

Abstract
BACKGROUND AND PURPOSE
P2X3 and P2X2/3 receptors are highly localized on the peripheral and central pathways of nociceptive signal transmission. The discovery of A-317491 allowed their validation as chronic inflammatory and neuropathic pain targets, but this molecule has a very limited oral bioavailability and CNS penetration. Recently, potent P2X3 and P2X2/3 blockers with a diaminopyrimidine core group and better bioavailability were synthesized and represent a new opportunity for the validation of P2X3-containing receptors as targets for pain. Here we present a characterization of three representative diaminopyrimidines.
EXPERIMENTAL APPROACH
The activity of compounds was evaluated in intracellular calcium flux and electrophysiological recordings from P2X receptors expressed in mammalian cells and in a in vivo model of inflammatory pain (complete Freund's adjuvant (CFA) in rat paws).
KEY RESULTS
Compound A potently blocked P2X3 (pIC50 = 7.39) and P2X2/3 (pIC50 = 6.68) and showed no detectable activity at P2X1, P2X2, P2X4 and P2X7 receptors (pIC50 < 4.7). Whole-cell voltage clamp electrophysiology confirmed these results. Compounds showed good selectivities when tested against a panel of different classes of target. In the CFA model, compound B showed significant anti-nociceptive effects (57% reversal at 3 mg·kg−1).
CONCLUSIONS AND IMPLICATIONS
The diaminopyrimidines were potent and selective P2X3 and P2X2/3 receptor antagonists, showing efficacy in vivo and represent useful tools to validate these receptors as targets for inflammatory and neuropathic pain and provide promising progress in the identification of therapeutic tools for the treatment of pain-related disorders.
Keywords: purinergic, P2X, antagonist, pain, patch clamp, FLIPR
Introduction
The ATP neurotransmitter system is recognized as one of the most important pathways of the transmission of nociceptive signals (Burnstock, 1996; Burnstock and Wood, 1996; Cook and McCleskey, 2002). ATP is the endogenous agonist for the P2X ligand–gated ion channel family (receptor and channel nomenclature follows Alexander et al., 2009). Peripheral injections of P2X receptor agonists can enhance nociception in animal models of chemical (formalin) and inflammatory (carrageenan) pain (Sawynok and Reid, 1997; Hamilton et al., 2001). Previous studies with non-selective antagonists have confirmed a significant role for P2X receptors across a range of animal models of pathological nociception. Studies using suramin, PPADS (pyridoxal-5-phosphate-6-azophenyl-2′,4′-disulphonic acid) and TNP-ATP (2′,3′-O-(2,4,6- trinitrophenyl) ATP) have demonstrated P2X-related anti-nociception in animal models of neuropathic, inflammatory and chemogenic pain (Driessen et al., 1994; Tsuda et al., 1999; Fukuhara et al., 2000; Stanfa et al., 2000; Tsuda et al., 2000; Zheng and Chen, 2000; Jarvis et al., 2001).
The cloning and characterization of the P2X3 receptor subunit, which is selectively localized on peripheral and central processes of sensory afferent neurons (Chen et al., 1995; Lewis et al., 1995; Vulchanova et al., 1997), has generated much interest in the role of this receptor in nociceptive signaling (Burnstock and Williams, 2000). The P2X3 subunit is expressed both as a functional homomer and as a heteromultimeric combination with the P2X2 subunit, forming the P2X2/3 receptor subtype (Lewis et al., 1995; Lynch et al., 1999). Moreover, P2X3 and P2X2/3 receptors are expressed on the isolectin B4 (IB4) binding subpopulations of small nociceptive neurons. The decreased sensitivity to noxious stimuli, associated with the loss of IB4 binding neurons expressing P2X3 receptors indicates that these sensory neurons are essential for the signaling of acute pain (Vulchanova et al., 2001).
Despite mounting evidence for the importance of these receptors, progress in this research field was hampered by the lack of potent small molecule antagonists. The discovery of A-317491 (Jarvis et al., 2002) marked a significant breakthrough, being a truly selective, non-nucleotide small molecule that blocks both P2X3 and P2X2/3 receptors. The use of this compound allowed the validation of these targets in multiple in vivo chronic inflammatory and neuropathic pain models. Despite its impressive activity in vivo, A-317491 presented major limitations as a potential new drug due to limited oral bioavailability and CNS penetration (Carter et al., 2009).
More recent data on a diaminopyrimidine series paved the way for further progress in the validation of P2X3 and P2X2/3 receptors as pain targets and in the identification of new drugs for the treatment of neuropathic and inflammatory pain, which represents a current unmet need in the clinic (Carter et al., 2009; Jahangir et al., 2009). In particular, AF-353 (previously known as RO-4) is a diaminopyrimidine that has recently been shown to bear a favourable pharmacokinetic profile and excellent antagonist potency and selectivity for P2X3 and P2X2/3 receptors (Gever et al., 2010). These characteristics suggest that AF-353 could be an excellent in vivo tool compound to enable studies of the role of these channels in animal models and, ultimately, increase confidence in a novel class of therapeutics for the treatment of pain-related disorders. The same compound, by acting on both the peripheral and central terminals of nociceptors, contributes to analgesic efficacy in a model of bone cancer pain behaviour in rats (Kaan et al., 2010).
In the present study, a fluorescence-based calcium imaging assay has been configured to characterize three compounds belonging to the diaminopyrimidine series (Figure 1), against P2X3 and P2X2/3 receptors. Potency and selectivity against other P2X receptor subtypes were assessed by means of the same assay platform and confirmed by whole-cell patch clamp electrophysiology experiments in recombinant, stably transfected cells. When tested in a panel of assays, the compounds were selective over the majority of a wide range of G-protein-coupled receptors (GPCR), enzymes, transporters, kinases and ion channels targets. Drug metabolism and pharmacokinetic (DMPK) studies identified one compound as more suitable for subsequent in vivo studies (complete Freund's adjuvant (CFA) model of inflammatory pain in rats) where it demonstrated efficacy.
Figure 1.
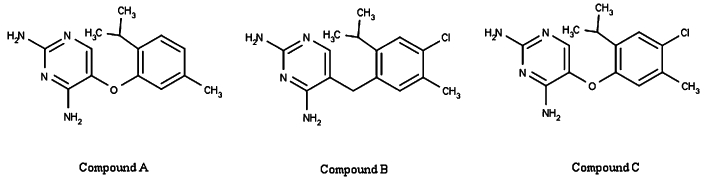
Chemical structures of the three diaminopyrimidines examined in the present study.
In summary, compounds A, B and C are potent and selective P2X3 and P2X2/3 receptor antagonists with compound B showing efficacy in an in vivo pain animal model. The results reported add to the growing repertoire of evidence supporting the important role for P2X3 and P2X2/3 receptors in the pain therapeutic area.
Materials and methods
In vitro studies
Cells
A stable Chinese Hamster Ovary (CHO) cell line co-expressing human P2X2 and P2X3 receptors was generated following electroporation of CHO-K1 cells (ATCC, Manassas, VA, USA, Cat. No. CCL-61) with expression plasmids pCIH-P2X2 and pCIN5-P2X3 (Rhodes et al., 1998), selection in 100 µg·mL−1 Hygromycin-B (Invitrogen, Cat. no. 10687-010) and 400 µg·mL−1 Geneticin G418 (Invitrogen, Carlsbad, CA, USA, Cat. no. 10131-027) and single cell dilution cloning. Electroporations were carried out as described in Chen et al. (2000). Stable 1321N1 cell lines independently expressing P2X2 and P2X3 were generated following electroporation of 1321N1 cells (ECACC, Salisbury, Wiltshire, UK, Cat. no. 86030402) with each expression vector and clonal lines selected as for the CHO-K1 cell line. In vitro fluorometric imaging plate reader (FLIPR) experiments on the human P2X7 receptor were carried out on U2OS cells (ATCC, Cat. No. HTB-96) transduced with a P2X7 expressing BacMam virus as previously described (Michel et al., 2009). A stable HEK cell line expressing the human P2X1 receptor was generated as described in Evans et al. (1996). A stable HEK293 cell line expressing the human P2X4 receptor was generated as previously described (Jones et al., 2000) Electrophysiological recordings on the human P2X7 receptorwere performed on a stable HEK cell line generated as previously described in Rassendren et al. (1997). The CHO cell line stably co-expressing the rat P2X2 and P2X3 receptors was generated as described in Kawashima et al. (1997).
Cell lines stably expressing human P2X1, P2X2, P2X3 or P2X7 receptors were grown in Dulbecco's modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum and non essential amino-acids (NEAA) and were maintained at 37°C in a humidified atmosphere containing 5% CO2. Human and rat P2X2/3 CHO cells were grown in Iscove's modified Dulbecco's medium supplemented with heat-inactivated fetal bovine serum, NEAA and hypoxanthine and thymidine (HT) supplement and were maintained at 37°C in a humidified atmosphere containing 5% CO2. Trypsin–EDTA or Versene™ (Invitrogen, Cat. no.:15040-066) were used to detach cells during cell passages. For electrophysiological recordings, cells were plated onto poly-d-lysine–coated glass coverslips (BD Biosciences, Franklin Lakes NJ) from 2 to 4 days prior to use and kept at 37°C. For measurements of intracellular Ca2+ levels, cells were grown to 70–80% confluency in a 384-well black plate with clear bottom [Falcon (BD Biosciences), Greiner (Greiner Bio-one, Frickenhausen, Germany) or Costar (Corning Costar, Lowell, MA, USA)]. For experiments on human P2X1 and P2X7 receptors, poly-d-lysine pre-treated plates were used.
In vitro FLIPR experiments on the 5HT3 receptor were carried out using a CHO stable cell line expressing the human 5HT3A receptor. This stable cell line was generated using a pCIN5-5HT3A expression vector in a similar manner to that described for the CHO-P2X cell lines. Experiments on the α3β4 and α1 nicotinic acetylcholine receptor (nAChR) were carried out using stable cell lines co-expressing the human nAChR α3 and β4 subunits and the human nAChR α1, β1, δ and ε subunits obtained from Merck-Millipore (Billerica, MA, USA, Cat. nos.: CYL3057 and CYL3052 respectively). Experiments on the α7 nAChR were carried out on a GH4C1 cell line stably expressing the human α7 subunit (generated as previously described for rat α7 in Virginio et al., 2002).
Intracellular calcium flux assays
P2X receptor function was determined on the basis of agonist-induced increase of cytosolic Ca2+ concentration measured using the Ca2+ chelating dye Fluo-4-acetoxymethyl ester (Fluo-4 AM; Molecular Probes, Invitrogen) in conjunction with the FLIPR instrument (Molecular Devices, Sunnyvale, CA, USA). Briefly, cells were loaded with 1–2 µM Fluo-4 AM in assay buffer for 45–60 min. Extracellular Fluo-4 AM was removed before the assay run by washing the cells three times with 100 µL of assay buffer and leaving 30 µL·per well at the end of this operation. All compound solutions were prepared in assay buffer. Fluorescence data were collected for a total time of 15 min with sample intervals of 1–6 s. Antagonists were added 5–10 min before the agonist solution. The assay buffer composition for human and rat P2X2/3 and human P2X2 receptors was (in mM) as follows: NaCl 145, KCl 5, CaCl2 2, MgCl2 1, HEPES 10, glucose 10, probenecid 2.5–3, pH 7.3 with NaOH. For P2X4 receptors, the assay buffer composition was Tyrode's solution supplemented with thapsigargin to avoid any P2Y receptor-mediated response to ATP. For the P2X7 channel, the concentration of divalent cations in the assay buffer was reduced to 1 mM CaCl2 and 0.5 mM of MgCl2, as for electrophysiological recordings. For human P2X3 receptors, NaCl was substituted by N-methyl-D-glucamine in the assay buffer. For all assays the percentage of block was calculated as [(C − control A) / (control B − control A)] × 100, where control A is the 0% of block corresponding to the average of fluorescence counts obtained after the addition of the agonist at a concentration close to the EC80[α,β-methylene-ATP (αβ-Me-ATP) for P2X1,P2X3 or P2X2/3 receptors, ATP for P2X2 receptors and 2′ and 3′-O-(4-benzoylbenzoyl)-ATP (Bz-ATP) for P2X7 receptors]; control B is 100% of block and is the average of fluorescence counts obtained after the addition of agonist plus an overmaximal concentration of antagonist, and C are the fluorescent counts measured in the presence of compound at different concentrations.
Electrophysiological recordings
Experiments were carried out at room temperature. Whole cell–perforated patch clamp recordings and, for P2X7 receptors, conventional whole-cell recordings (Hamill et al., 1981 and Shermann-Gold, 1993), were performed using the EPC9 patch clamp system and the Pulse program (HEKA elektronik, Dr. Schulze, GmbH, Lambrecht/Pfalz, Germany). Patch pipettes were pulled from thin-wall borosilicate glass capillary (1.5 mm outer diameter) using a Sutter Instrument P-97 pipette puller and had resistances of 2–5 MΩ. Cells were placed in a recording chamber and continuously superfused with the following solution (in mM): 135 NaCl, 2 KCl, 2 CaCl2, 1 MgCl2, 12 HEPES, D-glucose 10, pH 7.35 (with NaOH). For the different P2X receptor subtypes, the currents were elicited by application of different agonist as described for FLIPR experiments. For perforated whole-cell recordings, amphotericin B (240 mg·mL−1) was added to the intracellular solution that contained (in mM) 150 CsCl, 2 EGTA, 10 HEPES, pH 7.3 (with CsOH). Fast perfusion systems were used for local application of solutions (U-tube delivery system; Fenwick et al., 1982; or RSC-160 delivery system from Biologic Science Instruments, Claix, France). Antagonist pre-incubation of at least 2 min preceded the agonist and antagonist solution co-application. Cells were stimulated with 0.5–2 s pulses of agonist as indicated by horizontal bars above each trace in the Figures and delivered at intervals ≥2 min. The holding potential was −80 mV throughout the experiment. For P2X7 receptors, the concentration of divalent cations in the extracellular solution was reduced to 1 mM CaCl2 and 0.5 mM MgCl2 in order to reduce the pore formation during the recordings (Virginio et al., 1997). The intracellular solution was (in mM) as follows: 150 NaCl; 10 BAPTA; 10 HEPES; pH 7.3. Peak or steady-state current amplitudes were measured online. Percentage of block was calculated as [1 − (I / Ictrl)] × 100, where I is the current amplitude obtained in the presence of antagonist, and Ictrl is the amplitude of the control response.
Data analysis
For patch experiments, percentage of block values obtained for each antagonist concentration were averaged and plotted against the corresponding concentration. Data points were fitted using the four parameters logistic equation (Prism v.4, GraphPad Software, San Diego, CA) of the form:
where X is the logarithm of concentration, Y is the percentage of block relative to the control response and starts at 0 and goes to 100 with a sigmoid shape. Electrophysiology data stated in the text are best fit value ± standard error from the fitting for pIC50 values and mean ± SEM for percentage of block values.
For FLIPR experiments, data were analysed using ActivityBase software (IDBS, ID Business Solutions Ltd., Guilford, Surrey, UK) based on the four parameters logistic equation, and data stated in the text are mean ± SEM from individual test occasions.
Materials
All culture media and supplements were purchased from Invitrogen. All chemical reagents were purchased from SIGMA (Sigma-Aldrich, St. Louis, MO, USA). Compounds A, B and C were synthesized in GlaxoSmithKline laboratories (Harlow, Essex, UK).
In vivo studies
Animals
All animal care and experimental procedures were performed in accordance with the Animals (Scientific Procedures) Act 1986 and under the authority of a Home Office Project Licence. The inflammatory pain study was performed using adult male Random Hooded rats weighing 180–220 g (B & K Universal Ltd, Grimston, Aldbrough, Hull, UK). Rats were housed in groups of five to six and given ad libitum access to both water and RMI chow pelleted diet (SDS, Witham, UK). The holding and procedure rooms were maintained under conditions of constant temperature (21 ± 1°C), humidity (40–50%) and lighting (lights on 0700–1900). Rats were allowed a period of at least 5 days of adaptation to their environment before experiments commenced.
Inflammatory pain model protocol
Acute inflammatory pain was induced by injecting 100 µL of 1 mg·mL−1 CFA (Sigma, F-5881) sub-plantar into the left hind paw of rats. Hind limb weight bearing differences, as detailed below, were measured before CFA treatment (‘pre-CFA’), 23 h following CFA treatment (‘post-CFA’) to confirm the presence of an inflammatory response, and at a time point following drug administration (‘post-drug’). After the ‘post-CFA’ readings, rats were ranked and randomised into groups, such that the mean difference between baseline and ‘post CFA’ readings were approximately equal between groups, with n = 7 per dose group. The study was conducted with the operator unaware of the treatment each rat received. P2X2/3 receptor antagonist, compound B (3, 10, 30 mg·kg−1) or vehicle (1% DMSO, 99% PEG400) was injected subcutaneously (s.c.) 23.5 h after administration of CFA. Animals were re-assessed for weight bearing at 1 h after drug administration. For the assessment of weight bearing capacity, the distribution of weight between the two hind paws was measured using an incapacitance tester (Dual channel weight averager; Linton Instrumentation, Palgrave, Diss, Norfolk, UK). Normal rats distribute their body weight equally between the two hind paws, but when a hind paw is inflamed and/or painful, the weight is re-distributed so that less weight is put on the affected paw. Assessment of this change using the incapacitance tester is a sensitive method for measuring the development of incident pain. The two hind paws were placed on separate sensors and the drug induced reversal of CFA-induced weight bearing deficit is calculated over a period of 4 s (Clayton et al., 1997). Data are expressed as weight bearing deficit between hind paws in grams and the percentage reversal of CFA-induced weight bearing deficit on the inflamed paw, as calculated below:
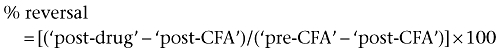 |
Data from this study were analysed using Statistica v5.1 software package (Statsoft, Inc., Tulsa, OK, USA). Data was analysed by anova followed by post hoc Fisher's LSD test comparing ‘post-drug’ responses with ‘post-CFA’ responses (for weight bearing deficit data) and comparing with vehicle responses (% reversal data). Statistically significant differences are expressed as P-values less than 0.05.
Pharmacokinetics and bioanalysis
Following weight-bearing measurements 1 h ‘post-CFA’, blood and brain samples were collected from all study animals for pharmacokinetic analysis. Blood samples were taken from the tail vein (approximately 80 µL) in K3EDTA coated tubes (Sarstedt Ltd., Leics, UK), from which an accurate 50 µL aliquot was transferred to Micronic tubes (Micronic BV, Lelystad, The Netherland) along with a 50 µL aliquot of deionized water. Rats were killed by means of CO2 inhalation; the brain were excised and then rinsed in ice-cold saline. Diluted blood and brains were frozen at −80°C awaiting analysis. On the day of analysis, blood and brain samples were thawed, and the brains were weighed and then homogenized with a Tomtec Autogiser (Receptor Technologies, Addenbury, Oxon, UK) in an equivalent weight of denionized water (ELGA maxima), and a 100 µL of homogenate was transferred to fresh 1.8 mL Micronic tubes. Standard curves and quality control samples were prepared in diluted blood (1:1) and brain homogenate (1:1) to cover the concentration range 0.018 to 18 µM. Quality controls were present in triplicate at 0.018, 0.036, 6.8 and 14.4 µM to assess the bias and precision of the analytical run. Blood and brain samples were extracted using 400 µL of acetonitrile containing a structural analogue as an internal standard (c. 0.8 µM), vortex mixed for 10 min and then centrifuged for 20 min at 4°C (Eppendorf 5810R, VWR, Poole, Dorset, UK). Sample analysis was performed by means of liquid chromatography-tandem mass spectrometry (LC-MS/MS) using an Agilent HP1100 integrated binary LC pump (Agilent Technologies, Stockport, Chesire, UK) and an API4000 tandem quadrupole mass spectrometer (PESciex, Ontario, Canada) equipped with a TurboIonspray™ interface operate in positive ion mode at 650°C. A small aliquot of extracted supernatant (2 µL) was injected onto a Hypersil Elite C18 (2.1 × 50 mm, 5µ, Thermo, Runcorn, Cheshire, UK) column at 40°C via a CTC PAL autosampler (Presearch, Hitchin, UK). A linear gradient was employed to elute compound B and internal standard, using 1 mM ammonium acetate acidified with 0.1 v/v formic acid as solvent A and acetonitrile as solvent B, using the gradient profile of 95% to 5% solvent B over 1.5 min. The total run time was 2.5 min to allow column equilibration at the initial and final solvent compositions. Chromatograms were processed (integrated) using Analyst 1.4 software (PE Sciex, Ontario, Canada) with a 1/y2-weighted regression, and data acceptance were based on the pre-defined criteria that measured concentration of standards were within 20% of the nominal value, and that two out of three quality control samples were within 20% of nominal for each QC level.
The methodology for estimating brain tissue binding has been reported in detail previously (Summerfield et al., 2006). Compound B was added to rat brain homogenate at a concentration of 1 µg·mL−1 and dialysed against PBS (pH 7.4) for 6 h at 37°C in a HTDialysis 96-well equilibrium dialysis system (HT Dialysis LLC, Gales Ferry, CT). Aliquots of PBS and brain homogenate were subsequently extracted with acetonitrile containing a proprietary internal standard and analysis by means of LC-MS/MS. The unbound fraction was determined as the concentration in PBS to that in brain tissue at equilibrium. The resultant unbound fraction for compound B was 0.028 ± 0.001 µg·mL−1, indicating that approximately 3% of the total brain concentration was present in the extracellular fluid and available for receptor binding (Jeffrey and Summerfield, 2007).
Results
In vitro activities of diaminopyrimidine compounds
Structures of three representative compounds of the diaminopyrimidine series, here named compound A, compound B and compound C are illustrated in Figure 1.
Potency and selectivity assessed by FLIPR assays
The studies were conducted in more than one type of transfected cell line. We demonstrated that the expression systems used did not adversely influence the pharmacology of the P2X receptors expressed, through the characterisation of known pharmacological standards (data not shown). The diaminopyrimidine compounds were tested against a panel of ionotropic purinergic receptors through use of FLIPR intracellular Ca2+ imaging technology (Table 1). All three compounds proved to be potent blockers of human P2X3 receptors (IC50 values in the nanomolar range). Compound A and compound C were assayed against the human and the rat P2X2/3 receptors, and their potencies were in the submicromolar range against the human orthologue (pIC50 values of 6.68 ± 0.02, n = 191 for compound A and 6.37 ± 0.12, n = 9 for compound C) and in the micromolar range against the rat orthologue (pIC50 values of 6.04 ± 0.03, n = 29 for compound A and 5.36 ± 0.13, n = 4. for compound C). The potencies of compound A and compound C were five- and sevenfold higher when tested on the human P2X3 homomeric receptor. Compound B showed a pIC50 value for the human P2X3 receptor of 6.31 ± 0.09, n = 9. All three compounds were demonstrated to be highly selective for P2X2/3 and P2X3 as they did not show blocking activity against human P2X1, P2X2, P2X4 and P2X7 receptors at the concentrations tested (100 µM for P2X2 and P2X4 receptors and 20 µM for the others).
Table 1.
FLIPR assay results for compound A, compound B and compound C assayed against human and rat P2X2/3 and human P2X3, P2X1, P2X2, P2X4 and P2X7 receptors
| Receptor subtype (cell background) | Compound A | Compound B | Compound C |
|---|---|---|---|
| hP2X2/3 (CHO) | 6.68 ± 0.02 (191) | NT | 6.37 ± 0.12 (9) |
| rP2X2/3 (CHO) | 6.04 ± 0.03 (29) | NT | NT |
| hP2X3 (1321N1) | 7.39 ± 0.06 (9) | 6.31 ± 0.09 (9) | 7.24 ± 0.18 (9) |
| hP2X1 (HEK) | <4.7 (17) | <4.7 (6) | <4.7 (5) |
| hP2X2 (1321N1) | <4 (9) | <4 (9) | <4 (9) |
| hP2X4 (CHO) | <5 (4) | <5 (4) | <5 (4) |
| hP2X7 (U2OS) | <4.7 (36) | <4.7 (4) | <4.7 (4) |
Data are mean ± SEM of pIC50 with the number of measurements in parenthesis. NT, not tested.
Electrophysiological recordings
In order to confirm the compound activities obtained by FLIPR technology, patch clamp experiments were performed on human P2X2/3 and P2X3 receptors and on rat P2X2/3 receptors, in concentration–response curve or single concentration experiments, whereas for P2X1, P2X2 and P2X7 receptors, the compounds were tested at the concentration of 10 µM.
In cells co-expressing human or rat P2X2 and P2X3 subunits, αβ-Me-ATP induced currents showed two components: a peak current corresponding to the fast desensitising homomeric P2X3 receptor and a steady-state current corresponding to the heteromeric P2X2/3 receptor (Figures 2 and 6). αβ-Me-ATP, at the concentrations used (7.5–10 µM), was not able to activate the P2X2 receptor, as this agonist displays low potency for P2X2 (North and Suprenant, 2000). All three series representatives were confirmed to be potent P2X2/3 receptor antagonists. Compound A and compound B were able to block the peak (P2X3) current and the sustained (P2X2/3) current, the latter with pIC50 values of 7.22 ± 0.04 (Figure 3) and 6.34 ± 0.08 respectively (n from three to five different cells per antagonist concentration). Compound C was tested in a single concentration (300 nM) and significantly blocked the human P2X2/3 induced current by 66.3 ± 5.0%, n = 3. In Figure 3C and D, the current traces recorded from two cells are reported as representative examples showing a significant block of the agonist-evoked current in the presence of 1 µM and 300 nM of compounds B and C respectively. Upon compound wash-out, a partial recovery of the control current was observed.
Figure 2.
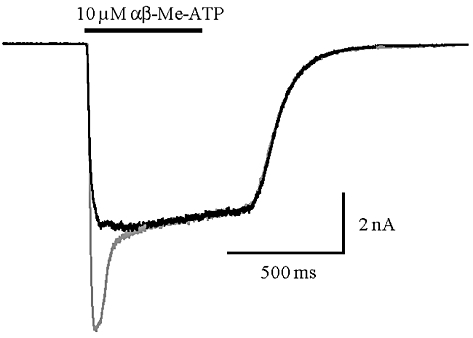
αβ-Me-ATP induced currents in a cell co-expressing human P2X3 and P2X2 receptor subunits. The peak and the steady-state components can be separated by a double application protocol where, in the first application, all receptor populations are activated (grey line) and in the second one, delivered 30 s later (black line), only the non-desensitising population (P2X2/3) are available for activation because the homomeric P2X3 receptor populaion have yet to recover from desensitisation.
Figure 6.
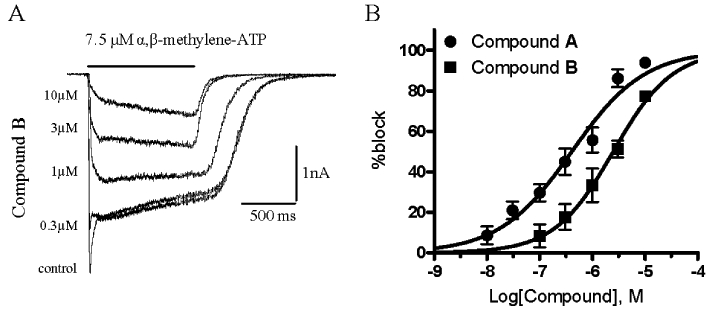
Compounds A and B are active at rat P2X2/3 receptor. Currents recorded from one representative cell, evoked by an αβ-Me-ATP concentration close to the EC80 value (7.5 µM) in the absence (control) and presence of increasing concentrations of compound A as indicated in the left side of the current traces (A). Percentage block of steady-state (P2X2/3) current responses evoked by the application of an EC80 concentration of αβ-Me-ATP (7.5 µM) were plotted against the logarithm of compound A or compound B concentration. Data points were fitted to the four parameter logistic equation. Each data point represents the mean ± SEM of at least three cells (B).
Figure 3.
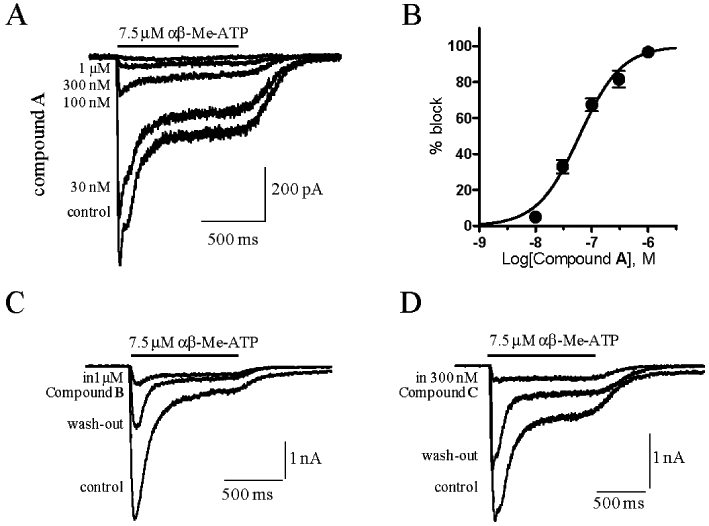
Compound A, B and C block of αβ-Me-ATP evoked current in cells expressing the human P2X2 and P2X3 receptor subunits. Representative examples of currents recorded from one cell, evoked by an αβ-Me-ATP concentration close to the EC80 value (7.5 µM) in the absence (control) and presence of increasing concentrations of compound A as indicated to the left of the current traces (A). Percentage block of steady-state (P2X2/3) current responses were plotted against the logarithm of compound A concentration. Data points were fitted to the four parameter logistic equation. Each data point represents the mean ± SEM of at least three cells (B). Representative examples of P2X2/3 or P2X3 currents recorded in the absence and presence of 1 µM Compound B (C) or 300 nM Compound C (D). The compounds were pre-incubated for at least 2 min before agonist co-application. After 5 to 15 min of wash-out, a partial recovery of control current was achieved.
In order to assess compound A activity against the human homomeric P2X3 receptor by direct measurement of the channel activity and to confirm the potency found in the FLIPR assay, electrophysiological recordings were performed using the 1321N1-human P2X3 cell line. These assays confirmed that this compound was a potent antagonist of the human homomeric P2X3 receptor with a pIC50 value of 6.94 ± 0.03 (n from two to four different cells per antagonist concentration; Figure 4B). Moreover, the effect of the compound was completely reversible after 15 min of wash-out as shown in Figure 4A.
Figure 4.
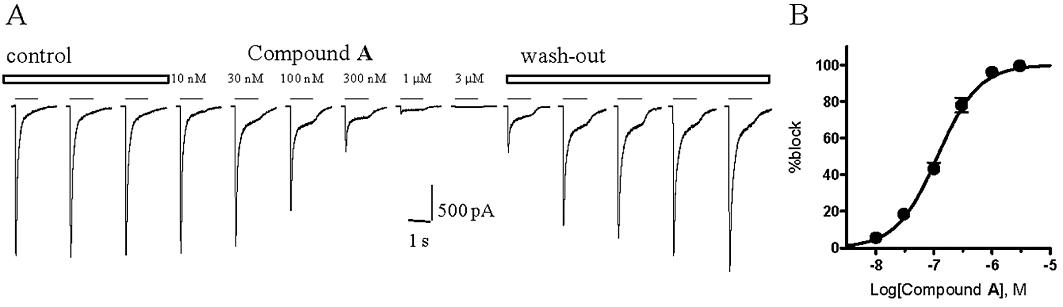
Compound A blocks αβ-Me-ATP evoked current in cells expressing the human P2X3 receptor. Representative currents recorded from one cell, evoked by an αβ-Me-ATP concentration close to the EC80 value (5 µM, applied for the time indicated by the horizontal bar above each current trace), in the absence (control), in the presence of increasing concentrations of compound A (as indicated above each current trace) and during the wash-out (A). Percentage block of peak current responses were plotted against the logarithm of compound A concentration. Data points were fitted to the four parameter logistic equation. Each data point represents the mean ± SEM from two to four cells (B).
The selectivity of the diaminopyrimidine compounds for P2X3 and P2X2/3 receptors was investigated by assessing their capacity to block human P2X1, P2X2 and P2X7 receptor subtypes. The 1321N1 cell line is characterized by the lack of any endogenous response to ATP, representing the ideal background to express the human P2X2 receptor to avoid any non-specific response to ATP (Bradley and Bradley, 2001). In 1321N1-hP2X2 cells, 30 µM ATP evoked a non-desensitizing current that was not significantly blocked by 10 µM compound A or by 10 µM compound C co-applied with the agonist after a compound pre-incubation of 2 min (percentage of block < 10%). Compound A was also assayed against human P2X1 and human P2X7 receptors demonstrating its inability to block agonist evoked currents at the test concentration of 10 µM (percentage of block < 10%; Figure 5C and D).
Figure 5.
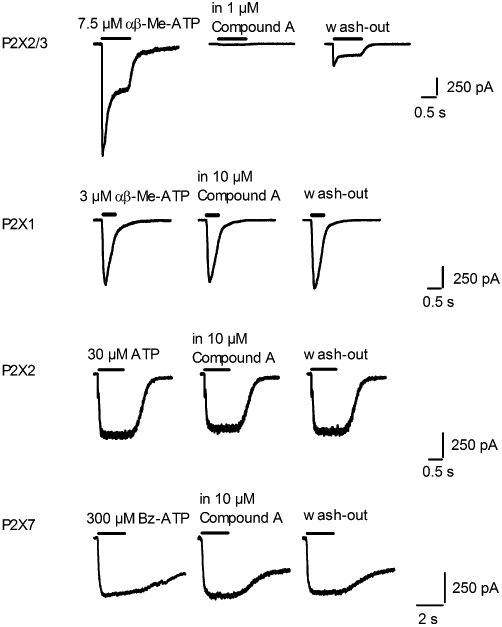
Compound A is a selective P2X2/3 antagonist. Representative currents recorded from cells expressing human P2X2/3, P2X1, P2X2 or P2X7 receptors, evoked by a concentration of agonist close to the EC80 value and applied for the time indicated by the horizontal bar above each current trace, in the absence and presence of the indicated concentration of compound A. Traces shown were recorded from the same cells after at least 3 min wash-out.
Before proceeding with in vivo studies, compound A and compound B were assayed on the rat P2X2/3 receptor in order to confirm their activity. Both compounds proved active against this receptor and demonstrating the same rank order of potency (pIC50 values of 6.39 ± 0.07, n from three to six different cells per antagonist concentration for compound A and 5.6 ± 0.07, n from three to four for compound B; Figure 6). However, compared with the human orthologue, approximately seven- and fivefold decreases in potency were observed for compounds A and B respectively.
Broad selectivity in vitro experiments
All three molecules were assessed against a panel of human target in vitro assays in order to assess their selectivity and to identify potential liabilities/ side effects. Compound A was active at the 5HT3 receptor, α1 and α3β4 nAChRs with pIC50 values ranging from 5.3 to 6.2 and at the α1A adrenoceptor with pKb = 6.4, and it showed no activity on a panel of 40 kinases (<50% at 25 µM). Compound B was active at the 5HT3 receptor, α1, α3β4 and α7 nAChRs with pIC50 values ranging from 5.2 to 6.4 and at the α1A adrenoceptor with pKb = 6.2. Compound C showed pIC50 = 6 at α1 nAChR, pKb values ranging from 5.7 to 6.8 at the α1A and α1B adrenoceptors and pIC50 = 5.6 at the organic anion-transporting polypeptide OATP1B1. Compounds B and C showed no activity in a panel of 25 kinases (<50% at 25 µM). All three compounds were not active (<50% inhibition at 10 µM) against the remaining targets (around 40 other GPCRs, enzymes, ion channels or transporters; data not shown).
In vivo studies of diaminopyrimidines
Pharmacokinetics of compounds A, B and C in rats
The intravenous pharmacokinetics of compounds A, B and C were assessed in a preliminary study in male Sprague-Dawley rats (1 mg·kg−1, n = 3). Despite the higher potencies of compounds A and C, both were eliminated extremely rapidly from plasma, with clearances of 213 ± 22 and 130 ± 17 mL·min−1·kg−1 respectively. These clearance values are well in excess of rat liver blood flow (Pollack et al., 1990), indicating a substantial contribution to their elimination by extra-hepatic metabolism and/or active transporter processes. In contrast, compound B was characterized by a moderate plasma clearance of 40.9 ± 3.5 mL·min−1·kg−1 and a steady-state distribution volume of 3.48 ± 0.33 L·kg−1, indicating distribution into tissues. The resultant plasma half-life was approximately 1 h. Hence, only compound B was deemed to have the appropriate pharmacokinetic properties for progression into the in vivo CFA model.
In vivo CFA model
As expected, before CFA there was little difference between the weight distribution of rat's hind limbs. After CFA, however, the difference in weight bearing between hind paws was approximately 80 g. This deficit between paws was significantly reduced 1 h after all doses of compound B by approximately 40 g, whereas the vehicle had no effect (Figure 7A). Representing the data as percent reversal of weight-bearing deficit, there was a significant effect at 3 and 10 mg·kg−1, producing reversals of 57 ± 12% and 55 ± 21% respectively (Figure 7B). Although there was some evidence of reversal at the top dose, producing a reversal of 36 ± 10%, this effect was not significant.
Figure 7.
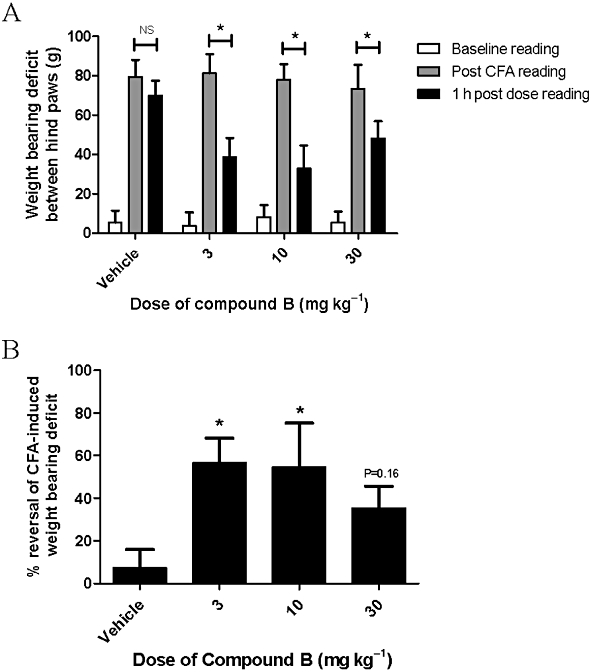
Effect of the compound B (3–30 mg·kg−1, s.c.) on CFA-induced hind limb weight deficit. Weight bearing deficit between hind limb raw data (A), and data are represented as % of reversal (B). Weight bearing of hind limbs was measured before and 1 h post-drug administration. Each point represents mean ± SEM from seven rats. *P < 0.05 compared with baseline reading (A) and vehicle treatment (B) by anova followed by Fisher's LSD test. NS, non significant.
Pharmacokinetic study of compound B
The blood and brain concentrations of compound B immediately following weight-bearing measurements (about 1 h after dosing) are summarized in Table 2. The concentration of compound B in blood increased in a dose-related manner from 0.47 µM (3 mg·kg−1) to 3.79 µM (30 mg·kg−1). Brain concentrations of compound B were seven- to ninefold higher than those in the blood, indicating extensive distribution into CNS tissue. Supporting in vitro experiments using equilibrium dialysis in brain tissue (see Methods section) showed that the unbound brain concentrations were approximately 0.12, 0.44 and 0.74 µM at 3, 10 and 30 mg·kg−1 respectively. Given the sub-micromolar and micromolar pIC50 values of compound B against P2X3 and P2X2/3 receptors, the unbound brain concentrations achieved in this model 1 h after dosing were in comparable concentration ranges.
Table 2.
Summary of pharmacokinetic data for compound B obtained from rats
| Dose | |||
|---|---|---|---|
| 3 mg·kg−1 | 10 mg·kg−1 | 30 mg·kg−1 | |
| Blood concentration (µM) | 0.47 ± 0.16 | 2.08 ± 0.45 | 3.79 ± 0.79 |
| Brain concentration (µM) | 4.29 ± 1.05 | 15.99 ± 2.30 | 26.78 ± 2.00 |
| Brain : blood ratio | 9.3 ± 1.8 | 7.9 ± 1.9 | 7.3 ± 1.2 |
Data are mean ± SD of seven observations.
Discussion
The three diaminopyrimidine compounds described here were functionally characterized for their pharmacological activity at ionotropic purinergic receptors using two in vitro assay methodologies. All three compounds were found to be potent antagonists at P2X3 and P2X2/3 receptors, being able to block agonist-induced increases in cytosolic Ca2+ concentration and current flow at nanomolar concentration ranges in cell lines expressing human P2X3 or P2X2/3 receptors. The compounds were also assessed using the same assays on rat P2X2/3 receptors expressed in the same cell background used for the human receptors. The rat P2X3 expressing cell line was not available for the present study; however, considering that cells expressing both the rat P2X3 and P2X2 receptor subunits show, in response to the agonist, the P2X3-mediated peak, we can extrapolate that at concentrations ≥1 µM compounds A, B and C would be able to block rat homomeric P2X3 receptors. The rank order of their potency was the same, but we observed a consistent decrease in potency for the rat orthologue (up to 10-fold), which is not surprising because within the P2X receptor family, a species specificity in pharmacology is recognized, which seems particularly pronounced for P2X7 receptor antagonists (Surprenant and North, 2009). A decrease in potency between rat and human P2X2/3 receptors, similar to that observed in the present study, has been described by Jarvis et al. (2002) for the compound A-317491 when assessed in a Ca2+ influx assay. The diaminopyrimidine compound AF-353 may inhibit the human P2X2/3 receptor in a non-competitive fashion (Gever et al., 2010). Since compounds A, B and C are also diaminopyrimidine compounds, a similar mechanism of action could be inferred, but experimental data would be required to demonstrate this. The diaminopyrimidine compounds showed no detectable activity against other P2X receptors, namely human P2X1, P2X2, P2X4 and P2X7, but some activity at 5HT3 and nACh receptors, at the α1 adrenoceptors and at the transporter OATP1B1. No activity was identified against a wide range of other targets belonging to ion channel, transporter, enzyme, GPCR or kinase classes. Similar selectivity results were found for compounds belonging to the same chemical class, RO-4 and RO-51 (Carter et al., 2009; Jahangir et al., 2009).
The diaminopyrimidine compounds presented in this paper, despite having a high to moderate clearance after intravenous administration, have potential for good CNS penetration partially overcoming the major problems of A-317491 as a drug candidate. In particular, DMPK studies on compound B demonstrated a moderate clearance and an extensive distribution into CNS tissue supported by a significant effect in the in vivo CFA model, a chronic inflammatory pain model, and thus validating P2X3 and P2X2/3 receptors as inflammatory pain targets.
It is worth pointing out that the lack of activity against P2X4 and P2X7 receptor subtypes shown by the diaminopyrimidines increases confidence that the action of compound B in the in vivo model can be attributed mainly to its activity at the P2X2/3 and P2X3 receptors. P2X4 and P2X7 receptor subtypes have been reported to play a role in chronic inflammatory or neuropathic pain. Thus, P2X4 receptors expressed by activated spinal microglia after peripheral nerve injury promote neuropathic pain (Tsuda et al., 2003). P2X7 receptors are expressed by immune cells, including mast cells, macrophages and T lymphocytes and have a key role in the secretion of IL-1β. Chessell et al. (2005) showed that disruption of the P2X7 purinoreceptor gene abolished neuropathic pain and that the receptor was up-regulated in human dorsal root ganglion cells and injured nerves from neuropathic pain patients.
Pain is not a unique sensory phenomenon, but rather a series of complex neurophysiological responses to acute or persistent injury and distinctive mechanisms contribute to acute nociceptive states, to pain arising from persistent or chronic tissue damage (inflammatory pain) and to pain arising from injury to the nervous system (neuropathic pain) (Woolf and Salter, 2000). Clinically, pain resulting from nerve damage is refractory to many current pharmacological treatments (Dworkin, 2002; McQuay, 2002). Increasing numbers of negative clinical trials of pharmacological treatments for neuropathic pain and ambiguities in the interpretation of these negative trials brings as consequence a great unmet medical need for patients with neuropathic pain who do not have adequate pain control (Dworkin et al., 2010). A diaminopyrimidine compound (Sharp et al., 2006) inhibited Aδ and C fibre-evoked responses, thus demonstrating a possible involvement of P2X3 or P2X2/3 receptors at the level of the spinal cord and supporting the role for P2X3 or P2X2/3 receptor antagonists in the modulation of neuropathic pain.
These observations and the results presented here demonstrate that future studies with diaminopyrimidine compounds will yield further breakthroughs in our understanding of the contributions of P2X3-containing receptors to the different states of pathological pain and ultimately will contribute to the identification of clinically useful compounds.
Glossary
Abbreviations
- αβ-Me-ATP
α,β-methylene-ATP
- Bz-ATP
2′ and 3′-O-(4-benzoylbenzoyl)-ATP
- CFA
complete Freund's adjuvant
- Compound A
5-{[5-methyl-2-(1-methylethyl)phenyl]oxy}-2,4-pyrimidinediamine
- Compound B
5-{[4-chloro-5-methyl-2-(1-methylethyl)phenyl]methyl}-2,4-pyrimidinediamine
- Compound C
5-{[4-chloro-5-methyl-2-(1-methylethyl)phenyl]oxy}-2,4-pyrimidinediamine
- DMPK
drug metabolism and pharmacokinetics
- FLIPR
fluorometric imaging plate reader
- Fluo-4 AM
fluo-4-acetoxymethyl ester
- PPADS
pyridoxal-5-phosphate-6-azophenyl-2′,4′-disulphonic acid
- TNP-ATP
(2′,3′-O-(2,4,6- trinitrophenyl)-ATP
Conflicts of interest
None.
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley KK, Bradley ME. Purine nucleoside-dependent inhibition of cellular proliferation in 1321N1 human astrocytoma cells. J Pharmacol Exp Ther. 2001;299:748–752. [PubMed] [Google Scholar]
- Burnstock G. A unifying purinergic hypothesis for the initiation of pain. Lancet. 1996;347:1604–1605. doi: 10.1016/s0140-6736(96)91082-x. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Williams M. P2 purinergic receptors: modulation of cell function and therapeutic potential. J Pharmacol Exp Ther. 2000;295:862–869. [PubMed] [Google Scholar]
- Burnstock G, Wood JN. Purinergic receptors: their role in nociception and primary afferent neurotransmission. Curr Opin Neurobiol. 1996;6:526–532. doi: 10.1016/s0959-4388(96)80060-2. [DOI] [PubMed] [Google Scholar]
- Carter DS, Alam M, Cai H, Dillon MP, Ford APDW, Gever JR, et al. Identification and SAR of novel diaminopyrimidines. Part 1: the discovery of RO-4, a dual P2X3/P2X2/3 antagonist for the treatment of pain. Bioorg Med Chem Lett. 2009;19:1628–1631. doi: 10.1016/j.bmcl.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Chen CC, Akopian AN, Sivilotti L, Colquhoun D, Burnstock G, Wood JN. A P2X purinoceptor expressed by a subset of sensory neurones. Nature. 1995;377:428–431. doi: 10.1038/377428a0. [DOI] [PubMed] [Google Scholar]
- Chen YH, Dale TJ, Romanos MA, Whitaker WRJ, Xie XM, Clare JJ. Cloning, distribution and functional analysis of the type III sodium channel from human brain. Eur J Neurosci. 2000;12:4281–4284. [PubMed] [Google Scholar]
- Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, et al. Disruption of the P2X(7) purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–396. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Clayton NM, Oakley I, Thompson S, Wheeldon A, Sargent B, Bountra C. Validation of the dual weight averager as an instrument for the measurement of clinically relevant pain. Br J Pharmacol. 1997;120:219. [Google Scholar]
- Cook SP, McCleskey EW. Cell damage excites nociceptors through release of cytosolic ATP. Pain. 2002;95:41–47. doi: 10.1016/s0304-3959(01)00372-4. [DOI] [PubMed] [Google Scholar]
- Driessen B, Reimann W, Selve N, Friderichs E, Bultmann R. Antinoci-ceptive effect of intrathecally administered P2-purinoceptor antagonists in rats. Brain Res. 1994;666:182–188. doi: 10.1016/0006-8993(94)90770-6. [DOI] [PubMed] [Google Scholar]
- Dworkin RH. An overview of neuropathic pain: syndromes, symptoms, signs and several mechanisms. Clin J Pain. 2002;18:343–349. doi: 10.1097/00002508-200211000-00001. [DOI] [PubMed] [Google Scholar]
- Dworkin RH, O'Connor AB, Audette J, Baron R, Gourlay GK, Haanpää ML, et al. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc. 2010;85:S3–S14. doi: 10.4065/mcp.2009.0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RJ, Lewis C, Virginio C, Lundstrom K, Buell A, Suprenant A, et al. Ionic permeability of, and divalent cation effects on, two ATP-gated cation channels (P2X receptors) expressed in mammalian cells. J Physiol. 1996;497.2:413–422. doi: 10.1113/jphysiol.1996.sp021777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick EM, Marty A, Neher E. A patch-clamp study of bovine chromaffin cells and of their sensitivity to acetylcholine. J Physiol-London. 1982;331:577–597. doi: 10.1113/jphysiol.1982.sp014393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara N, Imai Y, Sakakibara A, Morita K, Kitayama S, Tanne K, et al. Regulation of the development of allodynia by intrathecally administered P2 purinoceptor agonists and antagonists in mice. Neurosci Lett. 2000;292:25–28. doi: 10.1016/s0304-3940(00)01427-0. [DOI] [PubMed] [Google Scholar]
- Gever JR, Soto R, Henningsen RA, Martin RS, Hackos DH, Panicker S, et al. AF-353, a novel, potent and orally bioavailable P2X3/P2X2/3 antagonist. Br J Pharmacol. 2010;160:1387–1398. doi: 10.1111/j.1476-5381.2010.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflüg Arch Eur J Phy. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hamilton SG, McMahon SB, Lewin GR. Selective activation of nociceptors by P2X receptor agonists in normal and inflamed rat skin. J Physiol. 2001;534:437–445. doi: 10.1111/j.1469-7793.2001.00437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahangir A, Alam M, Carter DS, Dillon MP, Du Bois DJ, Ford APDW, et al. Identification and SAR of novel diaminopyrimidines. Part 2: the discovery of RO-51, a potent and selective, dual P2X3/P2X2/3 antagonist for the treatment of pain. Bioorg Med Chem Lett. 2009;19:1632–1635. doi: 10.1016/j.bmcl.2009.01.097. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Wismer CT, Schweitzer E, Yu H, van Biesen T, Lynch KJ, et al. Modulation of BzATP and formalin induced nociception: attenuation by the P2X receptor antagonist, TNP-ATP and enhancement by the P2X(3) allosteric modulator, cibacron blue. Br J Pharmacol. 2001;132:259–269. doi: 10.1038/sj.bjp.0703793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MF, Burgard EC, McGaraughty S, Honore P, Lynch K, Brennan TJ, et al. A-317491, a novel potent and selective nonnucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc Natl Acad Sci U S A. 2002;99:17179–17184. doi: 10.1073/pnas.252537299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey P, Summerfield SG. Challenges for blood-brain barrier (BBB) screening. Xenobiotica. 2007;37:1135–1151. doi: 10.1080/00498250701570285. [DOI] [PubMed] [Google Scholar]
- Jones A, Chessel IP, Simon J, Barnard EA, Miller KJ, Michel AD, et al. Functional characterization of the P2X4 receptor orthologues. Br J Pharmacol. 2000;129:388–394. doi: 10.1038/sj.bjp.0703059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaan TK, Yip PK, Patel S, Davies M, Marchand F, Cockayne DA, et al. Systemic blockade of P2X3 and P2X2/3 receptors attenuates bone cancer pain behaviour in rats. Brain. 2010;133:2549–2564. doi: 10.1093/brain/awq194. [DOI] [PubMed] [Google Scholar]
- Kawashima E, Estoppey D, Virginio C, Fahmi D, Rees S, Suprenant A, et al. A novel and efficient method for the stable expression of heteromeric ion channels in mammalian cells. Receptor Channel. 1997;5.2:53–60. [PubMed] [Google Scholar]
- Lewis C, Neidhart S, Holy C, North RA, Buell G, Suprenant A. Coexpression of P2X2 and P2X3 receptor subunits can account for ATP-gated currents in sensory neurons. Nature. 1995;377:432–435. doi: 10.1038/377432a0. [DOI] [PubMed] [Google Scholar]
- Lynch KJ, Touma E, Niforatos W, Kage KL, Burgard EC, Van Biesen T, et al. Molecular and Functional Characterization of Human P2X2 Receptors. Mol Pharmacol. 1999;56:1171–1181. doi: 10.1124/mol.56.6.1171. [DOI] [PubMed] [Google Scholar]
- McQuay HJ. Neuropathic pain: evidence matters. Eur J Pharmacol. 2002;6:11–18. doi: 10.1053/eujp.2001.0316. [DOI] [PubMed] [Google Scholar]
- Michel AD, Ng S-W, Roman S, Clay WC, Dean DK, Walter DS. Mechanism of action of species-selective P2X7 receptor antagonists. Br J Pharmacol. 2009;156:1312–1325. doi: 10.1111/j.1476-5381.2009.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North A, Suprenant A. Pharmacology of cloned P2X Receptors. Annu Rev Pharmacol Toxicol. 2000;40:563–580. doi: 10.1146/annurev.pharmtox.40.1.563. [DOI] [PubMed] [Google Scholar]
- Pollack GM, Brouwer KLR, Demby KB, Jones JA. Determination of hepatic blood flow in the rat using sequential infusions of indocyanine green and galactose. Drug Metab Dispos. 1990;18:197–202. [PubMed] [Google Scholar]
- Rassendren F, Buell GN, Virginio C, Collo G, North RA, Surprenant A. The permeabilizing ATP receptor, P2X7. Cloning and expression of a human cDNA. J Biol Chem. 1997;272:5482–5486. doi: 10.1074/jbc.272.9.5482. [DOI] [PubMed] [Google Scholar]
- Rhodes AD, Bevan N, Patel K, Lee M, Rees S. Mammalian expression of transmembrane receptors for pharmaceutical applications. Biochem Soc T. 1998;26:699–704. doi: 10.1042/bst0260699. [DOI] [PubMed] [Google Scholar]
- Sawynok J, Reid A. Peripheral adenosine 5X-triphosphate enhances nociception in the formalin test via activation of a purinergic P2X receptor. Eur J Pharmacol. 1997;330:115–121. doi: 10.1016/s0014-2999(97)01001-7. [DOI] [PubMed] [Google Scholar]
- Sharp CJ, Reeve AJ, Collins SD, Martindale JC, Summerfield SG, Sargent BS, et al. Investigation into the role of P2X3/P2X2/3 receptors in neuropathic pain following chronic constriction injury in the rat: an electrophysiological study. Br J Pharmacol. 2006;148:845–851. doi: 10.1038/sj.bjp.0706790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shermann-Gold R. Recording from perforated patches and perforated vesicles. In: Shermann-Gold R, editor. The Axon Guide. Foster City, CA: Axon Instruments, Inc; 1993. pp. 114–121. [Google Scholar]
- Stanfa LC, Kontinen VK, Dickenson AH. Effects of spinally administered P2X receptor agonists and antagonists on the responses of dorsal horn neurones recorded in normal, carrageenan-inflamed and neuropathic rats. Br J Pharmacol. 2000;129:351–359. doi: 10.1038/sj.bjp.0703047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerfield SG, Stevens AJ, Cutler L, del Carmen Osuna M, Hammond B, Tang SP, et al. Improving the in vitro prediction of in vivo central nervous system penetration: integrating permeability, P-glycoprotein efflux, and free fractions in blood and brain. J Pharmacol Exp Ther. 2006;316:1282–1290. doi: 10.1124/jpet.105.092916. [DOI] [PubMed] [Google Scholar]
- Surprenant A, North RA. Signaling at purinirgic P2X receptors. Annu Rev Physiol. 2009;71:333–359. doi: 10.1146/annurev.physiol.70.113006.100630. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Koizumi S, Kita A, Shigemoto Y, Ueno S, Inoue K. Mechanical allodynia caused by intraplantar injection of P2X receptor agonist in rats: involvement of heteromeric P2X2/3 receptor signaling in capsaicin-insensitive primary afferent neurons. J Neurosci. 2000;20:RC90. doi: 10.1523/JNEUROSCI.20-15-j0007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Ueno S, Inoue K. Evidence for the involvement of spinal endogenous ATP and P2X receptors in nociceptive responses caused by formalin and capsaicin in mice. Br J Pharmacol. 1999;128:1497–1504. doi: 10.1038/sj.bjp.0702960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virginio C, Church D, North RA, Suprenant A. Effects of divalent cations, protons and calmidazolium at the rat P2X7 receptor. Neuropharmacology. 1997;36:1285–1294. doi: 10.1016/s0028-3908(97)00141-x. [DOI] [PubMed] [Google Scholar]
- Virginio C, Giacometti A, Aldegheri L, Rimland JM, Terstappen GC. Pharmacological properties of rat alpha 7 nicotinic receptors expressed in native and recombinant cell systems. Eur J Pharmacol. 2002;445:153–161. doi: 10.1016/s0014-2999(02)01750-8. [DOI] [PubMed] [Google Scholar]
- Vulchanova L, Riedl MS, Shuster SJ, Buell G, Suprenant A, North RA, et al. Immunocytochemical study ofthe P2X2 and P2X3 receptor subtypes in rat and monkey sensory neurons on their central terminals. Neuropharmacology. 1997;36:1229–1242. doi: 10.1016/s0028-3908(97)00126-3. [DOI] [PubMed] [Google Scholar]
- Vulchanova L, Olson TH, Stone LS, Riedl MS, Elde R, Honda CN. Cytotoxic targeting of isolectin IB4-binding sensory neurons. Neuroscience. 2001;108:143–155. doi: 10.1016/s0306-4522(01)00377-3. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Zheng JH, Chen J. Modulatory roles of the adenosine triphosphate P2X-purinoceptor in generation of the persistent nociception induced by subcutaneous bee venom injection in the conscious rat. Neurosci Lett. 2000;278:41–44. doi: 10.1016/s0304-3940(99)00896-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.