

Abstract
We have performed quantum mechanical calculations for retinal model compounds to establish the rotational energy barriers for the C5-, C9-, and C13-methyl groups known to play an essential role in rhodopsin activation. Intraretinal steric interactions as well as electronic effects lower the rotational barriers of both the C9- and C13-methyl groups, consistent with experimental 2H NMR data. Each retinal methyl group has a unique rotational behavior which must be treated individually. These results are highly relevant for the parameterization of molecular mechanics force fields which form the basis of molecular dynamics simulations of retinal proteins such as rhodopsin.
Accurate modeling of rhodopsin is essential to studying G protein-coupled receptors (GPCRs), which play a central role in pharmacological and biomembrane research (1). Rhodopsin is the primary model for GPCR research due to its photoactive intermediates (2) and distinction as the only crystallized GPCR (3) until recently (4). Upon photon absorption retinal, the covalently bound ligand, undergoes an 11-cis → all-trans isomerization (Fig. 1), leading to a series of rhodopsin photoproducts and subsequent G protein activation (2). The retinylidene C5-, C9-, and C13-methyl (C13-Me) groups have unique interactions that are crucial to rhodopsin function, shifting the Meta I-Meta II equilibrium toward the activated Meta II state (5,6).
Figure 1.
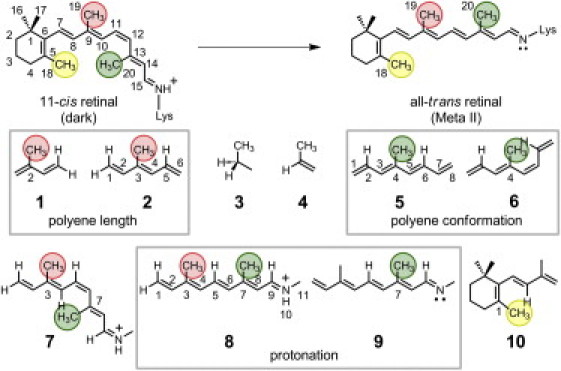
Molecular structure of retinal in the dark state (11-cis) and the Meta II intermediate (trans). Structures of model compounds used are 1: 2-methyl-1,3-butadiene; 2: 3-methyl-1,3E,5-hexatriene; 3: propane; 4: propene; 5: 4-methyl-1,3E,5E,7-octatetraene (4MOT) (trans); 6: 4-methyl-1,3E,5Z,7-octatetraene (cis); 7: 3,7-dimethylnon-1,3E,5Z,7E-tetraen-1-ylidene (N-methyl)amine (37DM+) (cis); 8: 37DM+ (trans); 9: 37DM; and 10: 1,3,3-trimethyl-2-[1′-(3′-methyl-1′,3′-butadienyl)]-cyclohexene. (Equivalent methyl positions corresponding to retinal are enclosed in circles and by parentheses in text.)
Here we address the intramolecular rotational behavior of the retinylidene methyl groups using quantum chemical calculations of the potential energy surface at the MP2 level of theory with a 6-31G∗∗ basis set (see the Supporting Material). The typical rotational barrier for methyl groups is ≈12 kJ mol–1 (3 kcal mol–1) (7). However, we show the methyl energy barriers in retinal are surprisingly complex, depending sensitively on the conformation and protonation state of the ligand. Our results have immediate implications for the dihedral force field components used in rhodopsin molecular dynamics simulations, which have typically treated the methyl groups uniformly (8,9).
When examining retinal methyl rotations, steric interactions with the polyene chain figure prominently. Methyl groups in an all-trans polyene chain experience 1-6 interactions between the methyl hydrogens and those hydrogen atoms bound to neighboring vinyl groups (Fig. 1). Such a 1-6 interaction exists in model compound 1. Note that in 1, and all branched model compounds with methyl groups, the methyl torsion angle (ϕ) equals zero when the methyl C–H bond and vinyl C=C bond are coplanar. In contrast to 1, which possesses a single methyl group-vinyl 1-6 hydrogen interaction, compound 2 possesses two such interactions. The extra 1-6 interaction raises the energies of both the minimum energy conformation and the eclipsed state; the net effect is to reduce the energy barrier versus 1. In Fig. 2 A this effect is seen in the torsional potential energy surfaces of 1 and 2, whereas in Fig. 2 B it is demonstrated by the 1-6 H-H distances computed as a function of torsion angle. Fig. 2 B shows that 2 is nearly symmetrical over the course of a methyl rotation.
Figure 2.
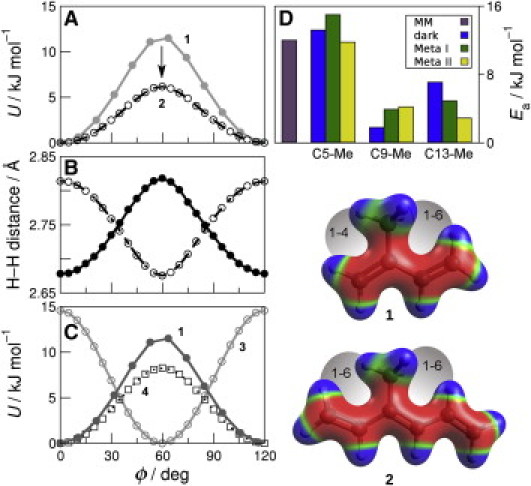
Torsional potential energy surfaces show that 1-6 interactions affect retinal methyl rotation. (A) Comparison of QM (circles) and MM (lines) methyl torsion angle energies in 1 and 2. (B) 1–6 distance in 2 between methyl hydrogen and C1 vinyl hydrogen (solid) and C5 vinyl hydrogen (open). (C) QM energy as a function of methyl torsion angle in 1, 3, and 4. (D) Activation energies (Ea) for C5-, C9-, and C13-Me groups from 2H NMR data for the dark, Meta I, and Meta II states of rhodopsin compared to a typical methyl dihedral energy barrier in a molecular mechanics force field. Steric interactions and electrostatic potentials are shown mapped to surfaces of 1 and 2.
Notably, the preceding results suggest that 2 is the minimum size for a model compound to represent the retinal C9-Me group. Because quantum mechanics (QM) calculations on bound retinal are currently prohibitively expensive, a more approximate but still effective approach is molecular mechanics (MM). Using the form of the torsional energy term in the MM package CHARMM (10), U(ϕ)=Kϕ[1+cos(nϕ+δ)], we fit the QM results for 1 and 2. The QM and MM results are compared in Fig. 2 A, where the remaining force field terms (bond lengths, angles, improper dihedrals, electrostatics, and dispersion interactions) were unmodified from the CHARMM distribution. Numerical values are provided in Table S1 (see the Supporting Material). An improved MM force field will allow future investigations of effective barrier heights in the full rhodopsin environment.
We also performed control QM calculations on two smaller model compounds, propane 3 and propene 4 (Fig. 2 C). Although the energy barrier of 3 is ≈15 kJ mol–1, the barrier of 4 is only ≈8 kJ mol–1. The observed barrier lowering is due to the very different orbital interactions in these two molecules. The methyl group in 4 is most stable at ϕ = 0°, where the methyl C–H bond is cis to the vinyl C=C bond. This conformation places the remaining methyl hydrogen atoms with the vector connecting them perpendicular to the C=C bond axis. It is thus situated to stabilize the conformation through hyperconjugation involving the filled π-like CH2 orbitals of the methyl group and the unoccupied π∗ molecular orbital of the double bond. In contrast, the stable propane conformer 3 leaves the methyl group in the staggered orientation typical of alkanes.
Due to the threefold symmetry of the methyl group, when one of the methyl hydrogens is eclipsed (ϕ = 180°), the other methyl hydrogens lie at 60°. Thus the maximum steric repulsion coincides with the highest energy state arising from orbital interactions, with the net effect of raising the energy barrier by ∼50% in 1 (Fig. 2 C). This steric effect would predict a rotational barrier for 2 that is slightly higher than 4 whereas QM calculations give the opposite (Fig. 2, A and C). We ascribe this phenomenon to altered π-orbital characteristics accompanying the increased polyene chain unsaturation from one double bond (4) to three (2).
The C13-Me group requires a longer polyene chain capable of mimicking both 11-cis- and all-trans-retinal. We chose the model compound 4MOT in both its trans 5 and cis 6 conformations (Fig. 1) to examine the retinal C13-Me energy barrier. To directly compare QM and MM results, we restrained the polyene torsions to planar conformations. Notably, the methyl rotation barrier is dramatically increased for the cis conformer 6, whereas the trans conformer 5 is similar to that for 2. The MM results (Fig. 3 A) suggest the increase arises from steric effects, because the MM force field uses the identical torsional energy term for both 5 and 6. Thus, the MM result (≈8 kJ mol–1 increase) comes solely from nonbonded terms in the force field. The good agreement between QM and MM results indicates the force field parameters should apply well to neutral methyl-substituted polyenes.
Figure 3.
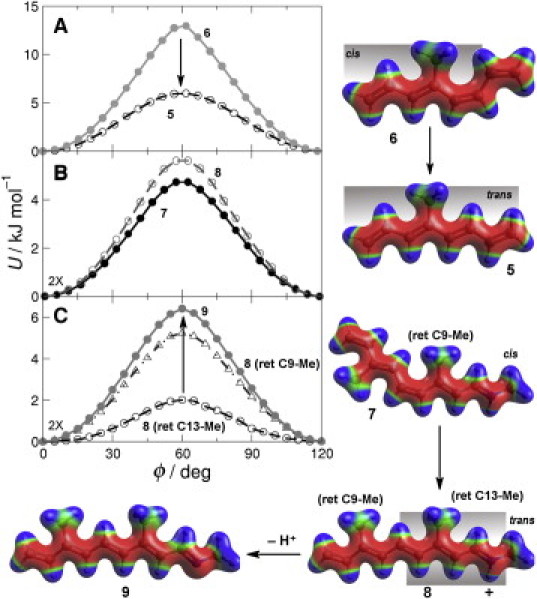
Polyene chain conformation and Schiff base protonation significantly affect retinal methyl rotation. (A) Comparison of QM (circles) and MM (lines) energy as a function of 4-Me (retinal C13-Me) torsion angle in 5 and 6. (B) QM energy as a function of C3-Me (retinal C9-Me group) torsion angle in 7 and 8. (C) QM energy versus methyl torsion angle for C7-Me (retinal C13-Me group) in 8 and 9 and C3-Me (retinal C9-Me group) in 8. Electrostatic potentials mapped to the surfaces of 6–9.
An additional consideration was to examine the retinal protonation state. Although retinal initially undergoes an 11-cis isomerization, the Meta I-Meta II transition involves Schiff base deprotonation (11). The protonated model compounds, 7 (cis) and 8 (trans), were first used to examine the effect of cis-trans isomerization on the torsional potential of the 3-Me (equivalent to retinal C9-Me group). In contrast to the C4-Me of 5 (retinal C13-Me group), which has a dramatic reduction in the rotational energy barrier upon isomerization (Fig. 3 A), the 3-Me of compound 8 (retinal C9-Me group) exhibits a modest increase of ≈1 kJ mol–1 versus 7 (Fig. 3 B). This change in polyene conformation corresponds to the change in retinal structure that accompanies the transition from the dark state to the Meta I state.
Next, Fig. 3 C shows the dramatic effect of protonation on the retinal C13-Me group rotation barrier for an all-trans chain. The protonated all-trans state 8, corresponding to lumirhodopsin and Meta I intermediates, exhibits an extremely low C7-Me (retinal C13-Me group) rotation barrier of ≈2 kJ mol–1, whereas deprotonation in 9, synonymous with the Meta II state, raises this barrier more than threefold. This effect stems from delocalization of positive charge in the protonated state, altering orbital interactions between the methyl group and the polyene chain. The positive charge should lower the π∗ energy and thereby promote interactions with the filled π-like CH2 orbital. The charge delocalization, however, is nonuniform along the polyene chain, favoring the nitrogen-end with minor effects on the C3-Me of 8. The C3-Me (retinal C9-Me group) has a barrier >5 kJ mol–1, substantially more than the C7-Me (retinal C13-Me group), and within 1 kJ mol–1 of the deprotonated molecule 9.
Previous Fourier-transform infrared (5) and 2H NMR (6) experiments demonstrate the crucial role retinal methyl groups play in rhodopsin activation (Fig. 2 D). The retinal C5-Me group Ea remains relatively constant, consistent with results for the C1-Me of 10 (see the Supporting Material). This suggests few steric changes between the C5-Me and the polyene chain and negligible protonation effects. Both retinal C9- and C13-Me groups experience steric effects during cis-trans isomerization (Fig. 3), reflecting the Ea changes from the dark state to Meta I (Fig. 2 D). Protonation effects are less clear; whereas the small increase in the retinal C9-Me rotation barrier upon deprotonation is consistent with QM and 2H NMR results (Fig. 3 C), the C13-Me barrier behaves oppositely. This suggests greater binding pocket rearrangement at the Schiff base end during Meta II, a reasonable expectation because deprotonation occurs there.
The QM results described here will have immediate application in the design of improved MM force fields for large-scale molecular dynamics simulations of the rhodopsin proteolipid assembly (12). The retinal moiety is a particularly challenging target for molecular mechanics-based approaches. Despite well-characterized protonation effects on polyene chain conjugation (13), this work reveals these effects significantly extend to the retinylidene methyl groups. We have shown that each retinal methyl substituent must be individually analyzed in the MM force field, dependent on conformation, proximity to the Schiff base, and protonation state. As increasing timescales and system complexity become more accessible to simulations, these force field developments can potentially yield key insights into the coupling between ps-ns and μs-ms events that are currently being revealed through various biophysical techniques.
Acknowledgments
We thank L. Adamowicz, R. S. Glass, R. W. Pastor, A. Sanov, and A. V. Struts for discussions. This work was supported by the National Science Foundation (MCB0950258 to S.E.F.) and the National Institutes of Health (EY012049 and EY018891 to M.F.B. and EY019614 to B.M.).
Supporting Material
References and Footnotes
- 1.Filmore D. It's a GPCR world. Mod. Drug Disc. 2004;7:24–28. [Google Scholar]
- 2.Lewis J.W., Kliger D.S. Absorption spectroscopy in studies of visual pigments: spectral and kinetic characterization of intermediates. Methods Enzymol. 2000;315:164–178. doi: 10.1016/s0076-6879(00)15842-2. [DOI] [PubMed] [Google Scholar]
- 3.Palczewski K., Kumasaka T., Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 4.Rosenbaum D.M., Zhang C., Kobilka B.K. Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogel R., Lüdeke S., Sheves M. Agonists and partial agonists of rhodopsin: retinal polyene methylation affects receptor activation. Biochemistry. 2006;45:1640–1652. doi: 10.1021/bi052196r. [DOI] [PubMed] [Google Scholar]
- 6.Struts A.V., Salgado G.F.J., Brown M.F. Retinal dynamics underlie its switch from inverse agonist to agonist during rhodopsin activation. Nat. Struct. Mol. Biol. 2011;18:392–394. doi: 10.1038/nsmb.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Widmalm G., Pastor R.W., Bull T.E. Molecular dynamics simulation of methyl group relaxation in water. J. Chem. Phys. 1991;94:4097–4098. [Google Scholar]
- 8.Röhrig U.F., Guidoni L., Rothlisberger U. Solvent and protein effects on the structure and dynamics of the rhodopsin chromophore. ChemPhysChem. 2005;6:1836–1847. doi: 10.1002/cphc.200500066. [DOI] [PubMed] [Google Scholar]
- 9.Sugihara M., Hufen J., Buss V. Origin and consequences of steric strain in the rhodopsin binding pocket. Biochemistry. 2006;45:801–810. doi: 10.1021/bi0515624. [DOI] [PubMed] [Google Scholar]
- 10.Brooks B.R., Brooks C.L., III, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahalingam M., Martínez-Mayorga K., Vogel R. Two protonation switches control rhodopsin activation in membranes. Proc. Natl. Acad. Sci. USA. 2008;105:17795–17800. doi: 10.1073/pnas.0804541105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martínez-Mayorga K., Pitman M.C., Brown M.F. Retinal counterion switch mechanism in vision evaluated by molecular simulations. J. Am. Chem. Soc. 2006;128:16502–16503. doi: 10.1021/ja0671971. [DOI] [PubMed] [Google Scholar]
- 13.Zhou H., Tajkhorshid E., Elstner M. Performance of the AM1, PM3, and SCC-DFTB methods in the study of conjugated Schiff base molecules. Chem. Phys. 2002;277:91–103. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.