band visible in the S100 fraction probed with anti-GFP is a cross-reacting endogenous protein.
A possible explanation would be that only the GTP-bound form of Ypt6p associates with membranes. To test this, we fractionated the cells by lysis and centrifugation at 13 000 g and then 100 000 g and analysed the pellets and supernatant by immunoblotting (Figure 8C). As controls, we checked the soluble enzyme phosphoglycerokinase (Pgk1p), which was found primarily in the supernatant, and the late Golgi/early endosomal SNARE Tlg1p, which was found partly in the P13 fraction and partly in the P100 fraction. In wild-type cells, GFP–Ypt6p was distributed between all three fractions, with ∼80% being present in the combined membrane pellets and the remainder in the supernatant. Strikingly, however, this distribution was almost identical whether or not Ric1p was present. Thus, the change in distribution of GFP–Ypt6p is unlikely to be due to its release from membranes.
To summarize, in the absence of its exchange factor complex, Ypt6p changes its punctate Golgi localization to a haze, yet appears to be still membrane bound. It is possible that in a ric1 mutant, Ypt6p is bound to small endosome-derived transport vesicles, which, because they are numerous and scattered, appear as a haze. This could occur if Ypt6p binds initially to endosomal membranes and is delivered to the Golgi on vesicles, or if it is permanently membrane associated and cycles through early endosomes in a similar manner to resident late Golgi membrane proteins. By the same argument, the fact that the distribution of Ric1p is unaltered in ypt6Δ cells implies that it does not cycle through early endosomes.
Discussion
In this paper we have shown that Ric1p and Rgp1p form a complex that is peripherally associated with Golgi membranes and that has the ability to catalyse, in vitro, guanine nucleotide exchange on Ypt6p. This in vitro evidence is supported by the observations that small amounts of Ric1p and Rgp1p can be isolated from cells in a complex with Ypt6p, which dissociates upon addition of GTP. Furthermore, ric1 and rgp1 mutants have similar phenotypes to a ypt6 mutant, and like ypt6Δ, a ric1Δ mutant shows synthetic lethality with an imh1Δ mutation.
A ric1 mutant was originally isolated in a screen for genes affecting ribosome synthesis, and Ric1p was postulated to be a transcription factor (Mizuta et al., 1997). However, the same screen yielded a ypt6 mutant, and it has been established that a variety of secretory mutants affect ribosome synthesis (Mizuta and Warner, 1994; Li and Warner, 1996). RIC1 also shows genetic interactions with clathrin mutants (Bensen et al., 2000). Hence, the effects of Ric1p on ribosome synthesis are likely to be secondary to its effects on Ypt6p. Removal of Ric1p from a ypt6Δ strain does not result in any additional growth impairment, and the growth defects of ric1 and rgp1 mutants can be fully rescued by overexpression of YPT6. This argues that the major and perhaps only role of Ric1p and Rgp1p is in the activation of Ypt6p.
Why two proteins should be required for GEF function is unclear. However, the single proteins seem relatively unstable, and preliminary results indicate that neither Ric1p nor Rgp1p is able to localize to Golgi membranes efficiently in the absence of its partner. Thus, the complex forms a functional unit. Further analysis will be required to determine the precise residues responsible for the GEF activity.
Neither Ric1p nor Rgp1p shows any detectable homology to other known GEFs, including those active on rab-like GTPases such as Sec2p (a GEF for Sec4p), Vps9p (a GEF for Vps21p) or the rab5 GEF rabex 5. However, database searches show that Ric1p contains a C-terminal region that is clearly homologous to proteins in Schizosaccharomyces pombe and other species, including humans (Figure 9). The proteins from higher eukaryotes that contain this domain are obvious candidates for rab6 exchange factors.

Fig. 9. Multiple sequence alignment of the C-terminal domain of Ric1p with related sequences found in proteins from other species: Drosophila melanogaster (DDBJ/EMBL/GenBank accession No. AF181626.1); Homo sapiens (AB037853.1; brackets in the residue number indicate that it is a partial sequence); Caenorhabditis elegans (Q09417); S.pombe (AL133361.1). Sequences were aligned with Clustal W1.8 (Thompson et al., 1994) and displayed using the program Boxshade (http://www.ch.embnet.org/software/BOX_form.html).
The phenotypes of ypt6, ric1 and rgp1 mutants are fully consistent with a role for Ypt6p in transport from early endosomes to the Golgi complex. Indeed, the properties of cells lacking Ypt6p are strikingly similar to those of cells lacking the SNARE Tlg1p, which has an established role in this process (Holthuis et al., 1998b). The most characteristic features are loss of late Golgi markers, such as Kex2p, to the vacuole and a failure to recycle Snc1p, but the mutants also share temperature-sensitive growth, partial mis-sorting of carboxypeptidase Y and similar ultrastructural abnormalities (Holthuis et al., 1998a; Tsukada et al., 1999; Lewis et al., 2000). Interestingly, in one yeast strain (W303), both Tlg1p and Ypt6p are also required for transport of secretory proteins through the Golgi (Li and Warner, 1996; Coe et al., 1999). The reason for this strain-specific effect is unclear, but it may be that delivery of components from endosomes to the Golgi is important for maintenance of secretory function.
The localization of Ric1p–Rgp1p on Golgi membranes suggests that the GTP form of Ypt6p acts on the Golgi, to catalyse fusion of vesicles with it. This fits well with previous observations on Ypt1p, the GTPase that catalyses ER–Golgi transport. Though the GEF for Ypt1p has not been identified, its activity cofractionates with the Golgi (Jones et al., 1998). Furthermore, although Ypt1p itself can be found on both Golgi and ER, and is likely to be present on vesicles that move between them, recent in vitro experiments show that its function requires it to be only on Golgi membranes, not on the transport vesicles themselves (Cao and Barlowe, 2000). Thus, both Ypt1p and Ypt6p may be activated to their GTP-bound state on Golgi membranes, and act there. Since Ric1p colocalizes quite well with Sec7p, which seems to be associated mostly with late Golgi cisternae (Rossanese et al., 1999; Lewis et al., 2000), Ypt6p may preferentially catalyse fusion with these later compartments.
Overexpression of Ypt6p allows it to function in the absence of its GEF, perhaps through spontaneous GTP binding or interaction with another GEF such as the unidentified one for Ypt1p. Under these circumstances, Ypt6p must perform its function through effectors that are distinct from Ric1p and Rgp1p. Ypt1p is thought to recruit the tethering factor Uso1p to membranes (Cao et al., 1998), and it is tempting to speculate that Ypt6p performs a similar function for Imh1p, which shows some similarity with Uso1p. Indeed, Imh1p contains a GRIP domain, which has been shown to target proteins to the Golgi and has been reported to bind to rab6 on blots (Barr, 1999; Kjer-Nielsen et al., 1999; Munro et al., 1999). However, we have not been able to detect any interaction between Ypt6p and Imh1p by coprecipitation, and the fact that Imh1p can function (and indeed is essential for growth) in the absence of Ypt6p, and vice versa, argues that any such interaction is of minor functional importance. The genetic relationships are nevertheless striking, and suggest that Imh1p either contributes to the same fusion event as Ypt6p, albeit independently, or is involved in an alternative pathway from endosomes to Golgi. But the strong conclusion remains that Ypt6p must have an effector other than Imh1p, which remains to be identified.
Removal of its GEF did not prevent Ypt6p from binding to membranes, as judged by fractionation of the cells, suggesting that GDP–GTP exchange is not needed for membrane association of Ypt6p. This is in agreement with previous studies that detected transient membrane-bound rab–GDP intermediates, indicating that membrane binding precedes and is distinct from nucleotide exchange (Soldati et al., 1994; Ullrich et al., 1994). Similarly, in the absence of its GEF, Sec2p, Sec4p still associates with late secretory vesicles (Walch-Solimena et al., 1997). Surprisingly, however, Ypt6p has a dispersed, presumably vesicular distribution in ric1Δ cells, suggesting that it may be bound to small vesicles rather than to Golgi membranes. One possibility is that Ypt6p in its GDP-bound state binds initially to early endosomes or to vesicles that have budded from them (which is in agreement with our observation that, even in wild-type cells, a substantial proportion of the Ypt6p does not colocalize with Sec7p). Interaction of vesicle-bound Ypt6p with the Ric1p–Rgp1p complex could then occur as vesicles dock with the late Golgi, leading to the generation of Ypt6p–GTP and activation and/or recruitment of other components of the docking and fusion machinery. Of course, free Ypt6p may also be recruited directly to Golgi membranes by the Ric1p–Rgp1p complex, if this is present. Once fusion is complete, a Ypt6p GTPase-activating protein, Gyp6p (Strom et al., 1993), presumably returns Ypt6p to the GDP state. It might then dissociate from the Golgi membrane and rebind to endosomes, or share the same transport pathway as other late Golgi membrane proteins that recycle through early endosomes.
Such a mechanism would ensure that active Ypt6p is generated where and when it is needed. However, it is not clear whether precise control of this is actually necessary for Ypt6p to function, and, since the site at which Ypt6p hydrolyses GTP is unknown, an additional function for Ypt6p in early endosomes cannot be ruled out. In practice, it has proven very difficult to distinguish early endosomal from late Golgi membranes in yeast (Lewis et al., 2000).
It is striking that the distribution of Ric1p in a ypt6 mutant remains punctate rather than dispersed. This argues that Ric1p–Rgp1p itself does not cycle through endosomes, as one would expect it to be trapped in vesicles under these conditions if it did. The Ric1p–Rgp1p complex may therefore be one component that functionally differentiates Golgi membranes from endosomes, perhaps recognizing directly some characteristic feature of the membrane, such as its lipid composition. Indeed, attempts to fractionate membranes containing the complex showed that it was readily released in vitro, implying that it is not firmly anchored to some integral protein (our unpublished observations). Interestingly, the rab GEFs identified so far are all peripheral membrane proteins. These could, in principle, identify even continuously maturing compartments, binding and leaving as their membrane composition changes.
One remaining question concerns the relationship between the function of Ypt6p in yeast cells and that of rab6 in animal cells. Rab6 is found on the Golgi as well as on mobile structures that move around the cytoplasm (White et al., 1999). It has been argued that it is required for retrograde transport of toxin molecules from the cell surface to the ER, and that it may mediate a novel pathway direct from the trans-Golgi network to the ER (Girod et al., 1999). However, our results suggest that at least some of the rab6-positive structures may in fact be en route from early endosomes to the Golgi. The situation is complicated by the fact that there are at least three different isoforms of rab6 in humans; it is possible that these have distinct functions, some of which are unique to animal cells. Further insight may be provided by examining the location and function of the mammalian protein related to Ric1p.
Materials and methods
Yeast strains and plasmids
The yeast strains used in this work are listed in Table I. The following plasmids were used: YCplac111 and Ycplac33, ARS1/CEN4 vectors with the LEU2 and the URA3 marker, respectively (Gietz and Sugino, 1988); YEplac181, 2µ vector with the LEU2 marker (Gietz and Sugino, 1988); pRS316-ADE3-IMH1, a pRS316 ARS4/CEN6 plasmid (Sikorski and Hieter, 1989) with the ADE3 gene (inserted at the SmaI–SpeI sites) and the IMH1 gene (inserted at the SacI–XbaI sites); pUN100-(PNOP1)PtA, an ARS1/CEN4 plasmid expressing the protein A tag (Senger et al., 1998). The integrative (PTPI1)YFP–SEC7 construct was kindly provided by Drs S.Reichelt and T.Levine. The yeast genomic library was from ATCC (ATCC no. 77164).
Table I. Yeast strains used in this study.
| Strain | Genotype | Reference/source |
|---|---|---|
| SEY6210 | MATα ura3 his3 leu2 trp1 suc2 lys2 | S.Emr |
| IAY25 | MATa ura3 his3 leu2 trp1 can1 ade2 ade3 | J.Kilmartin and I.Adams |
| MHY002 | MATα ura3 his3 leu2 trp1 suc2 lys2 ypt6::HIS3 | Pelham laboratory |
| BNY01 | MATα ura3 his3 leu2 trp1 suc2 lys2 imh1::HIS5 (Sp) | Pelham laboratory |
| SSY01 | MATa ura3 his3 leu2 trp1 ade2 ade3 imh1::HIS5(Sp) [pRS316-URA3-ADE3-IMH1] | this study |
| SSY02 | MATα ura3 his3 leu2 trp1 suc2 lys2 ric1::TRP1 | this study |
| SSY03 | MATα ura3 his3 leu2 trp1 suc2 lys2 rgp1::HIS3 | this study |
| SSY04 | MATα ura3 his3 leu2 trp1 suc2 lys2 ypt6::HIS5(Sp) ric1::TRP1 | this study |
| SSY05 | MATα ura3 his3 leu2 trp1 suc2 lys2 ypt6::HIS5(Sp) rgp1::TRP1 | this study |
| SSY06 | MATα ura3 his3 leu2 trp1 suc2 lys2 ypt6::HIS5(Sp) [YCplac111-LEU2-GFP-YPT6] | this study |
| SSY07 | MATα ura3 his3 leu2 trp1 suc2 lys2 ric1::TRP1 [YCplac111-LEU2-RIC1-GFP] | this study |
| SSY08 | MATα ura3 his3 leu2 trp1 suc2 lys2 ric1::TRP1 rgp1::HIS3 | this study |
| SSY09 | MATa ura3 leu2 trp1 ade2 sec7::(PTPI1)YFP-SEC7 | this study |
| SSY10 | cross between SSY09 and SSY06 | this study |
| SSY11 | cross between SSY09 and SSY07 | this study |
Synthetic lethal screen with an imh1 deletion mutant
To perform a synthetic lethal screen with the imh1Δ deletion mutant, the red/white colony sectoring assay (Kranz and Holm, 1990) was used. The screening strain was constructed by mating the imh1Δ strain with the IAY25 strain, selecting after tetrad dissection the ade2 ade3 imh1Δ haploid progeny and transforming it with a pRS316-ADE3-IMH1 plasmid. This sectoring strain was grown in selective medium (–Ura) and UV-mutagenized (with a killing rate ranging between 97 and 99%). Thirteen-thousand-and-one-hundred colonies were analysed for a red, non-sectoring phenotype at 30°C, on YEPD plates containing 4% glucose. After three successive rounds of selection, six mutagenized colonies that were synthetically lethal with imh1Δ, but not IMH1, were isolated (sl6, 9, 61, 68, 78 and 233). Sl233 was transformed with a TRP1/CEN-based yeast genomic library and seven transformants that exhibited red/white sectoring and could grow on 5-FOA plates were isolated. Restriction analysis of the recovered plasmids demonstrated that five of them contained overlapping genomic inserts that did not correspond to the IMH1 gene. Sequencing of the inserts, followed by subcloning of the ORFs present, demonstrated that the RIC1 gene complemented the sl233 mutant. The synthetic lethality between imh1Δ and the ric1Δ deletion mutants was verified by deleting the RIC1 gene in the starting IMH1 sectoring strain. Sl6, 9 and 68, but not sl78, regained both the sectoring phenotype and growth on 5-FOA plates upon transformation with RIC1. Transformation of sl78 with the genomic library, followed by similar analysis as described above, demonstrated that three sectoring transformants were complemented by the same overlapping genomic fragments and that the complementing activity resided in the YPT6 gene.
Deletion of the RIC1, RGP1, YPT6 and IMH1 genes
For the deletion of the RIC1 gene, an internal NheI–XhoI fragment (Ala100 to Leu1052) was removed and replaced by a filled-in fragment coding for the TRP1 gene. The ric1::TRP1 allele, with 5′ and 3′ flanking sequences, was excised as an EcoRI fragment and transformed into either the SEY6210 wild-type strain or the IMH1 sectoring strain used for the synthetic lethality screen. The complete RGP1 ORF was deleted by generating two unique BamHI sites via PCR-mediated mutagenesis, one just after the start codon and the other just before the stop codon, removal of the DNA between start and stop codon and insertion of a BamHI fragment containing either the HIS3 or the TRP1 gene. The rgp1::HIS3, or rgp1::TRP1 alleles, carrying 5′ and 3′ flanking sequences, were excised as SacI–SphI fragments and transformed into the SEY6210 strain. The ypt6::HIS5 and imh1::HIS5 strains were kindly provided by M.van Horssen and B.Nichols. Deletion of YPT6 and IMH1 in these strains was achieved by replacing the entire coding regions of these genes by the HIS5 gene of S.pombe.
Construction of RIC1, RGP1, YPT6 and YPT1 fusion genes
In order to tag RIC1 with protein A, a naturally occurring XhoI site present three codons before the stop codon of the RIC1 ORF was used to insert an XhoI fragment coding for the three last residues of Ric1p, followed by the TEV protease cleavage site and two IgG-binding domains from Staphylococcus aureus protein A. The same C-terminal XhoI site was used to insert in-frame the S65T/V163A variant of GFP and construct the RIC1–GFP fusion. To tag RGP1 with protein A or GFP, a BamHI site was introduced just before the stop codon by PCR and a BamHI fragment coding for TEV-PtA, or GFP, respectively, was inserted. The GFP–YPT6 fusion protein was constructed by introducing a BamHI site after the start codon of YPT6 by PCR and in-frame insertion of a BamHI fragment coding for GFP. Most of the RIC1 and RGP1 fusion proteins and the GFP–YPT6 fusion were expressed under the control of their respective authentic promoters from centromeric plasmids with the LEU2 (YCplac111) or URA3 (YCplac33) markers and could complement the phenotypes of the ric1, rgp1 or ypt6 deletion mutants, respectively. In order to obtain sufficient amounts of Ric1p and Rgp1p from the ric1 rgp1 mutant, the RIC1–PtA and RGP1–PtA fusions were also subcloned into high-copy vectors (YEplac181). To construct the PtA–YPT6 and PtA–YPT1 fusions, the YPT6 or YPT1 open reading frames were cloned into the NdeI and BamHI sites of the pUN100-(PNOP1)PtA plasmid. The PtA–YPT6 and PtA–YPT1 fusions were expressed under the control of the NOP1 promoter from a centromeric plasmid with the LEU2 marker (pUN100).
Antibodies
The antibody used to detect protein A fusions was from DAKO (catalogue no. Z0113). Anti-GFP polyclonal serum was a gift from R.Arkowitz (MRC LMB), the anti-Tlg1p polyclonal antibody as previously described (Holthuis et al., 1998a) and the anti-Pgk1p monoclonal from Molecular Probes (catalogue no. A-6457).
Affinity purification of RIC1-PtA, RGP1-PtA and PtA-YPT6
For affinity purification of Ric1p–PtA, Rgp1p–PtA and PtA–Ypt6p, yeast strains expressing the corresponding protein A fusion proteins (Table I) were grown in selective medium (–Leu) (OD600 2.0–2.5). Cells were spheroplasted and kept frozen at –20°C. One gram of frozen spheroplast pellet was lysed in 16 ml ice-cold lysis buffer (150 mM KCl, 20 mM Tris–HCl pH 8.0, 5 mM MgCl2, 1% Triton X-100) supplemented with a cocktail of protease inhibitors (complete EDTA-free; Boehringer Mannheim). The extract was centrifuged for 15 min at 15 000 r.p.m. (SS-34 rotor, Sorvall) and the supernatant was first precleared by incubation with Sepharose Fast-flow beads (Pharmacia) and then loaded on to a column packed with 200 µl IgG–Sepharose beads (Pharmacia). Beads were washed with 20 ml of lysis buffer. For low pH elution of protein A fusions, beads were washed with 1 ml 5 mM ammonium acetate and bound proteins were eluted with 1 ml 0.5 M acetic acid/ammonium acetate pH 3.4. For TEV protease-dependent elution, beads were washed with 10 ml of TEV buffer [150 mM NaCl, 50 mM Tris–HCl pH 8.0, 0.5 mM EDTA, 0.8 mM dithiothreitol (DTT), 0.01% Triton X-100], then incubated with 20–40 U of TEV protease (Life Technologies) at room temperature for 90 min. Beads were then spun down and the soluble fraction containing the released proteins was collected. In all cases, both low pH and TEV digestion elution released most of the fusion proteins (>80% as judged by Coomassie staining), except for the PtA–Ypt6p TEV elution, where the efficiency was ∼50%. Where indicated, lysis of the PtA–YPT6 spheroplasts was carried out in the presence of 1 mM GDP or GTP-γS. Extracts were incubated for 15 min on ice and then processed, in the presence of GDP or GTP-γS, as described above.
Mass spectrometric analysis
Protein identification by mass spectrometry of tryptic fragments was performed as previously described (Jensen et al., 1997). Samples were analysed with a Voyager-DE-STR MALDI-TOF mass spectrometer (PerSeptive Biosystems). Database searches using peptide masses were performed with the MS-Fit program (http://prospector.ucsf.edu/ucsfhtml3.2/msfit.htm). Fifteen tryptic peptides were obtained from Rgp1p (sequence coverage 31%) derived from the Ric1p–PtA purification. Thirteen tryptic peptides were obtained from Ric1p (sequence coverage 14%) derived from the Rgp1p–PtA purification. In the case of the PtA–Ypt6p purification, 26 tryptic peptides matched the protein identified as Rgp1p (sequence coverage 49%) and 12 peptides matched Ric1p (sequence coverage 14%). In all cases, the minimum required mass accuracy was 50 p.p.m.
Cell fractionation
Cells expressing the GFP–Ypt6p fusion protein were grown in selective medium, harvested at mid-logarithmic phase and spheroplasted as described above. Then, 0.5 g of spheroplasts were lysed in 1 ml of hypo-osmotic buffer (50 mM Tris–HCl pH 7.4, 200 mM sorbitol, 1 mM EDTA) at 4°C, in the presence of a cocktail of protease inhibitors. The cell lysate was centrifuged sequentially at 500 g (5 min), 13 000 g (10 min) and 100 000 g (60 min). Samples of each fraction were collected and analysed by SDS–PAGE followed by western blotting using anti-GFP, anti-Tlg1p and anti-Pgk1p antibodies.
Confocal microscopy
To view live cells expressing the various GFP fusions, cells were grown in the appropriate selective medium at 30°C to early log phase, and examined with an MRC-600 confocal microscope. For double-label GFP/YFP analysis, diploid cells expressing the Ric1p–GFP/YFP–Sec7p and GFP–Ypt6p/YFP–Sec7p pairs were grown to early log phase, fixed with 4% formaldehyde for 5 min, washed once with 0.1% Triton X-100 and twice with medium and then mounted on slides coated with concanavalin A. A Bio-Rad Radiance confocal microscope was used for imaging. YFP images were first collected using the 514 nm argon laser line for excitation and a 590/70 nm emission filter, and the cells then bleached by repeated slow scans with the same laser line until the YFP signal was no longer detectable. Finally, GFP images were collected using the 488 nm laser line and a 515/30 nm emission filter. This procedure was necessary because 488 nm light excites both YFP and GFP. Controls consisting of cells expressing the same YFP and GFP constructs singly confirmed that GFP was neither imaged nor bleached by the 514 nm laser line under these conditions, and that negligible YFP fluorescence could be detected using the 488 nm line following bleaching with 514 nm light. These controls were important for optimization of the scanning conditions.
Nucleotide exchange assay
The Rgp1p–PtA and the PtA–Ypt6p fusions were used to affinity purify the Ric1p–Rgp1p complex and Ypt6p from yeast cells, respectively, as described above. To ensure that no Ric1p–Rgp1p would be present in the Ypt6p preparation prior to the exchange assay, PtA–Ypt6p was isolated from a ric1 rgp1 deletion strain. Proteins were released from the IgG–Sepharose beads by TEV protease digestion. For GDP exchange assay, 450 µl of Ypt6p (270 pmol) in 110 mM NaCl, 50 mM Tris–HCl pH 8.0, 1 mM EDTA, 0.8 mM DTT, 0.005% Triton X-100 was incubated with 1.5 µM [3H]GDP (10.1 Ci/mmol, Amersham) for 60 min at 30°C. One-hundred-and-fifty microlitres of [3H]GDP–Ypt6p was mixed with an equal volume of 110 mM NaCl, 50 mM Tris–HCl pH 8.0, 12 mM MgCl2, 0.8 mM DTT, 2 mM GDP, in the presence or absence of Ric1p–Rgp1p complex and incubated at 24°C. At the times indicated, aliquots were removed, diluted into ice-cold stop buffer (110 mM NaCl, 50 mM Tris–HCl pH 8.0, 6 mM MgCl2) and filtered through nitrocellulose membranes (0.45 µm, Millipore). Membrane-bound radioactivity was measured by a liquid scintillation counter. Ypt1p purification and GDP exchange in the presence of the Ric1p–Rgp1p complex was carried out in the same way. To test the activities of Ric1p or Rgp1p separately, Ric1p–PtA or Rgp1p–PtA was expressed from high-copy vectors and purified independently from the ric1 rgp1 double mutant as described above. Ypt6p preloaded with [3H]GDP was incubated with Ric1p, Rgp1p or the Ric1p–Rgp1p complex and nucleotide exchange was assayed as above. In all cases protein concentrations were determined both by the Bio-Rad assay and by comparison of the Coomassie staining of purified proteins with a BSA standard.
Acknowledgments
Acknowledgements
We thank Steffi Reichelt for advice and assistance with the two-colour confocal microscopy, and Mike Black, Mike Lewis, Sean Munro and Fulvio Reggiori for helpful comments on the manuscript. We are grateful to Drs M.van Horssen, B.Nichols, S.Reichelt and T.Levine for gifts of yeast strains. S.S. was supported by long-term postdoctoral fellowships from EMBO and the Human Frontiers Science Program.
References
- Aguilera A., Moskowitz,P. and Klein,H.L. (1990) Molecular analysis of RGP1, a new yeast gene required for proper mitotic growth. Nucleic Acids Res., 18, 1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr F.A. (1999) A novel Rab6-interacting domain defines a family of Golgi-targeted coiled-coil proteins. Curr. Biol., 9, 381–384. [DOI] [PubMed] [Google Scholar]
- Bensen E.S., Costaguta,G. and Payne,G.S. (2000) Synthetic genetic interactions with temperature-sensitive clathrin in Saccharomyces cerevisiae. Roles for synaptojanin-like Inp53p and dynamin-related Vps1p in clathrin-dependent protein sorting at the trans-Golgi network. Genetics, 154, 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X. and Barlowe,C. (2000) Asymmetric requirements for a Rab GTPase and SNARE proteins in fusion of COPII vesicles with acceptor membranes. J. Cell Biol., 149, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X., Ballew,N. and Barlowe,C. (1998) Initial docking of ER-derived vesicles requires Uso1p and Ypt1p but is independent of SNARE proteins. EMBO J., 17, 2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavrier P. and Goud,B. (1999) The role of ARF and Rab GTPases in membrane transport. Curr. Opin. Cell Biol., 11, 466–475. [DOI] [PubMed] [Google Scholar]
- Coe J.G., Lim,A.C., Xu,J. and Hong,W. (1999) A role for Tlg1p in the transport of proteins within the Golgi apparatus of Saccharomyces cerevisiae. Mol. Biol. Cell, 10, 2407–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R.D. and Sugino,A. (1988) New yeast–Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene, 74, 527–534. [DOI] [PubMed] [Google Scholar]
- Girod A., Storrie,B., Simpson,J.C., Johannes,L., Goud,B., Roberts,L.M., Lord,J.M., Nilsson,T. and Pepperkok,R. (1999) Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nature Cell Biol., 1, 423–430. [DOI] [PubMed] [Google Scholar]
- Gotte M., Lazar,T., Yoo,J.S., Scheglmann,D. and Gallwitz,D. (2000) The full complement of yeast Ypt/Rab-GTPases and their involvement in exo- and endocytic trafficking. Subcell. Biochem., 34, 133–173. [DOI] [PubMed] [Google Scholar]
- Holthuis J.C., Nichols,B.J., Dhruvakumar,S. and Pelham,H.R. (1998a) Two syntaxin homologues in the TGN/endosomal system of yeast. EMBO J., 17, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis J.C., Nichols,B.J. and Pelham,H.R. (1998b) The syntaxin Tlg1p mediates trafficking of chitin synthase III to polarized growth sites in yeast. Mol. Biol. Cell, 9, 3383–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen O.N., Podtelejnikov,A.V. and Mann,M. (1997) Identification of the components of simple protein mixtures by high-accuracy peptide mass mapping and database searching. Anal. Chem., 69, 4741–4750. [DOI] [PubMed] [Google Scholar]
- Jones S., Richardson,C.J., Litt,R.J. and Segev,N. (1998) Identification of regulators for Ypt1 GTPase nucleotide cycling. Mol. Biol. Cell, 9, 2819–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjer-Nielsen L., Teasdale,R.D., van Vliet,C. and Gleeson,P.A. (1999) A novel Golgi-localisation domain shared by a class of coiled-coil peripheral membrane proteins. Curr. Biol., 9, 385–388. [DOI] [PubMed] [Google Scholar]
- Kranz J.E. and Holm,C. (1990) Cloning by function: an alternative approach for identifying yeast homologs of genes from other organisms. Proc. Natl Acad. Sci. USA, 87, 6629–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar T., Gotte,M. and Gallwitz,D. (1997) Vesicular transport: how many Ypt/Rab-GTPases make a eukaryotic cell? Trends Biochem. Sci., 22, 468–472. [DOI] [PubMed] [Google Scholar]
- Lewis M.J., Nichols,B.J., Prescianotto-Baschong,C., Riezman,H. and Pelham,H.R. (2000) Specific retrieval of the exocytic SNARE Snc1p from early yeast endosomes. Mol. Biol. Cell, 11, 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B. and Warner,J.R. (1996) Mutation of the Rab6 homologue of Saccharomyces cerevisiae, YPT6, inhibits both early Golgi function and ribosome biosynthesis. J. Biol. Chem., 271, 16813–16819. [DOI] [PubMed] [Google Scholar]
- Li B. and Warner,J.R. (1998) Genetic interaction between YPT6 and YPT1 in Saccharomyces cerevisiae. Yeast, 14, 915–922. [DOI] [PubMed] [Google Scholar]
- Mizuta K. and Warner,J.R. (1994) Continued functioning of the secretory pathway is essential for ribosome synthesis. Mol. Cell Biol., 14, 2493–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuta K., Park,J.S., Sugiyama,M., Nishiyama,M. and Warner,J.R. (1997) RIC1, a novel gene required for ribosome synthesis in Saccharomyces cerevisiae. Gene, 187, 171–178. [DOI] [PubMed] [Google Scholar]
- Munro S. and Nichols,B.J. (1999) The GRIP domain—a novel Golgi-targeting domain found in several coiled-coil proteins. Curr. Biol., 9, 377–380. [DOI] [PubMed] [Google Scholar]
- Nichols B.J. and Pelham,H.R. (1998) SNAREs and membrane fusion in the Golgi apparatus. Biochim. Biophys. Acta, 1404, 9–31. [DOI] [PubMed] [Google Scholar]
- Pelham H.R. (1999) SNAREs and the secretory pathway—lessons from yeast. Exp. Cell Res., 247, 1–8. [DOI] [PubMed] [Google Scholar]
- Pfeffer S.R. (1999) Transport-vesicle targeting: tethers before SNAREs. Nature Cell Biol., 1, E17–22. [DOI] [PubMed] [Google Scholar]
- Rossanese O.W., Soderholm,J., Bevis,B.J., Sears,I.B., O’Connor,J., Williamson,E.K. and Glick,B.S. (1999) Golgi structure correlates with transitional endoplasmic reticulum organization in Pichia pastoris and Saccharomyces cerevisiae. J. Cell Biol., 145, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman J.E. (1994) Mechanisms of intracellular protein transport. Nature, 372, 55–63. [DOI] [PubMed] [Google Scholar]
- Senger B., Simos,G., Bischoff,F.R., Podtelejnikov,A., Mann,M. and Hurt,E. (1998) Mtr10p functions as a nuclear import receptor for the mRNA-binding protein Npl3p. EMBO J., 17, 2196–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R.S. and Hieter,P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldati T., Shapiro,A.D., Svejstrup,A.B. and Pfeffer,S.R. (1994) Membrane targeting of the small GTPase Rab9 is accompanied by nucleotide exchange. Nature, 369, 76–78. [DOI] [PubMed] [Google Scholar]
- Strom M., Vollmer,P., Tan,T.J. and Gallwitz,D. (1993) A yeast GTPase-activating protein that interacts specifically with a member of the Ypt/Rab family. Nature, 361, 736–739. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M. and Gallwitz,D. (1996) Isolation and characterization of SYS genes from yeast, multicopy suppressors of the functional loss of the transport GTPase Ypt6p. J. Cell Sci., 109, 2471–2481. [DOI] [PubMed] [Google Scholar]
- Tsukada M., Will,E. and Gallwitz,D. (1999) Structural and functional analysis of a novel coiled-coil protein involved in Ypt6 GTPase-regulated protein transport in yeast. Mol. Biol. Cell, 10, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O., Horiuchi,H., Bucci,C. and Zerial,M. (1994) Membrane association of Rab5 mediated by GDP-dissociation inhibitor and accompanied by GDP/GTP exchange. Nature, 368, 157–160. [DOI] [PubMed] [Google Scholar]
- Walch-Solimena C., Collins,R.N. and Novick,P.J. (1997) Sec2p mediates nucleotide exchange on Sec4p and is involved in polarized delivery of post-Golgi vesicles. J. Cell Biol., 137, 1495–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters M.G. and Pfeffer,S.R. (1999) Membrane tethering in intracellular transport. Curr. Opin. Cell Biol., 11, 453–459. [DOI] [PubMed] [Google Scholar]
- White J. et al. (1999) Rab6 coordinates a novel Golgi to ER retrograde transport pathway in live cells. J. Cell Biol., 147, 743–760. [DOI] [PMC free article] [PubMed] [Google Scholar]