

Abstract
A catalytic, enantioselective method for the C–H functionalization of indoles by diazo compounds has been achieved. With catalytic amounts of Rh2(S-NTTL)4, the putative Rh-carbene intermediates from α-alkyl-α-diazoesters react with indoles at C(3) to provide α-alkyl-α-indolylacetates in high yield and enantioselectivity. From DFT calculations, a mechanism is proposed that involves a Rh-ylide intermediate with oxocarbenium character.
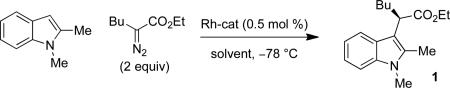
Indoles are important structural motifs in a myriad of biologically interesting natural products and pharmaceutical targets.1 Accordingly, several methods have been developed for the generation of highly functionalized indoles.2 Among these strategies is the selective functionalization by metal carbenes derived from α-diazocarbonyl compounds,3 a reactivity pattern that has been utilized in various total syntheses4 as well as selective tryptophan modification in peptides and proteins.5 However, the only catalytic enantioselective reaction of indoles and transient metal carbenes is Davies’ [3+2] annulation of indoles with styryldiazoacetates (eq. 1).6 While indol-3-yl acetate derivatives with stereogenic centers positioned α- to C-3 have high medicinal value,1e,f only one example of an enantioselective C–H functionalization reaction of an indole has been reported, and the ee was < 5%.6 Described herein is a general Rh-catalyzed method for enantioselective C–H functionalization of indoles by carbenoids derived from α-alkyl-α-diazoesters (eq. 2).
 |
(1) |
 |
(2) |
During the course of our studies on the development of Rh-catalyzed reactions of α-alkyl-α-diazoesters that are selective over β-hydride elimination,7 we reported a method for enantioselective cyclopropanation of olefins.7a The most effective catalyst for enantioselective cyclopropanation was dirhodium(II) tetrakis[N-phthaloyl-(S)-tert-leucinate] [Rh2(SPTTL)4], a catalyst first described by Hashimoto.8 We7a and Charette9 have independently observed that Rh2(S-PTTL)4 and several other phthalimide-derived complexes crystallize in the “chiral crown” conformation. This conformation, in which the four phthalimide groups are projected on one face of the complex, has also been recently observed in crystalline Cu2(S-PTTL)410 and dirhodium(II) tetrakis[N-(1,8-naphthaloyl)-(S)-tert-leucinate [Rh2(S-NTTL)4].10,11 Models for asymmetric induction based on chiral crown conformations have been proposed7a,9 and debated,12 and factors that create bias for the chiral crown configuration over competing conformations have been discussed.7a,9,10 With this foundation, we hypothesized that enantioselective reactions between indoles and α-alkyl-α-diazoesters could be catalyzed by Rh-complexes proposed to adopt chiral crown conformations.
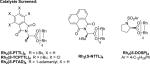
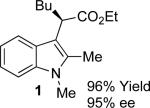
We began our investigation with the reaction of 1,2-dimethylindole with a 2-fold excess of ethyl 2-diazohexanoate (Table 1). A variety of Rh-complexes derived from t-leucine were screened, as were Rh2(S-DOSP)4 and Rh2(S-PTAD)4. While many of the catalysts screened gave 1 with good enantioselectivity, it was found that Rh2(S-NTTL)4 in toluene at −78 °C was optimal both in terms of yield and enantioselectivity, as 1 was formed in 95% yield and 95% ee. In line with our previous observations,7 the use of low temperature was critical to the success of the reaction: the analogous reaction at higher temperature (0 °C) gave 1 in only 36% yield and 85% ee (entry 7).
Table 1.
Selected Enantioselective Indole Functionalization Optimization Experimentsa
 | ||||
|---|---|---|---|---|
| entry | solvent | catalyst | yield(%)b | ee(%)c |
| 1 | PhMe | Rh2(S-PTTL)4 | 73 | 85 |
| 2 | PhMe | Rh2(S-TCPTTL)4 | 20 | 76 |
| 3 | PhMe | Rh2(S-NTTL)4 | 95 | 95 |
| 4 | PhMe | Rh2(R-PTAD)4 | 54 | –91 |
| 5 | PhMe | Rh2(S-DOSP)4 | 24 | 20 |
| 6 | CH2Cl2 | Rh2(S-NTTL)4 | 56 | 92 |
| 7d | PhMe | Rh2(S-NTTL)4 | 36 | 85 |
Conditions: indole (0.2 M), Rh-cat (0.5 mol %) at –78 °C, α-diazoester (0.67 M) added via syringe pump.
Isolated yield.
Determined by chiral HPLC analysis.
Reaction run at 0 °C. Optimal conditions in bold.
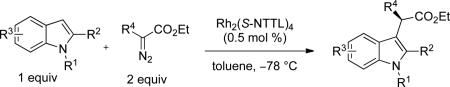
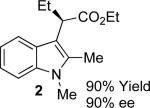
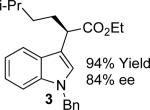
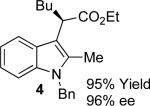
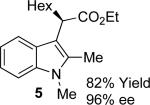
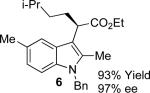
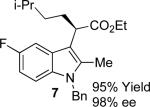
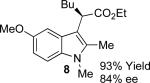
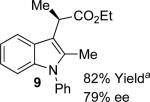
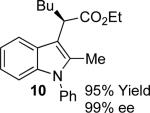
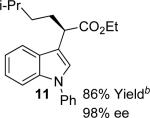
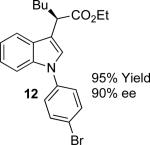
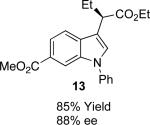
With the optimized reaction conditions in hand, the scope of this transformation was then explored, and the results are summarized in Table 2. High yields (82-96%) and enantioselectivities (79-99% ee) are obtained across a wide array of substrates. Diazoesters bearing a variety of α-alkyl substituents, covering the range of methyl, ethyl, butyl, and isopentyl, led to functionalized indoles with high yield and enantioselectivity. Diazoesters with primary α-alkyl substitutents gave higher enantioselectivity than ethyl α-diazopropionate (compare 9 and 10). The transformation is successful for reactions of indoles with a range of functionality, including fluoro-, bromo-, methoxy, siloxy, Boc-protected aniline and ester functional groups. Methyl (compounds 1-2, 5, 8), benzyl (compounds 3-4, 6-7), and aryl (compounds 9-15) groups on nitrogen were well tolerated, while indole itself gave the product of N-H insertion in low enantiomeric excess.13a An attempt to react ethyl α-diazo-5-methylhexanoate with 1,3-dimethylindole was also unsuccessful: only intramolecular β-elimination was observed.
Table 2.
Scope of the Reactionc
 | ||
|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
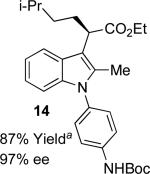
|
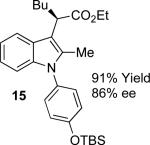
|
a 5 equiv. of diazoester was used.
b Contained < 5% of the product of alkene cyclopropanation.
All yields and ee's refer to the average of two runs.
The nucleophilicity of the nitrogen atom plays a critical role. While the reaction is successful for N-aryl or N-alkyl indole derivatives, substituting strongly electron withdrawing groups on nitrogen (e.g. acetyl or Boc) completely shut down the intermolecular reaction and only the products of β-hydride elimination of the carbenoid were observed. The reaction was also highly sensitive to the steric bulk at the indole C(2) position. High yields and enantioselectivities were obtained when R2 is small (R2 = H, Me); highest enantioselectivities were generally observed when R2 = Me. Experiments with larger R2 substituents (Et, n-Bu, CF3, Ph, I) were unsuccessful. However, indoles with N-to-C(2) ring fusion were excellent substrates, and compounds 16 and 17 were obtained in good yield and high enantioselectivity13b (Scheme 1).
Scheme 1.

Reactions of Fused Indoles
Previous mechanistic proposals for the indole C–H functionalization reaction include a cyclopropanation/fragmentation pathway3b,c or an ylide formation/proton transfer pathway.3d,e,6 To rule out the former pathway, we carried out the reaction between indole 18 and ethyl α-diazohexanoate in the presence of racemic 19 and 20. Compounds 19 and 20 were prepared as an inseparable 1:1 mixture from the Rh2(Piv)4 catalyzed reaction between 1-phenylindole and ethyl α-diazo-5-methylhexanoate. In the event, Rh2(S-NTTL)4 catalyzed the formation of 8 in 89% yield and 84% ee, and compounds 19 and 20 were isolated in a 1:1 ratio with 97% mass recovery (Scheme 2). That cyclopropane 20 did not rearrange under the reaction conditions provides evidence against the cyclopropanation/fragmentation mechanism.
Scheme 2.

Evidence against a cyclopropanation/fragmentation mechanism
To investigate the plausibility of mechanism that involves a Rh-ylide, calculations were carried out for the reaction between 2-methylindole and Et(EtO2C)C=Rh2(O2CH)4 using B3LYP method with two basis sets: lanl2dz for Rh, and 6-311+G(d,p) for other atoms. A lanl2dz effective core potential was utilized. The calculations support the mechanism shown in Figure 1a, in which the intermediate ylide E is a stabilized oxocarbenium ion formed via transition state D. Relative to a pre-reaction complex between the carbene and indole, transition state D has a barrier of ΔE(ZPE)‡ = 8.8 kcal/mol (Figure 1b), and the formation of ylide E is exothermic by E(ZPE) = 16.0 kcal/mol. Transition state D is formed by end-on approach14 of the indole to the carbene, with only a minor change in the Rh-Rh bond length in D (2.498 Å) relative to the carbene (2.490 Å). While both C–C and C–O bonds are formed in this process, the advancement of these bond formations is not synchronous. Thus, the C–C distance (2.457 Å, labeled a in Scheme 2b) in D is considerably shorter than the C–O bond distance (2.815 Å, labeled b in Figure 1b) in transition state D. By contrast, the corresponding distances in ylide E (Scheme 2c) are 1.542 Å and 1.501 Å, respectively.
Figure 1.

(a) Proposed mechanism. (b) Calculated transition state (D) for the reaction between 2-methylindole and Et(EtO2C)C=Rh2(O2CH)4. (c) Calculated structure of ylide E.
In transition state D, the C-1 methyl group projects toward one of the formate ligands (3.299 Å separation, labeled c in Figure 1b), whereas the nitrogen substituent projects away from the Rh-carboxylate core. Thus, this model is consistent with the observed sensitivity toward steric effects for substitution at C(2), but tolerance of a broad range of substitution on the indole nitrogen. IRC analysis indicates that transition state D leads to ylide E, the structure of which is shown in Figure 1c.
Computation was also used to consider the formation of an ylide intermediate via an electrophilic aromatic substitution-type mechanism. A transition state that does not possess oxocarbenium character was also located (see Supporting Information), and was found to be higher in energy than D by ΔΔE(ZPE)‡ = 1.8 kcal/mol. In this higher energy transition state, the benzene ring of the indole is positioned above the ester functionality.
In our prior work on asymmetric cyclopropanation, we proposed that alignment of the carbenoid in the chiral crown cavity of Rh2(S-PTTL)4 leaves the si face of the carbenoid more accessible for reactivity.7a Rh2(S-NTTL)4 also has a crown structure10,11 (Figure 2a), and it is possible that a similar model for asymmetric induction may be in operation for the formation of ylide E (Fig 2b); aromatization and stereoretentive protonation of the C–Rh bond would then provide the indole product F. We consider that the conversion of E to F is stepwise, as computations suggest that an intramolecular 1,2-hydride shift for the conversion of E to F is not plausible (ΔE(ZPE)‡ = 30.2 kcal/mol) at –78 °C. An alternative possibility is that asymmetry is induced via dynamic kinetic resolution of intermediate Rh-enolates (e.g. G and G’ in Figure 2c) that are not configurationally stable.15 In this scenario, the enantiodetermining step would involve a dynamic equilibrium between G and G’ that is faster than the rate of protonation (Figure 2c).
Figure 2.

(a) x-ray crystal structure of Rh2(S-NTTL)4. (b) Asymmetric induction may be explained by approach of the indole to the si-face of the Rh-carbene, with subsequent aromatization and stereoretentive protonation. (c) Alternatively, asymmetric induction may occur via dynamic kinetic resolution, provided that equilibrium between diastereomeric Rh-enolates G and G’ is fast relative to the rate of protonation.
To conclude, we have developed an enantioselective, Rh2(S-NTTL)4 catalyzed method for C–H functionalization of indoles by α-alkyl-α-diazoesters. From DFT calculations, a mechanism is proposed that involves a Rh-ylide intermediate with oxocarbenium character. Asymmetric induction may be explained by approach of the indole to the si-face of the Rh-carbene, with subsequent aromatization and stereoretentive protonation. Alternatively, asymmetric induction may occur via dynamic kinetic resolution of a rhodium enolate intermediate. Efforts to distinguish these mechanisms are ongoing.
Supplementary Material
Acknowledgement
For financial support we thank NIGMS (NIH R01 GM068650). NMR spectra were obtained with instrumentation supported by NSF CRIF:MU, CHE 0840401. We thank Glenn Yap for x-ray crystallography.
Footnotes
Supporting Information Available: Full experimental details, 1H and 13C-NMR spectra, stereochemical assignments, computational details and crystallographic (CIF) data are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dewick PM. Medicinal Natural Products: A Biosynthetic Approach. John Wiley & Sons Inc.; Chichester: 2009. Barton DHR, Nakanishi K, MethCohn O, Kelly JW. Comprehensive Natural Products Chemistry. Pergamon Press; Oxford: 1999. Moody CJ, editor. Advances in Nitrogen Heterocycles. JAI Press Inc.; Greenwich: 1995. Kochanowska-Karamyan AJ, Hamann MT. Chem. Rev. 2010;110:4489. doi: 10.1021/cr900211p. (e) For medicinally active indol-3-yl acetate derivatives with stereocenters α- to C3, see: Batt DG, Qiao JX, Modi DP, Houghton GC, Pierson DA, Rossi KA, Luettgen JM, Knabb RM, Jadhav PK, Wexler RR. Biorg. Med. Chem. Lett. 2004;14:5269. doi: 10.1016/j.bmcl.2004.08.037. Black WC, Bayly C, Belley M, Chan CC, Charleson S, Denis D, Gauthier JY, Gordon R, Guay D, Kargman S, Lau CK, Leblanc Y, Mancini J, Ouellet M, Percival D, Roy P, Skorey K, Tagari P, Vickers P, Wong E, Xu L, Prasit P. Biorg. Med. Chem. Lett. 1996;6:725.
- 2.For recent reviews, see: Patil S, Patil R. Curr. Org. Synth. 2007;4:201. Humphrey GR, Kuethe JT. Chem. Rev. 2006;106:2875. doi: 10.1021/cr0505270. Cacchi S, Fabrizi G. Chem. Rev. 2005;105:2873. doi: 10.1021/cr040639b. Maryanoff BE, Zhang H, Cohen JH, Turchi IJ, Maryanoff CA. Chem. Rev. 2004;104:1431. doi: 10.1021/cr0306182. For recent examples, see: Guo C, Song J, Luo S, Gong L. Angew. Chem. Int. Ed. 2010;49:5558. doi: 10.1002/anie.201002108. Pathak TP, Gligorich KM, Welm BE, Sigman MS. J. Am. Chem. Soc. 2010;132:7870. doi: 10.1021/ja103472a. Ganesh M, Seidel D. J. Am. Chem. Soc. 2008;130:16464. doi: 10.1021/ja8063292. Stokes BJ, Dong H, Leslie BE, Pumphrey AL, Driver TG. J. Am. Chem. Soc. 2007;129:7500. doi: 10.1021/ja072219k.
- 3.Selected examples: Lian Y, Davies HML. Org. Lett. 2010;12:924. doi: 10.1021/ol9028385. Yadav JS, Reddy BVS, Satheesh G. Tetrahedron Lett. 2003;44:8331. Gibe R, Kerr MA. J. Org. Chem. 2002;67:6247. doi: 10.1021/jo025851z. Muthusamy S, Gunanathan C, Babu SA, Suresh E, Dastidar P. Chem. Commun. 2002:824. doi: 10.1039/b200412g. Johansen MB, Kerr MA. Org. Lett. 2010;12:4956. doi: 10.1021/ol1020948. (f) For a useful review, see: Davies HML, Hedley SJ. Chem. Soc. Rev. 2007;36:1109. doi: 10.1039/b607983k.
- 4.a Tamaki K, Huntsman EWD, Petsch DT, Wood JL. Tetrahedron Lett. 2002;43:379. [Google Scholar]; b Wood JL, Stoltz BM, Dietrich H, Pflum DA, Petsch DT. J. Am. Chem. Soc. 1997;119:9641. [Google Scholar]; c Wood JL, Stoltz BM, Dietrich H. J. Am. Chem. Soc. 1995;117:10413. [Google Scholar]
- 5.a Popp BV, Ball ZT. J. Am. Chem. Soc. 2010;132:6660. doi: 10.1021/ja101456c. [DOI] [PubMed] [Google Scholar]; b Antos JM, Francis MB. J. Am. Chem. Soc. 2004;126:10256. doi: 10.1021/ja047272c. [DOI] [PubMed] [Google Scholar]
- 6.Lian Y, Davies HML. J. Am. Chem. Soc. 2010;132:440. doi: 10.1021/ja9078094. [DOI] [PubMed] [Google Scholar]
- 7.a DeAngelis A, Dmitrenko O, Yap GPA, Fox JM. J. Am. Chem. Soc. 2009;131:7230. doi: 10.1021/ja9026852. [DOI] [PMC free article] [PubMed] [Google Scholar]; b DeAngelis A, Taylor MT, Fox JM. J. Am. Chem. Soc. 2009;131:1101. doi: 10.1021/ja807184r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Panne P, DeAngelis A, Fox JM. Org. Lett. 2008;10:2987. doi: 10.1021/ol800983y. [DOI] [PMC free article] [PubMed] [Google Scholar]; d DeAngelis A, Panne P, Fox JM. J. Org. Chem. 2008;73:1435. doi: 10.1021/jo702308f. [DOI] [PubMed] [Google Scholar]; e Panne P, Fox JM. J. Am. Chem. Soc. 2007;129:22. doi: 10.1021/ja0660195. [DOI] [PubMed] [Google Scholar]
- 8.Hashimoto S, Wantanabe W, Sato T, Shiro M, Ikegami S. Tetrahedron Lett. 1993;34:5109. [Google Scholar]
- 9.Lindsay VNG, Lin W, Charette AB. J. Am. Chem. Soc. 2009;131:16383. doi: 10.1021/ja9044955. [DOI] [PubMed] [Google Scholar]
- 10.DeAngelis A, Boruta DT, Lubin J, Plampin JN, Yap GPA, Fox JM. Chem. Commun. 2010:4541. doi: 10.1039/c001557a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghanem A, Gardiner MG, Williamson RM, Müller P. Chem.–Eur. J. 2010;16:3291. doi: 10.1002/chem.200903231. [DOI] [PubMed] [Google Scholar]
- 12.Nadeau E, Ventura DL, Brekan JA, Davies HML. J. Org. Chem. 2010;75:1927. doi: 10.1021/jo902644f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Known substrate limitations: indole with ethyl α-diazobutanoate gave the product of N-H insertion in 29% yield and <1% ee; 6-(hydroxymethyl)-N-methylindole with ethyl α-diazo-5-methylhexanoate gave the product of OH-insertion in 41% yield and <1% ee; 1,2-dimethyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole with ethyl α-diazo-5-methylhexanoate gave the desired CH functionalization product in 81% yield, but only 13% ee; 5-nitro-1,2-dimethylindole with ethyl α-diazobutanoate did not give any intermolecular products. (b) For the preparation of 16 and 17, it was important to use only one equivalent of diazoester. Enantioselectivity was lower and difficult to reproduce when 2 equivalents of diazoester were used. Azine side products7c were formed when excess amounts of diazoesters were employed. We believe that the azine acts as a base that epimerizes 16 and 17.
- 14.Nowlan DT, Gregg TM, Davies HML, Singleton DA. J. Am. Chem. Soc. 2003;125:15902. doi: 10.1021/ja036025q. [DOI] [PubMed] [Google Scholar]
- 15.Moss RJ, Wadsworth KJ, Chapman CJ, Frost CG. Chem. Commun. 2004:1984. doi: 10.1039/b406905f. Frost CG, Penrose SD, Lambshead K, Raithby PR, Warren JE, Gleave R. Org. Lett. 2007;9:2119. doi: 10.1021/ol070603g. Sibi MP, Tatamidani H, Patil K. Org. Lett. 2005;7:2571. doi: 10.1021/ol050630b. Nishimura T, Hirabayashi S, Yasuhara Y, Hayashi T. J. Am. Chem. Soc. 2006;128:2556. doi: 10.1021/ja058763f. For a review, see Mohr JT, Hong AY, Stoltz BM. Nature Chem. 2009;1:359. doi: 10.1038/nchem.297.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.