

Abstract
The proven candidate genes for amelogenesis imperfecta (AI) are AMELX, ENAM, MMP20, KLK4, FAM83H, and WDR72. We performed mutation analyses on seven families with hypomaturation AI. A novel WDR72 dinucleotide deletion mutation (g.57,426_57,427delAT; c.1467_ 1468delAT; p.V491fsX497) was identified in both alleles of probands from Mexico and Turkey. Haplotype analyses showed that the mutations arose independently in the two families. The disease perfectly segregated with the genotype. Only persons with both copies of the mutant allele were affected. Their hypomineralized enamel suffered attrition and orange-brown staining following eruption. Expression of WDR72 fused to green fluorescent protein showed a cytoplasmic localization exclusively and was absent from the nucleus. We conclude that WDR72 is a cytoplasmic protein that is critical for dental enamel formation.
Keywords: protein localization, enamel maturation, enamel, tooth, Amelogenesis imperfect
Introduction
Amelogenesis imperfecta (AI) is a heterogeneous group of inherited conditions featuring the pathological formation of dental enamel. One out of every 700 to 14,000 persons is affected, depending upon the population being studied (Witkop and Sauk, 1976; Chosack et al., 1979; Backman and Holm, 1986; Dummer et al., 1990). By the strict definition, the phenotype is limited to dental enamel and is not associated with more generalized defects (Witkop, 1971). The initial candidate genes for AI were those encoding enamel matrix proteins, and 4 have been implicated in its etiology: amelogenin (AMELX; MIM *300391) (Lagerström et al., 1991), enamelin (ENAM; MIM *606585) (Rajpar et al., 2001), kallikrein 4 (KLK4; MIM *603767) (Hart et al., 2004), and enamelysin (MMP20; MIM *604629) (Kim et al., 2005b).
In addition to candidate gene approaches, genome-wide scans combined with mutational analyses identified AI-causing genes not previously suspected to play a role in dental enamel formation. Family with sequence similarity 83 member H (FAM83H; MIM 611927) was shown to cause autosomal-dominant hypocalcified AI (ADHCAI) (Mendoza et al., 2007; Kim et al., 2008), a severe and common form of AI. The function of FAM83H is still obscure, but it is expressed by ameloblasts (Lee et al., 2009) and is associated with intracellular vesicles (Ding et al., 2009). The enamel of persons with FAM83H mutations is less mineralized than normal and undergoes extensive post-eruptive enamel loss, but sometimes retains focal areas of normal-looking enamel (El-Sayed et al., 2010).
WD repeat-containing protein 72 (WDR72; MIM 613214) was recently linked to AI by homozygosity mapping with a single nucleotide polymorphism (SNP) microarray followed by mutational analyses (El-Sayed et al., 2009). Three different AI-causing mutations in WDR72 were identified, and all showed a recessive pattern of inheritance. At the time of tooth eruption, the enamel was relatively smooth and creamy-brown-colored, but suffered rapid attrition and staining that left the enamel surface irregular and dark brown or orange (El-Sayed et al., 2009).
Hypomaturation AI is thought to be caused by malformations that specifically affect modulating ameloblasts in the late stages of enamel formation, after the enamel layer has reached its final dimensions, but before it has fully mineralized and hardened (Smith, 2008). In this study, we report a WDR72 mutation in two families with autosomal-recessive hypomaturation AI and test the hypothesis that WDR72 functions inside the cell by expressing the protein fused to green-fluorescent protein (GFR) in HEK293T cells and localizing it by fluorescent microscopy.
Materials & Methods
Patients
Seven families with AI phenotypes resembling those associated with WDR72 mutations were selected from the AI families we have recruited for genetic studies. Three families showed an autosomal-dominant pattern of inheritance, two were recessive, and two could not be determined. Clinical and radiological examinations were performed, and blood samples were collected with the understanding and written consent of each participant according to the Declaration of Helsinki. This study protocol was independently reviewed and approved by the Institutional Review Boards at Seoul National University Dental Hospital, the University of Istanbul, and the University of Michigan.
Primer Design, Polymerase Chain-reaction(PCR), and DNA Sequencing
Genomic DNA was extracted from peripheral whole blood. WDR72 exons and adjacent intron boundaries were amplified as described previously (El-Sayed et al., 2009). Primer pairs for exon 1 (sense, 5′-CGGTACCCGTGACCCTAAG; antisense, 5′-GGCCCAGCTGTATCCAAG) were designed with Primer 3 on the Web (http://frodo.wi.mit.edu/primer3/). PCR amplifications were performed with the HiPi DNA polymerase premix (Elpis Biotech, Taejeon, Korea) and purified with a PCR Purification Kit (Elpis Biotech). DNA sequencing was performed at the DNA sequencing center (Macrogen, Seoul, Korea). Sequences were analyzed with Mutation Surveyor (Softgenetics, Ann Arbor, MI, USA).
Expression Vector Construction
WDR72 coding sequence was amplified with synthetic oligonucleotide primers (sense, 5′-ggctcgagAAATGAGGACTTCCCT GCAGG; antisense, 5′-ggtctagaCACCTTGCAGGGGCAGAC CT) with a human cDNA clone (BC101614.1; Thermo Scientific Open Biosystems, Huntsville, AL, USA) as template. The amplification product was purified and subcloned into the pEGFP-C1 vector (Clontech, Mountainview, CA, USA) after both the plasmid and amplification product were double-digested with XhoI and XbaI restriction endonucleases. The engineering strategy placed the WDR72 translation initiation codon in-frame with an N-terminal green fluorescent protein (GFP) domain. The entire construct was characterized by DNA sequencing.
Analysis of Intracellular Localization
The pEGFP-C1-based plasmids that express green fluorescent protein (GFP) alone and WDR72 fused to GFP were purified with the AccuPrep Nano-Plus Plasmid Midi Extraction Kit (Bioneer, Taejeon, Korea). HEK293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS (GIBCO-BRL, Carlsbad, CA, USA) and Antibiotic-Antimycotic liquid (GIBCO-BRL) at 37°C under 5% CO2. For transfection, cells were grown on 6-well plates and transfected with Lipofectamine2000 (Invitrogen, Carlsbad, CA, USA). After 24 hrs, transfected cells were trypsinized and seeded onto poly-L-lysine-coated cover glass in 6-well plates. The next day, the cells were fixed with 4% paraformaldehyde, and nuclear DNA was stained in 20 µM bisBenzimide H 33342 trihydrochloride (Sigma, St. Louis, MO, USA) for 10 min. The samples were washed in PBS and mounted with Fluorescent Mounting Medium (DAKO, Glostrup, Denmark). Confocal laser scanning was performed with an OLYMPUS-FV300 fluorescence microscope.
Results
Mutation Results
Mutational analysis of probands from families 1 and 2 revealed the same dinucleotide deletional mutation in exon 12 in both alleles of WDR72 (Appendix Fig. 1). The standardized description of this mutation according to published nomenclature recommendations (den Dunnen and Antonarakis, 2003) for the cDNA sequence is c.1467_1468delAT, based upon the NCBI reference sequence NM_182758.2, where the A of the ATG translation initiation codon is nucleotide 1. The genomic description is g.57,426_57,427delAT and is based upon the NCBI reference sequence NC_000015.9, with the first nucleotide of the reference sequence (not the translation initiation codon) being nucleotide 1. The dinucleotide deletion leads to a predictable change in the translation product, which is described as p.V491fsX497. The normal WDR72 amino acid sequence in this region (GDLDSC VILWDIF…) is changed to GDLDSCDLVGYLY*. Instead of a WDR72 translation product having 1102 amino acids, the protein is correctly translated through Cys490, 7 random amino acids are added, and the mutant protein terminates after Tyr497. Despite certainty in predicting the effect of the mutation on translation, no evidence indicates that the mutant mRNA is translated at all, rather than being degraded by nonsense-mediated decay. The mutation segregated perfectly with the AI phenotype in both families, indicating that it is disease-causing. A summary of the WDR72 gene structure is provided in Appendix Fig. 2. If translated, the p.V491fsX497 mutation would abbreviate the protein in the sixth of seven WD40 repeat domains on the N-terminal side of WDR72 (Appendix Fig. 3).
Clinical Findings
Family 1 is a Hispanic family from Mexico. Nine family members spanning three generations were recruited and contributed DNA. Haplotype analysis revealed that the two grandmothers (I:2 and I:4) are related and carriers of the same disease allele. All three of the grandchildren are homozygous for the c.1467_1468delAT mutation and display the AI phenotype. The parents of the proband have normal dentitions (Appendix Fig. 4); the pattern of inheritance is autosomal-recessive.
The permanent dentition of the proband showed orange-brown staining throughout (Fig. 1). Enamel roughness was localized and more likely to be observed in the occlusal or incisal half of the tooth and is apparently due to attrition following eruption. Indeed, progressive deterioration of the dentition was documented photographically in the proband’s younger sister (III:2; Fig. 2). It is evident that the true phenotype of the WDR72 mutation is best observed during tooth eruption, because of the occurrence of rapid and extensive post-eruptive changes. The enamel layer has reduced radiodensity compared with normal enamel, an indication that the enamel layer is hypomineralized.
Figure 1.
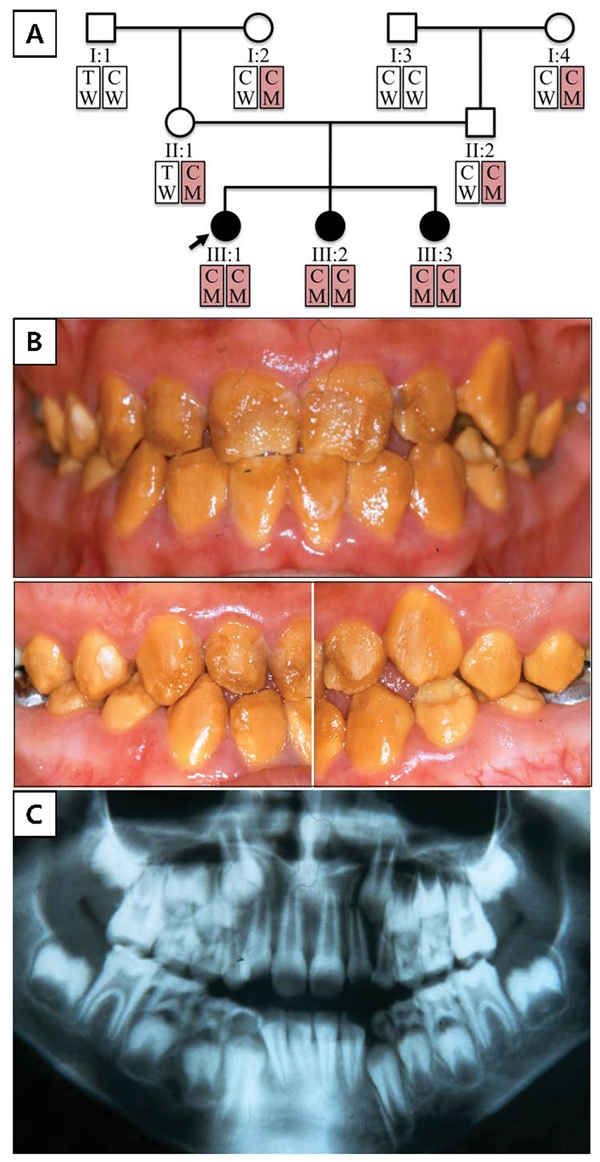
Family 1 pedigree and haplotypes; proband’s (III:1) oral photos and radiograph. (A) Pedigree of family 1. The haplotype of each individual is shown below the symbol. Red boxes indicate alleles harboring the mutation (key: T, thymine; C, cytosine; W, wild-type; M, mutant). (B) Frontal and lateral photographs of the proband (III:1) taken at age 10 yrs, 7 mos. (C) Dental panoramic radiograph of the proband (III:1) taken at age 6 yrs, 6 mos.
Figure 2.
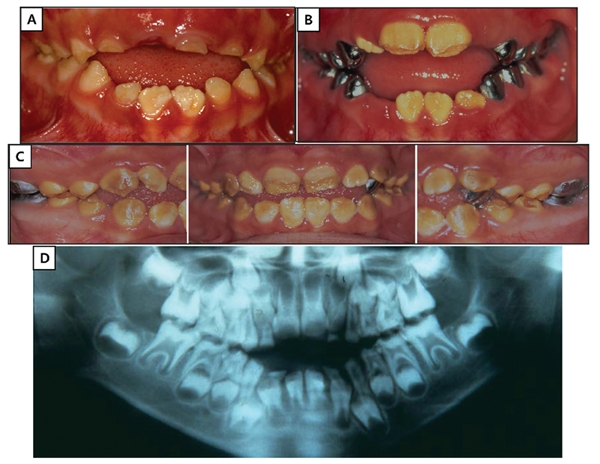
Family 1 affected sister (III:2). (A) Oral photo taken at age 5 yrs, 6 mos. (B) Oral photo taken at age 6 yrs, 6 mos. (C) Oral photos taken at age 8 yrs, 7 mos. (D) Panoramic radiograph taken at age 5 yrs, 6 mos. Note the progressive deterioration of the enamel crowns, especially in the occlusal and incisal regions following eruption into function.
Family 2 is from Turkey. Six family members spanning two generations were recruited and contributed DNA. The pattern of inheritance is autosomal-recessive; only the two sons who are homozygous for the c.1467_1468delAT mutation display the AI phenotype (Fig. 3A). The enamel phenotype appears to be more severe in family 1 compared with family 2 (Fig. 3B), although the genetic etiology is the same. This suggests that habits and diet might influence the pattern of degeneration (attrition and staining) following tooth eruption; it also suggests the possibility of a genetic modifier. Radiologically, the phenotype is similar between the two families, with the enamel layer showing reduced radiodensity (Fig. 3C).
Figure 3.
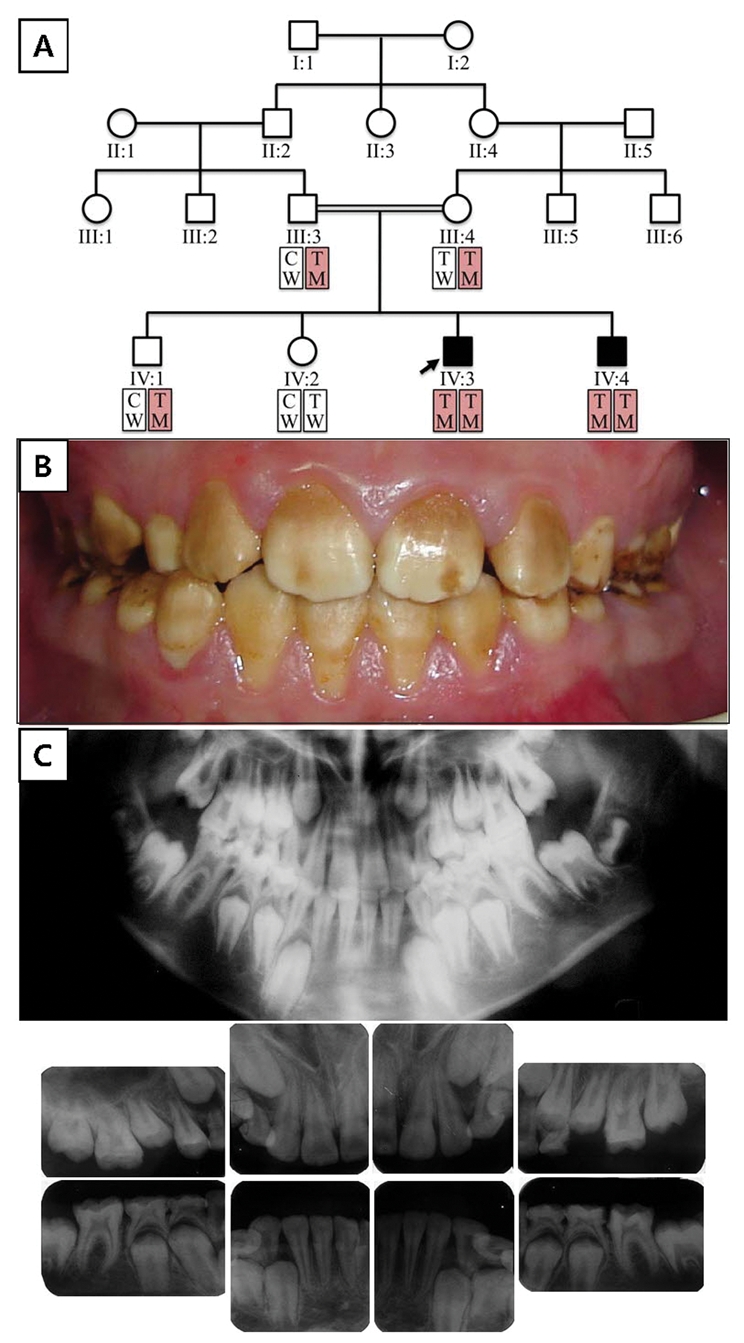
Family 2 pedigree and haplotypes; proband’s (IV:3) oral photo and radiographs. (A) Pedigree of family 2. Haplotype of each individual is shown below the symbol. Red boxes indicate alleles harboring the mutation (key: T, thymine; C, cytosine; W, wild-type; M, mutant). (B) Frontal photograph of the proband (IV:3) at age 11 yrs. (C) Dental panoramic and intra-oral radiographs of the proband (IV:3) at age 11 yrs.
An SNP in exon 12 of WDR72 (c.1407T>C, p.T469T, rs6416452) immediately adjacent to the AI-causing mutation site is a C in the diseased allele of family 1 and a T in the diseased allele of family 2. This finding demonstrates that the identical disease-causing mutation in the two families is not identical because of common ancestry (Figs. 1A, 3A), but arose independently in the two families. This confirms what is obvious from the various ethnic backgrounds and raises the possibility that this is a mutational hotspot in WDR72.
Analysis of Intracellular Localization
The wild-type WDR72 fused to GFP localized exclusively in the cytoplasm of the transfected HEK293T cells, in contrast to the widespread localization of pEGFP-C1 control plasmid (Fig. 4).
Figure 4.
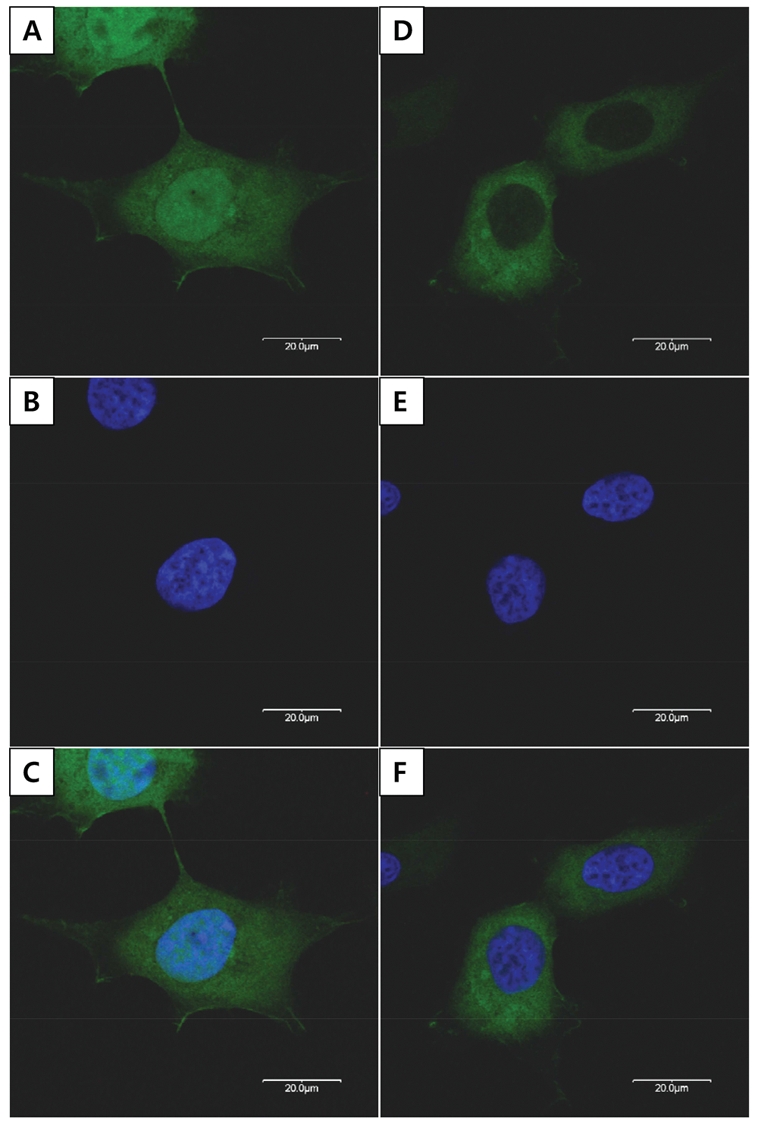
Analysis of intracellular localization. HEK293T cells transfected with pEGFP-C1 vectors expressing green fluorescent protein (GFP) alone and WDR72 fused to GFP (green). (A) Widespread GFP expression. (B) Nuclear Hoechst 33342 staining. (C) Overlapping image of (A) and (B). (D) Wild-type WDR72 localized in the cytoplasm. (E) Nuclear Hoechst 33342 staining. (F) Overlapping image of (D) and (E).
Discussion
The WD40 or WD-repeat domain family (pfam00400) includes 609 human genes (Speicher et al., 2010). The naming of human genes based upon the presence of WD-repeat domains extends up to WDR94. The WD40 designation reflects the approximate length (40 amino acids) of the conserved repeat domain and the high frequency of a tryptophan-aspartate (WD) dipeptide. WD40 domains are always found in a cluster of 4 to 16 repeats (Li and Roberts, 2001). The sequence repeat contains no absolutely conserved amino acids, but the basic pattern can be recognized by computer algorithms, such as REPv1.1 (Andrade et al., 2000). Analysis of WDR72 by this software identified 7 WD repeats, all on the N-terminal side of the protein. WDR7, the closest homologue of WDR72, showed the same 7 repeats, plus 2 additional repeats near its C-terminus (which is a part of the protein that is not homologous to WDR72). WD-repeats form highly symmetrical folds that combine into a circular ß propeller structure that has no catalytic activity, but serves as a platform upon which protein complexes reversibly assemble (Smith, 2008). This structural element is comprised of the N-terminal 600 amino acids of WDR72, and folds into a domain that is presumed to engage in protein-protein complexes with unknown protein partners. WDR72 does not contain a signal peptide, and the WD40 domains start only 15 amino acids from the N-terminus. Taken together with the results of our GFP fusion study, it is unlikely that these associations occur in the nucleus or extracellular space.
A key aspect of WD-repeat proteins is that the ß propeller structure that mediates protein-protein interactions is combined with a variable domain that conveys novel functions to individual members of the family. Among the 106 human proteins predicted to have 7 WD-repeats, 104 contain at least one significant additional non-WD domain (Smith, 2008). WDR7 shares strong homology with WDR72 up to Gly850 (Gly845 in WDR72), and then the two sequences diverge. WDR7 (from Thr863 to Leu974) has a conserved homology domain that resembles a PKC-like catalytic subunit. WDR72 has no recognizable homology domains in its C-terminal region.
Why mutations in both WDR72 alleles cause isolated enamel defects is puzzling. The expressed sequence tag profile for WDR72 indicates that WDR72 is expressed in multiple tissues, with the healthy tissues showing the highest number of transcripts per million being the bladder (165), kidney (145), and mouth (74). Genes that are specific for enamel formation, such as amelogenin, enamelin, and ameloblastin, tend to degenerate when tooth development is discarded during evolution (Deméré et al., 2008; Sire et al., 2008; Meredith et al., 2009). Even when both WDR72 alleles are defective, the only phenotype observed is in enamel, suggesting that WDR72 expression is only critical for dental enamel formation. WDR72, however, is well-conserved in birds, although it is shorter (927 vs. 1102 amino acids) than in mammals, due to a deletion of ~ 175 amino acids from its C-terminus. The selective pressures that maintain WDR72 in chickens are unclear. Perhaps WDR72 is important for other processes besides enamel formation, and the reported WDR72 mutations cause a subtle non-dental phenotype that has so far escaped detection. It seems that, among WD40 family members in non-dental tissues, there is functional redundancy which is not compensated for in ameloblasts.
Family 1 in this study was one of the original 24 families with AI whom we recruited for genetic studies (Kim et al., 2006). We have now identified two AMELX (Kim et al., 2004), two ENAM (Kim et al., 2005a, 2006), one MMP20 (Kim et al., 2005b), five FAM83H (Lee et al., 2008; Ding et al., 2009), and one WDR72 mutation in this group, bringing to 12 the number of families having the genetic etiology of their AI determined. None of the 24 families had a KLK4 mutation. The number of genes that cause amelogenesis imperfecta is not known, but the 6 proven candidate genes cause AI in half of the first 24 kindreds we characterized.
Supplementary Material
Acknowledgments
We thank all the family members for their cooperation.
Footnotes
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MEST) (M10646010003-08N4601-00310), by a Science Research Center grant to Bone Metabolism Research Center (2009-0063266) funded by the Korean Ministry of Education, Science and Technology, and by USPHS Research Grant DE015846 (NIDCR/NIH).
The author(s) declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Andrade MA, Ponting CP, Gibson TJ, Bork P. (2000). Homology-based method for identification of protein repeats using statistical significance estimates. J Mol Biol 298:521-537 [DOI] [PubMed] [Google Scholar]
- Backman B, Holm AK. (1986). Amelogenesis imperfecta: prevalence and incidence in a northern Swedish county. Community Dent Oral Epidemiol 14:43-47 [DOI] [PubMed] [Google Scholar]
- Chosack A, Eidelman E, Wisotski I, Cohen T. (1979). Amelogenesis imperfecta among Israeli Jews and the description of a new type of local hypoplastic autosomal recessive amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol 47:148-156 [DOI] [PubMed] [Google Scholar]
- Deméré TA, McGowen MR, Berta A, Gatesy J. (2008). Morphological and molecular evidence for a stepwise evolutionary transition from teeth to baleen in mysticete whales. Syst Biol 57:15-37 [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. (2003). Mutation nomenclature. Curr Protoc Hum Genet Chapter 7:Unit 7.13 [DOI] [PubMed] [Google Scholar]
- Ding Y, Estrella MR, Hu YY, Chan HL, Zhang HD, Kim JW, et al. (2009). Fam83h is associated with intracellular vesicles and ADHCAI. J Dent Res 88:991-996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummer PM, Kingdon A, Kingdon R. (1990). Prevalence and distribution by tooth type and surface of developmental defects of dental enamel in a group of 15- to 16-year-old children in South Wales. Community Dent Health 7:369-377 [PubMed] [Google Scholar]
- El-Sayed W, Parry DA, Shore RC, Ahmed M, Jafri H, Rashid Y, et al. (2009). Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am J Hum Genet 85:699-705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed W, Shore RC, Parry DA, Inglehearn CF, Mighell AJ. (2010). Ultrastructural analyses of deciduous teeth affected by hypocalcified amelogenesis imperfecta from a family with a novel Y458X FAM83H nonsense mutation. Cells Tissues Organs 191:235-239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons D, Hong S, et al. (2004). Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet 41:545-549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hu YY, Lin BP, Boyd C, Wright JT, et al. (2004). Amelogenin p.M1T and p.W4S mutations underlying hypoplastic X-linked amelogenesis imperfecta. J Dent Res 83:378-383 [DOI] [PubMed] [Google Scholar]
- Kim JW, Seymen F, Lin BP, Kiziltan B, Gencay K, Simmer JP, et al. (2005a). ENAM mutations in autosomal-dominant amelogenesis imperfecta. J Dent Res 84:278-282 [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hart TC, Hart PS, Ramaswami MD, Bartlett JD, et al. (2005b). MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet 42:271-275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Lin BP, Seymen F, Bartlett JD, Hu JC. (2006). Mutational analysis of candidate genes in 24 amelogenesis imperfecta families. Eur J Oral Sci 114 (Suppl 1):3-12 [DOI] [PubMed] [Google Scholar]
- Kim JW, Lee SK, Lee ZH, Park JC, Lee KE, Lee MH, et al. (2008). FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am J Hum Genet 82:489-494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerström M, Dahl N, Nakahori Y, Nakagome Y, Backman B, Landegren U, et al. (1991). A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1). Genomics 10:971-975 [DOI] [PubMed] [Google Scholar]
- Lee MJ, Lee SK, Lee KE, Kang HY, Jung HS, Kim JW. (2009). Expression patterns of the Fam83h gene during murine tooth development. Arch Oral Biol 54:846-850 [DOI] [PubMed] [Google Scholar]
- Lee SK, Hu JC, Bartlett JD, Lee KE, Lin BP, Simmer JP, et al. (2008). Mutational spectrum of FAM83H: the C-terminal portion is required for tooth enamel calcification. Hum Mutat 29:E95-E99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Roberts R. (2001). WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci 58:2085-2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza G, Pemberton TJ, Lee K, Scarel-Caminaga R, Mehrian-Shai R, Gonzalez-Quevedo C, et al. (2007). A new locus for autosomal dominant amelogenesis imperfecta on chromosome 8q24.3. Hum Genet 120:653-662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith RW, Gatesy J, Murphy WJ, Ryder OA, Springer MS. (2009). Molecular decay of the tooth gene Enamelin (ENAM) mirrors the loss of enamel in the fossil record of placental mammals. PLoS Genet 5:e1000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ. (2001). Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum Mol Genet 10:1673-1677 [DOI] [PubMed] [Google Scholar]
- Sire JY, Delgado SC, Girondot M. (2008). Hen’s teeth with enamel cap: from dream to impossibility. BMC Evol Biol 8:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TF. (2008). Diversity of WD-repeat proteins. Subcell Biochem 48:20-30 [DOI] [PubMed] [Google Scholar]
- Speicher M, Antonarakis SE, Motulsky AG, editors (2010). Vogel and Motulsky’s human genetics: problems and approaches. 4th ed. New York: Springer [Google Scholar]
- Witkop CJ., Jr (1971). Heterogeneity in inherited dental traits, gingival fibromatosis and amelogenesis imperfecta. South Med J 64(Suppl 1):16-25 [DOI] [PubMed] [Google Scholar]
- Witkop CJ, Jr, Sauk JJ., Jr (1976). Heritable defects of enamel. In: Oral facial genetics. Stewart RE, Prescott GH, editors. St. Louis, MO: C.V. Mosby Co, pp. 151-226 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.