

Abstract
Mutations in the transcription factors PAX9 and MSX1 cause selective tooth agenesis in humans. In tooth bud mesenchyme of mice, both proteins are required for the expression of Bmp4, which is the key signaling factor for progression to the next step of tooth development. We have previously shown that Pax9 can transactivate a 2.4-kb Bmp4 promoter construct, and that most tooth-agenesis-causing PAX9 mutations impair DNA binding and Bmp4 promoter activation. We also found that Msx1 by itself represses transcription from this proximal Bmp4 promoter, and that, in combination with Pax9, it acts as a potentiator of Pax9-induced Bmp4 transactivation. This synergism of Msx1 with Pax9 is significant, because it is currently the only documented mechanism for Msx1-mediated activation of Bmp4. In this study, we investigated whether the 5 known tooth-agenesis-causing MSX1 missense mutations disrupt this Pax9-potentiation effect, or if they lead to deficiencies in protein stability, protein-protein interactions, nuclear translocation, and DNA-binding. We found that none of the studied molecular mechanisms yielded a satisfactory explanation for the pathogenic effects of the Msx1 mutations, calling for an entirely different approach to the investigation of this step of odontogenesis on the molecular level.
Keywords: tooth agenesis, Msx1, Pax9, Bmp4, missense mutation
Introduction
To date, several mutations in the homeodomain transcription factor MSX1 and the paired domain transcription factor PAX9 have been identified in tooth agenesis patients (Vastardis et al., 1996; Stockton et al., 2000; Lidral and Reising, 2002; Das et al., 2003; Lammi et al., 2003; Mostowska et al., 2003, 2006; Jumlongras et al., 2004; Chishti et al., 2006; Kapadia et al., 2006; Xuan et al., 2008; Wang et al., 2009a). These findings had been inspired and supported by animal studies showing that Msx1 and Pax9 are co-expressed in dental mesenchyme during the early stages of tooth development, and that homozygous deletion of Msx1 or Pax9 results in an arrest of tooth development at the bud stage (Chen et al., 1996; Peters et al., 1998; Bei et al., 2000). Mesenchymal expression of bone morphogenetic protein 4 (Bmp4), a key signaling factor for tooth development, is down-regulated in the Msx1 or Pax9 knockout animal model. Furthermore, single heterozygous Pax9+/− or Msx1+/− mutant mice have a normal dentition, whereas the compound Pax9+/−Msx1+/− double-heterozygous mice lack lower incisors, and with incomplete penetrance, third molars (Chen et al., 1996; Nakatomi et al., 2010). This phenotype can be partially rescued by transgenic expression of BMP4 in dental mesenchyme, indicating a genetic interaction between Pax9 and Msx1 and confirming Bmp4 as a downstream target of these two transcription factors (Nakatomi et al., 2010).
Our previous in vitro studies supported this hypothesis: Using a proximal 2.4-kb Bmp4 promoter fragment, we have shown that Pax9 but not Msx1 can directly activate the expression of Bmp4. Msx1, however, can physically interact with Pax9 and potentiate Pax9’s activation of Bmp4 expression (Ogawa et al., 2006; Wang et al., 2009a), thus providing a tentative explanation for the activation of Bmp4 by Msx1. We have recently tested our in vitro molecular model using tooth-agenesis-causing PAX9 paired domain mutations (Wang et al., 2009a). In vitro testing showed that most of the Pax9 mutants had lost or reduced DNA binding ability and decreased transcriptional activities with Bmp4 promoter constructs. The extent of these functional defects correlated well with the severity of the dental phenotype seen in the families carrying the mutations. One of the Pax9 mutations had lost synergism with Msx1.
The aim of the present study was to investigate if MSX1 mutations result in tooth agenesis because the mutant proteins lose their ability to cooperate with PAX9, or if inadequacies in other functional properties of the mutant Msx1 proteins can be discovered by in vitro testing. Here, we analyzed all 5 tooth-agenesis-causing missense mutations in the MSX1 gene with our in vitro system to test our hypothesis about the molecular mechanism of Bmp4 activation in tooth bud mesenchyme. We conclude that the original hypothesis is untenable with regard to Msx1’s molecular mechanism of action.
Materials & Methods
Construction of Expression Plasmids and Site-directed Mutagenesis
The mammalian expression vector pCMV-Pax9 with a c-Myc epitope tag and pCMV-Msx1 with a FLAG epitope tag were used as previously described (Ogawa et al., 2006). In vitro site-directed mutagenesis was performed with the QuikChange mutagenesis kit (Stratagene, La Jolla, CA, USA) to construct pCMV-Msx1 with the M61K, A194V, R196P, A219T, and A221E mutations. Similar vectors were constructed with Msx2 and Dlx1, 2, and 5 coding sequences.
We used the pEYFP-C1 vector (Clontech Laboratories, Palo Alto, CA, USA) to construct YFP-Msx1 fusion proteins to determine subcellular localization. All constructs were sequenced entirely to confirm the correct reading frame and point mutation.
Protein Half-life Assay
We over-expressed FLAG-Msx1 wild-type or mutant proteins in COS7 cells. After overnight culture, cells were treated with 90 mM cycloheximide (CHX) for 1 to 6 hrs to inhibit cellular protein synthesis. The abundance of Msx1 proteins was subsequently examined by Western blot with samples taken at 0, 1, 2, 4, and 6 hrs after the start of CHX treatment. Actin was used as quantification control.
Western Blot Analysis and Co-immunoprecipitation
These were performed as described previously (Wang et al., 2009a). Primary antibodies included anti-FLAG M2 (Stratagene), anti-actin (Abcam, Cambridge, MA, USA), anti-Pax9 M-18 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-FLAG M2 affinity gel (Sigma-Aldrich, St. Louis, MO, USA). Visualization was achieved with mouse or goat horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) and enhanced chemiluminescence (GE Healthcare, Piscataway, NJ, USA). All blots were done in triplicate.
Gel Shift and Super Shift Assay
The oligonucleotide probe used for gel shift assays was the 26mer tgaccctatgtaattgcattcctgaa, which contains the previously established homo domain-binding site 12B (Catron et al., 1993). Electrophoretic mobility shift assays (EMSA) were performed with the LightShift Chemiluminescent EMSA Kit (Pierce Biotechnology, Rockford, IL, USA), according to the manufacturer’s instructions and in triplicate.
Subcellular Localization
To demonstrate in vivo expression of wild-type and mutant Msx1, we transfected COS7 cells (in triplicate) with wild-type Msx1 or mutant Msx1 in the pEYFP-C1 vector and examined them 24 hrs later with a Nikon Eclipse TE2000-U fluorescence microscope. Vectors containing only the Msx1 homeodomain or Msx1 sequences without homeodomain were used as controls.
Reporter Assay
Msx1 expression vectors were co-transfected with a Bmp4 promoter-reporter or a Msx1 promoter-reporter construct [p2.4Bmp4-Luc and p3.5Msx1-Luc, described previously (Ogawa et al., 2006)] into COS7 cells in a 12-well format. The pCMV-SPORT β-Gal plasmid (Invitrogen, Carlsbad, CA, USA) was used as an internal control. For each well, 500 ng of pCMV-Pax9, 250 ng of pCMV-Msx1, 150 ng of promoter-reporter construct, and 100 ng of pCMV-SPORT β-Gal were co-transfected. Transfection was performed in duplicate and repeated 3 times. Twenty-four hours after transfection, cell extracts were assayed by the luciferase assay system (Promega, Madison, WI, USA) and a β-gal assay kit (Invitrogen).
Results
Review of Phenotypes and Mutations
All 5 Msx1 missense mutations studied cause quite severe phenotypes, with great intra- and inter-familial variation (Table), in particular, the A219T substitution, which is inherited in an autosomal-recessive mode in two consanguineous families. The A194V mutation presented with incomplete penetrance, suggesting that other gene variants may be involved. Premolars are affected most often, followed by molars and incisors. The average number of missing teeth is comparable with the observations made in patients with PAX9 mutations.
Table.
Synopsis of the Characteristic Phenotypical and Functional Features of Wild-type Msx1 and Tooth-agenesis-causing Msx1 Mutants
| Wild-type | M61K | A194V | R196P | A219T | A221E | |
|---|---|---|---|---|---|---|
| Inheritance | ADa | AD/I.P.a | AD | ARa | AD | |
| Tooth phenotypeb | 12 (8) | 14 (10) | 11 (7) | 18 (15.5) | 12 (8) | |
| Localization | Nuclear | Nuclear | Nuclear | Nucl clump | Nuclear | Nuclear |
| Co-IP | + | + | + | + | + | + |
| Half-life | +++ | ++ | +++ | + | + | +++ |
| Synerg, Bmp4c | Less | Slightly up | Like WT | Less | More | |
| Synerg, Msx1c | Like WT | Slightly up | Slightly up | Repressed | Slightly up | |
| DNA-binding | More | Like WT | None | None | Like WT | |
| Reference | Lidral and Reising, 2002 | Mostowska et al., 2006 | Vastardis et al., 1996 | Chishti et al., 2006 | Xuan et al., 2008 |
AD, autosomal-dominant; AR, autosomal-recessive; I.P., incomplete penetrance.
Tooth phenotype: showed average number of missing teeth, and the number in parentheses excludes third molars.
“Synerg, Bmp4” represents synergism with Pax9 on the Bmp4 promoter, and “Synerg, Msx1” represents synergism with Pax9 on the Msx1 promoter.
Sequence Alignment and Structure-based Predictions
Alignment of the Msx1, Msx2, and Msx3 amino acid sequences of human and mouse, as well as Msx1 sequences from diverse species, revealed that the 5 affected residues are conserved. Based on the crystal structure of the Msx1 homeodomain complex with DNA (Hovde et al., 2001), we compared the mutant and wild-type residues in the 3D model (Fig. 1). A194V and R196P are located in helix 2, A219T and A221E in helix 3, and M61K is positioned N-terminal of the homeodomain in a conserved stretch of amino acids known as one of the two groucho/TLE-binding domains in Msx1. The R196P mutation has the most severe effect; it will diminish DNA binding and destabilize the α-helix at the same time. The A219T and A221E mutations introduce a hydrophilic amino acid in place of the hydrophobic alanine; the A194V substitution has only a minor effect on structural stability.
Figure 1.
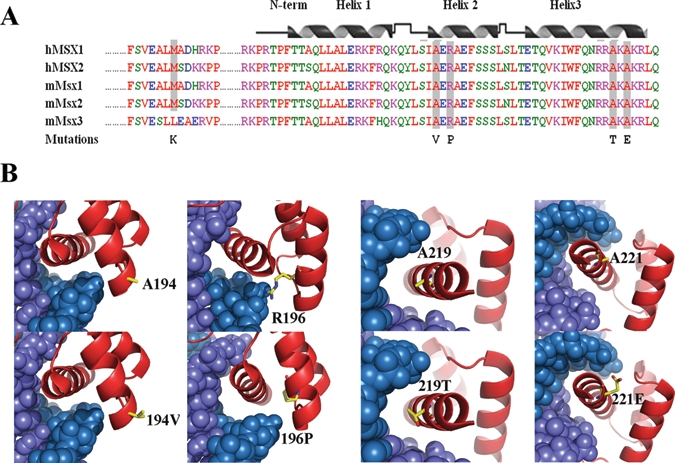
Sequence alignments and 3D structure prediction. (A) Protein sequence alignment of human MSX1/2 and mouse Msx1/2/3. The positions of mutated amino acids are indicated with grey shading. The secondary structure of the Msx1 homeodomain is shown above the sequence. Two of the mutations are located within helix 2 and the other two in helix 3. (B) Protein-DNA interaction models of the Msx1 homeodomain with and without the missense mutations. The homeodomain protein chain is red, and the DNA double helix is shown in blue. The wild-type and mutant amino acids stand out as yellow stick models.
Effects of Mutations on the Half-life of Msx1 Proteins
The half-life of Msx1 was decreased by all mutations except A194V (Fig. 2A). The R196P and A219T mutants had the shortest half-life, dropping more than three-fold. The protein half-life of the M61K and A221E was reduced to less than half compared with that of the wild-type protein.
Figure 2.
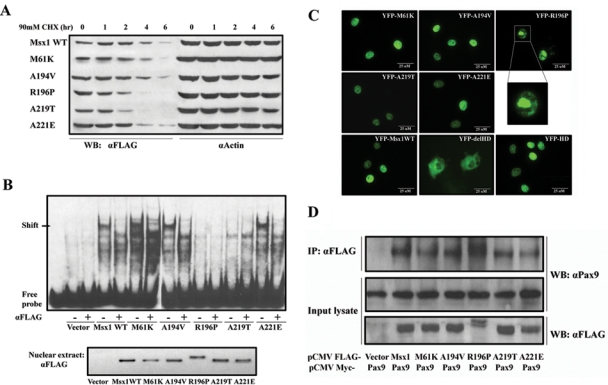
Characterizations of Msx1 protein. (A) Msx1 protein half-life: Cos7 cells were transfected with Flag-Msx1 expression vectors and treated with 90 mM cycloheximide (CHX) for 1 to 6 hrs. Western blot of cell lysates with anti-FLAG was used to assess the protein degradation speed; actin served as a control. (B) DNA-binding abilities: Electrophoretic mobility shift assay (EMSA) was used to test DNA-binding activity of nuclear extracts from Flag-Msx1-transfected COS7 cells with a double-stranded oligonucleotide probe containing a consensus binding-site for Msx1. Specificity of the complexes was confirmed by super-shift induction with anti-FLAG antibody. To verify that equal amounts of Msx1 protein were used for the EMSA, we performed a Western blot of nuclear extracts (lower panel). (Note that R196P migrates more slowly than the other proteins.) (C) Nuclear localization: YFP-Msx1 fusion proteins were expressed in COS7 cells. None of the missense mutations affected nuclear localization, in contrast to the YFP-Msx1 construct without homeodomain, which is found in the cytoplasm (YFP-delHD). The homeodomain by itself suffices for translocation to the nucleus (YFP-HD). YFP-R196P showed densely clumped distribution within the nuclei. These protein aggregates could be the consequence of protein interactions or of a secondary modification which also causes the R196P mutant to migrate more slowly than the other mutants and wild type Msx1. (D) Protein interaction: Co-immunoprecipitation with COS7 cells co-transfected with Myc-tagged Pax9 proteins and FLAG-tagged Msx1. Upper panel: Western blot analyses of immunoprecipitates of whole-cell lysates pulled down with anti-FLAG antibody and probed with an anti-Pax9 antibody. Middle panel: Whole-cell lysates probed with anti-Pax9 antibody or (lower panel) anti-Flag antibody. (Note that the Msx1-R196P migrates more slowly in both immunoprecipitations and input cell lysate.)
DNA Binding to an Msx1 Consensus Site
Gel-shift and supershift assays show that the DNA-binding ability of the Msx1 protein is abolished by the mutations R196P and A219T (Fig. 2B). Interestingly, R196P affects the same amino acid that is mutated to histidine in the MSX2 homeodomain (R172H) in a family diagnosed with parietal foramina (Wilkie et al., 2000) and has been reported to cause 85% reduction in DNA-binding. The loss of DNA-binding of the R196P mutation can be easily deduced from the structural prediction (Fig. 1B). The M61K, A194V, and A221E mutant proteins show the same patterns of shifted bands as the wild-type sequence. This finding is plausible, since M61K is located outside of the DNA-binding domain, A194V does not interact with the DNA helix, and A221E appears to interact only marginally with DNA (Fig. 1B).
Nuclear Localization of Msx1 Mutants
Msx1 protein without the homeodomain is mainly localized in the cytoplasm, while the Msx1 homeodomain by itself is located in the nucleus (Fig. 2C). These findings indicate that nuclear localization is mediated by the homeodomain of Msx1. All 5 mutant Msx1 proteins are found primarily in the nuclear compartment of transfected cells.
Physical Interactions between Pax9 and Msx Proteins
Co-transfection of pCMV-Myc Pax9 and pCMV-FLAG Msx1, followed by co-immunoprecipitation, showed that all of the mutant proteins are able to interact physically with wild-type Pax9 (Fig. 2D). The homeodomain of the Msx2 protein differs from Msx1’s homeodomain by only 2 conservative amino acid replacements, but Msx2 as well as the Dlx proteins did not co-immunoprecipitate with Pax9 (see Appendix Fig.). Co-IP does not coincide with transcriptional potentiation of Pax9 (see below).
Transcriptional Activation in the Presence of Pax9
Wild-type Msx1 represses transcription of Bmp4 and Msx1 promoter constructs in vitro, but in the presence of Pax9, it synergistically up-regulates transcription from both promoters (Ogawa et al., 2006; Wang et al., 2009a). Evaluating the 5 mutants, we found that M61K and A219T exhibited reduced synergistic cooperation with Pax9 on the Bmp4 promoter (Fig. 3A); on the Msx1 promoter construct, A219T even repressed Pax9’s activity on transcription (Fig. 3B). However, A194V, R196P, and A221E could synergistically interact with Pax9 on both promoters even more effectively than wild-type Msx1.
Figure 3.
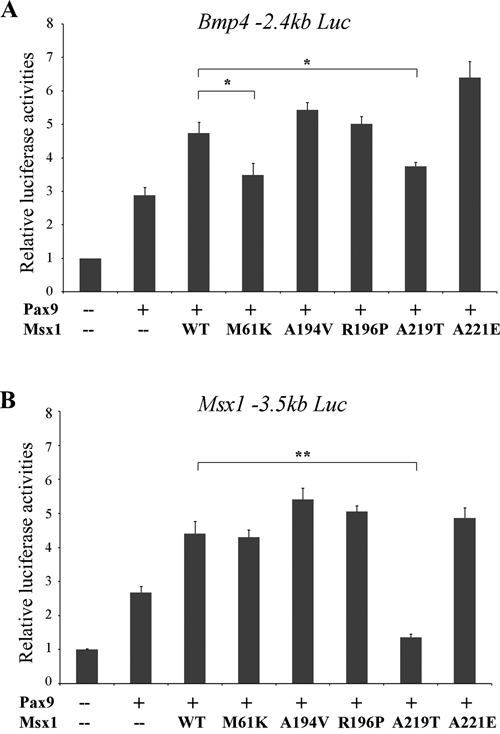
Luciferase reporter assays of synergistic promoter up-regulation. (A) Bmp4 promoter-reporter constructs (p2.4 Bmp4-luciferase) or (B) Msx1 promoter-reporter constructs (p3.5 Msx1-luciferase) were co-transfected with the different Msx1 expression vectors and the wild-type Pax9 expression vector at a 1:2 ratio. (Error bars represent standard error; *p < 0.05, **p < 0.01.)
We also tested Msx2 and the Dlx1 and 2 proteins in this assay. While Msx2 behaved like Msx1, the Dlx proteins either had no effect or inhibited Pax9’s transactivation ability (see Appendix Fig.).
Discussion
It has been known for more than a decade that Msx1 and Pax9 are required for Bmp4 expression in tooth bud mesenchyme, but the molecular basis for this inter-relationship is largely unidentified. Potentiation of Pax9 is currently the only known molecular mechanism of Bmp4-activation by Msx1 in tooth bud mesenchyme, since Msx1 by itself cannot activate the proximal Bmp4 promoter, and other potential mediators are presently unknown. Homeodomain transcription factors usually acquire their selective functional properties through interactions with a variety of other co-factors, including paired domain transcription factors; thus, it could have been possible that Pax9 was one of these co-factors.
Here we analyzed naturally arising, human hypodontia-causing missense mutations in MSX1 to examine their interaction with Pax9 and Bmp4 on a molecular level. Most of the mutant Msx1 proteins could still potentiate Pax9-induced Bmp4 and Msx1 promoter activation, suggesting that Msx1’s ability to cause human tooth agenesis may be independent of any synergism with Pax9. The severity of the families’ phenotypes cannot be reasonably explained by the relatively mild variations observed in our functional tests, suggesting again that a different molecular mechanism must be responsible for Msx1-caused tooth agenesis. Even the A194V mutation which showed incomplete penetrance and behaved like wild-type protein in all our tests cannot yet be excluded as a disease-causing mutation.
In conclusion, we infer from the functional evaluation of the 5 MSX1 missense mutations that disturbances in the interaction with Pax9 are probably not the basis of the human phenotypes caused by MSX1 mutations, unless another molecular mechanism for the observed dependence of Bmp4 expression on combined Msx1 and Pax9 activity is discovered. A recent study by Chandler et al. (2009) described the existence of an enhancer element far upstream of the Bmp4 promoter that drives Bmp4 expression in tooth bud mesenchyme. This enhancer could be located and tested for responsiveness to wild-type Msx1 and the Msx1 mutants, with and without Pax9 as co-activator.
Alternatively, an altogether different pathway could be involved. Early on, it had been shown that Msx1-deficient mice displayed reduced expression of not only Bmp4 but also of Lef1, syndecan-1, Runx2, and Fgf3 (Chen et al., 1996; Bei and Maas, 1998). In the dental mesenchyme, Fgf3 is another well-established signaling molecule besides Bmp4 that is known to promote the tooth morphogenetic process. Fgf3 appears to be expressed downstream of Wnt/Lef1 and of Runx2 (D’Souza et al., 1999). The Msx1 pathway resulting in Bmp4 expression seems to operate in parallel with the pathway that leads to Msx1-dependent Fgf3 expression in tooth bud mesenchyme. In each of these two pathways, Msx1 is likely to cooperate with different partner factors targeting different promoters. The fact that one of the tooth agenesis MSX1 mutations (M61K) is located in a Gro/Tle binding site points to a disturbance in transcriptional repression mechanisms.
Surprisingly, Msx1 may even be completely dispensable for odontogenesis: Activation of the Wnt pathway through Apc depletion (Wang et al., 2009a), but not β-catenin stabilization (Liu et al., 2008), could bypass Msx1-dependent signaling pathways for the formation of supernumerary tooth-like structures with enamel knots, normal mineralization, and signs of root growth. However, these teeth were rather small and dysmorphic, suggesting that the mesenchymal Msx1-dependent pathways play a significant role in determining proper size and shape of the normal endogenous tooth organ, possibly by delaying premature differentiation.
Translating experimental evidence from mice to humans is still a challenge, especially when the organ system in question shows several remarkable differences: The mouse dentition is monophyodont and consists of only molars and continually growing incisors, while tooth agenesis resulting from Pax9 and Msx1 mutations affects mostly the secondary dentition in humans. Also, a study with human embryonic tissue revealed a few differences in gene expression patterns—for example, PAX9 is not exclusively expressed in tooth bud mesenchyme but is also found in the dental epithelium (Lin et al., 2007). Ultimately, we will have to find valid in vitro assays that may help to interpret mouse data on a human background.
Supplementary Material
Acknowledgments
We thank Dr. Hitesh Kapadia for his advice on this study. This work is supported by National Institutes of Health/National Institute of Dental and Craniofacial Research (NIH/NIDCR) R01 award DE019471-01 to RDS.
Footnotes
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
References
- Bei M, Maas R. (1998). FGFs and BMP4 induce both Msx1-independent and Msx1-dependent signaling pathways in early tooth development. Development 125:4325-4333 [DOI] [PubMed] [Google Scholar]
- Bei M, Kratochwil K, Maas RL. (2000). BMP4 rescues a non-cell-autonomous function of Msx1 in tooth development. Development 127:4711-4718 [DOI] [PubMed] [Google Scholar]
- Catron KM, Iler N, Abate C. (1993). Nucleotides flanking a conserved TAAT core dictate the DNA binding specificity of three murine homeodomain proteins. Mol Cell Biol 13:2354-2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler KJ, Chandler RL, Mortlock DP. (2009). Identification of an ancient Bmp4 mesoderm enhancer located 46 kb from the promoter. Dev Biol 327:590-602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bei M, Woo I, Satokata I, Maas R. (1996). Msx1 controls inductive signaling in mammalian tooth morphogenesis. Development 122:3035-3044 [DOI] [PubMed] [Google Scholar]
- Chishti MS, Muhammad D, Haider M, Ahmad W. (2006). A novel missense mutation in MSX1 underlies autosomal recessive oligodontia with associated dental anomalies in Pakistani families. J Hum Genet 51:872-878 [DOI] [PubMed] [Google Scholar]
- D’Souza RN, Åberg T, Gaikwad J, Cavender A, Owen M, Karsenty G, et al. (1999). Cbfa1 is required for epithelial-mesenchymal interactions regulating tooth development in mice. Development 126:2911-2920 [DOI] [PubMed] [Google Scholar]
- Das P, Hai M, Elcock C, Leal SM, Brown DT, Brook AH, et al. (2003). Novel missense mutations and a 288-bp exonic insertion in PAX9 in families with autosomal dominant hypodontia. Am J Med Genet A 118:35-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovde S, Abate-Shen C, Geiger JH. (2001). Crystal structure of the Msx-1 homeodomain/DNA complex. Biochemistry 40:12013-12021 [DOI] [PubMed] [Google Scholar]
- Jumlongras D, Lin JY, Chapra A, Seidman CE, Seidman JG, Maas RL, et al. (2004). A novel missense mutation in the paired domain of PAX9 causes non-syndromic oligodontia. Hum Genet 114:242-249 [DOI] [PubMed] [Google Scholar]
- Kapadia H, Frazier-Bowers S, Ogawa T, D’Souza RN. (2006). Molecular characterization of a novel PAX9 missense mutation causing posterior tooth agenesis. Eur J Hum Genet 14:403-409 [DOI] [PubMed] [Google Scholar]
- Lammi L, Halonen K, Pirinen S, Thesleff I, Arte S, Nieminen P. (2003). A missense mutation in PAX9 in a family with distinct phenotype of oligodontia. Eur J Hum Genet 11:866-871 [DOI] [PubMed] [Google Scholar]
- Lidral AC, Reising BC. (2002). The role of MSX1 in human tooth agenesis. J Dent Res 81:274-278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Huang Y, He F, Gu S, Zhang G, Chen Y, et al. (2007). Expression survey of genes critical for tooth development in the human embryonic tooth germ. Dev Dyn 236:1307-1312 [DOI] [PubMed] [Google Scholar]
- Liu F, Chu EY, Watt B, Zhang Y, Gallant NM, Andl T, et al. (2008). Wnt/beta-catenin signaling directs multiple stages of tooth morphogenesis. Dev Biol 313:210-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostowska A, Kobielak A, Biedziak B, Trzeciak WH. (2003). Novel mutation in the paired box sequence of PAX9 gene in a sporadic form of oligodontia. Eur J Oral Sci 111:272-276 [DOI] [PubMed] [Google Scholar]
- Mostowska A, Biedziak B, Trzeciak WH. (2006). A novel c.581C>T transition localized in a highly conserved homeobox sequence of MSX1: is it responsible for oligodontia? J Appl Genet 47:159-164 [DOI] [PubMed] [Google Scholar]
- Nakatomi M, Wang XP, Key D, Lund JJ, Turbe-Doan A, Kist R, et al. (2010). Genetic interactions between Pax9 and Msx1 regulate lip development and several stages of tooth morphogenesis. Dev Biol 340:438-449 [DOI] [PubMed] [Google Scholar]
- Ogawa T, Kapadia H, Feng JQ, Raghow R, Peters H, D’Souza RN. (2006). Functional consequences of interactions between Pax9 and Msx1 genes in normal and abnormal tooth development. J Biol Chem 281:18363-18369 [DOI] [PubMed] [Google Scholar]
- Peters H, Neubüser A, Kratochwil K, Balling R. (1998). Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev 12:2735-2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockton DW, Das P, Goldenberg M, D’Souza RN, Patel PI. (2000). Mutation of PAX9 is associated with oligodontia. Nat Genet 24:18-19 [DOI] [PubMed] [Google Scholar]
- Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE. (1996). A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet 13:417-421 [DOI] [PubMed] [Google Scholar]
- Wang XP, O’Connell DJ, Lund JJ, Saadi I, Kuraguchi M, Turbe-Doan A, et al. (2009). Apc inhibition of Wnt signaling regulates supernumerary tooth formation during embryogenesis and throughout adulthood. Development 136:1939-1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Groppe JC, Wu J, Ogawa T, Mues G, D’Souza RN, et al. (2009a). Pathogenic mechanisms of tooth agenesis linked to paired domain mutations in human PAX9. Hum Mol Genet 18:2863-2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu H, Wu J, Zhao H, Zhang X, Mues G, et al. (2009b). Identification and functional analysis of two novel PAX9 mutations. Cells Tissues Organs 189:80-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie AO, Tang Z, Elanko N, Walsh S, Twigg SR, Hurst JA, et al. (2000). Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nat Genet 24:387-390 [DOI] [PubMed] [Google Scholar]
- Xuan K, Jin F, Liu YL, Yuan LT, Wen LY, Yang FS, et al. (2008). Identification of a novel missense mutation of MSX1 gene in Chinese family with autosomal-dominant oligodontia. Arch Oral Biol 53:773-779 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.