

Abstract
The transthyretin amyloidoses are a subset of protein misfolding diseases characterized by the extracellular deposition of aggregates derived from the plasma homotetrameric protein transthyretin (TTR2) in peripheral nerves and the heart. We have established a robust disease-relevant human cardiac tissue culture system to explore the cytotoxic effects of amyloidogenic TTR variants. We have employed this cardiac amyloidosis tissue culture model to screen 23 resveratrol analogs as inhibitors of amyloidogenic TTR-induced cytotoxicity and to investigate their mechanisms of protection. Resveratrol and its analogs kinetically stabilize the native tetramer preventing the formation of cytotoxic species. In addition, we demonstrate that resveratrol can accelerate the formation of soluble non-toxic aggregates and that the resveratrol analogs tested can bring together monomeric TTR subunits to form non-toxic native tetrameric TTR.
Keywords: Protein misfolding, amyloid, transthyretin, cytotoxicity, resveratrol, cardiomyopathy
INTRODUCTION
The systemic amyloidoses, characterized by the extracellular deposition of amorphous aggregates and amyloid fibrils derived from normal or mutant secreted proteins, are prototypic examples of protein misfolding disorders [1–3]. Transthyretin (TTR) is one of the 29 human proteins identified thus far, associated with systemic amyloidosis.
TTR is a 55 kDa homotetrameric protein that transports retinol and thyroxine (T4) in plasma and T4 in the cerebrospinal fluid. The capacity of TTR to bind a large variety of small molecules [4], other proteins and peptides like the Alzheimer’s disease related Aβ peptide [5], suggests that it may perform other biological functions, perhaps related to detoxification of unwanted metabolites and misfolded proteins [6].
The disassembly of the native homotetrameric TTR to its constituent monomers is required for aggregation and subsequent fibril formation in vitro and presumably in vivo [7]. TTR fibrillogenesis is accelerated by the presence of any of the approximately 100 different amyloidogenic mutations [8–11] which decrease the thermodynamic and/or kinetic stability of the mutant TTRs with respect to the wild type protein [8].
The clinical syndromes associated with TTR aggregation are senile systemic amyloidosis (SSA), characterized by deposition of wild type TTR (WT TTR) in the heart, and familial amyloidotic polyneuropathy (FAP) and cardiomyopathy (FAC) related to deposition of mutant forms of TTR in the peripheral nerves and the heart, respectively. The V122I TTR variant, found in 3–4% of African-Americans, is the most common amyloidogenic mutation worldwide and it is associated with FAC [11].
Herein, we introduce the human cardiac AC16 cell line as a robust tissue culture system to serve as a model of the TTR cardiomyopathies [12]. AC16 cells are derived from adult ventricular cardiomyocytes, the site of cardiac TTR deposition in SSA and FAC. They express primary cardiomyocyte biochemical markers like α– and β-myosin heavy chain, α-cardiac actin, troponin I, the gap junction proteins connexin-43 and -40, etc, which make them a relevant model for cardiac-specific tissue culture studies [12].
In the present study we show the effects of several amyloidogenic cardiotoxic TTR variants on cell viability, which are in stark contrast to the effects of a non-amyloidogenic, stable TTR variant. We used this model to evaluate 22 analogs of the plant polyphenol resveratrol (1) for their capacity to prevent TTR-induced cardiotoxicity. Most of the compounds selected for the screening are potent inhibitors of TTR aggregation and fibril formation in an acid-mediated in vitro assay [13–15]. We explore the correlation between TTR kinetic stabilization by these compounds and their capacity to prevent cell damage. We also demonstrate that these compounds may inhibit TTR-induced cytotoxicity by more than one mechanism.
MATERIALS AND METHODS
Preparation of recombinant TTR
The proteins were produced in an E. coli expression system [16; 17] and purified at 4°C, unless stated otherwise. The identity of the proteins was confirmed by liquid chromatography/mass spectrometry.
Cell culture
AC16 cells were grown in DMEM/F12 (1:1) (Cellgro) supplemented with 10% FBS, 2mM L-glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin at 37°C in a 5% CO2 incubator.
Cell Viability Assays
AC16 cells (70–90% confluent) were seeded in black wall clear bottom 96 well plates (250 cells/well) in Opti-MEM, supplemented with 5% FBS, 2mM L-glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin, 1 mM Hepes and 45 mM CaCl2 (seeding Opti-MEM medium) and incubated overnight at 37°C. The seeding Opti-MEM medium was removed and immediately replaced with 100 μL/well of the appropriate TTR solutions or vehicle (1:1 HBSS:Opti-MEM supplemented with glutamine and antibiotics and 0.4 mg/ml fatty acid-free BSA (Sigma, A6003)). The cells were incubated for 24h at 37°C and cell viability measured using the resazurin reduction assay [13; 18].
TTR-induced cytotoxicity inhibition
Resveratrol and its analogs were tested for their capacity to prevent TTR-induced cytotoxicity in AC16 cells as described in [14].
Glutaraldehyde cross-linking
Was performed as described in [19]. The samples were analyzed in 15% SDS-PAGE and stained with Coomassie Blue.
Analytical Ultracentrifugation
Sedimentation velocity profiles were obtained on a temperature-controlled Beckman Coulter XL-I analytical ultracentrifuge equipped with an An-50 Ti rotor and a photoelectric scanner. Data at 280 nm were collected at a rotor speed of 50,000 rpm in a continuous mode at 37°C, with a step size of 0.003 cm. The program SEDFIT was used to analyze the data using c(s) distributions.
Size-Exclusion Chromatography by FPLC
The samples to be analyzed were filtered through a 0.22 μm filter and analyzed on a calibrated 10/30 analytical column Superdex 75 in an Akta FPLC (GE Biosciences) at 4°C. The running buffer was 10 mM sodium phosphate pH 7.6, 100 mM KCl, 1 mM EDTA.
RESULTS
Human Cardiomyocytes are sensitive to TTR variants that deposit in human heart
Human cardiac AC16 cells were treated with several recombinant TTR variants at concentrations ranging from 2 to 16 μM (normal human TTR plasma concentration is 3–7 μM) for 24 h and cell viability was measured by resazurin reduction assay [18]. TTR variants associated with familial amyloid cardiomyopathy [10; 11; 20; 21] (V122I TTR, V30M TTR, V20I TTR and L111M TTR) were toxic to the cardiomyocytes in a concentration-dependent manner, whereas the T119M TTR variant, which is stable and non amyloidogenic [22; 23], was not (Fig 1). Thus, it appears that in AC16 cells, TTR proteotoxicity correlates with its amyloidogenic potential.
Fig 1. Amyloidogenic TTR variants are cytotoxic to the human cardiac cell line AC16.
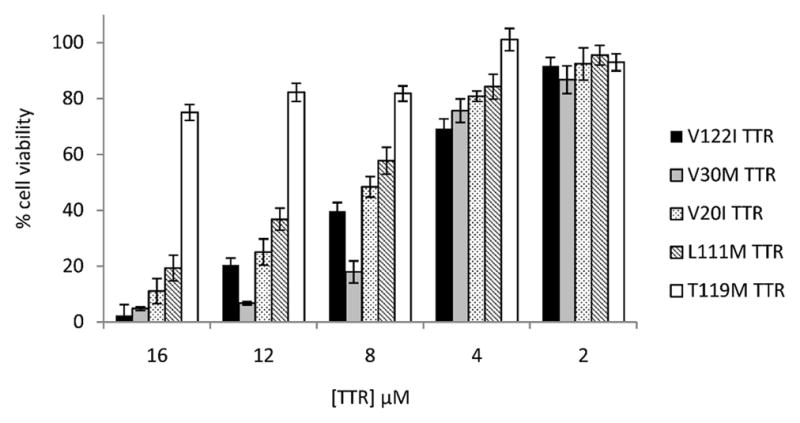
AC16 cells were treated with several concentrations of amyloidogenic (V122I TTR, black bars; V30M TTR, grey bars, V20I TTR, dotted bars, L111M TTR, dashed bars) or non-amyloidogenic TTR (T119M TTR, white bars) for 24h. Cell viability was measured by resazurin reduction assay and compared to cells treated with vehicle only (100% cell viability). Amyloidogenic TTR variants decrease cell viability in a concentration-dependent manner, whereas T119M TTR is not toxic to the cells.
Consistent with previous observations [19; 24; 25], WT TTR purified at room temperature was not toxic to AC16; however, when it was purified at 4°C, WT TTR was toxic to the cells in a concentration-dependent manner. In contrast, the amyloidogenic V122I TTR variant was cytotoxic to AC16 cells regardless of the temperature of purification (Supplemental Figure S1). In this work we used both WT TTR and V122I TTR purified at 4°C as cytotoxic insults to human cardiac AC16 cells.
In agreement with our previous observations in the FAP tissue culture model system, large soluble aggregates and TTR fibrils prepared as in [19] were not cytotoxic to AC16 cells (Supplemental Figure S2).
TTR-induced cytotoxicity in cardiac cells can be prevented with resveratrol (1) and its analogs
We used the TTR cardiac amyloidosis AC16 tissue culture model to test a library of compounds for their capacity to prevent TTR-induced cytotoxicity. In vitro studies have shown that the plant polyphenol resveratrol (1) is a potent and selective inhibitor of TTR aggregation [26; 27]. The use of resveratrol (1) as a possible pharmacologic agent is limited by its poor bioavailability, as it is rapidly metabolized to inactive glucoronate and sulfonate derivatives [28]. Thus, resveratrol analogs with better bioavailability are potential candidates as pharmacologic agents for the treatment of SSA and FAC.
We selected 23 compounds, 19 of which are extremely potent inhibitors of acid-mediated TTR aggregation in vitro [13; 14]. Furthermore, they are able to bind selectively to TTR in ex-vivo human plasma and show low binding affinity for the T4 receptor.
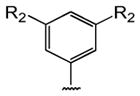
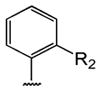
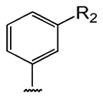
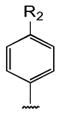
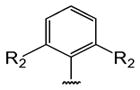
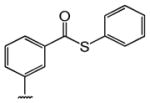
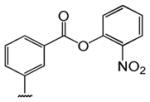
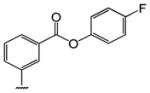
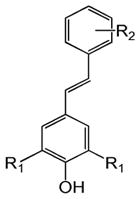
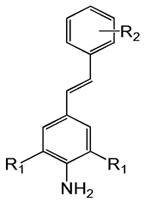
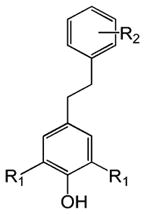
Table 1 shows the structures of resveratrol (1) and the analogs tested and the percentage of acid-mediated aggregation/amyloid formation of WT TTR in the presence of 2 molar equivalents of each compound in vitro, relative to WT TTR alone (100%). Compounds 1–18 are classical stilbenes with an unsaturated linker between the two aromatic rings, whereas compounds 19–23 are dihydrostilbene derivatives. Of the stilbene set, compounds 13–16 are esters or thioesters which act as leaving groups when the TTR ε-NH2 group of Lys15 –situated in the T4 binding pocket- attacks the carbonyl affording covalent kinetic stabilizers of the tetrameric state [13]. Compounds 17–18 have structures similar to 14–16, but do not bind covalently to TTR. Finally, compounds 9–12 are very poor inhibitors of acid-mediated fibril formation in vitro (Table 1) [14].
Table 1.
Structures of resveratrol (1) and resveratrol analogs tested for their capacity to prevent TTR-induced cytotoxicity in AC16 cells. Between parentheses is shown the compound number and between brackets are shown the values of acid-mediated WT TTR fibril formation in an in vitro assay measured by turbidity. 100% fibril formation corresponds to values of turbidity of WT TTR alone; 0% indicates no turbidity was detected thus, total inhibition of TTR amyloid fibril and aggregate formation. The details of the assay and most of the fibril formation data have been previously published and are shown here for reference.
| R1 | R2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
||
|
| |||||||||
|
H | OH (1) [6%] | |||||||
|
| |||||||||
| Br | -CH3 (2) [0%] | -OCH3 (3) [0%] | -OCH3 (4) [0%] | -OCH3 (5) [0%] | (13%) [2%] | ||||
| -Cl (9) [45%] | -NH2 (6) [0%] | -NH2 (7) [0%] | -NH2 (8) [0%] | ||||||
| -Br (10) [61%] | |||||||||
| -CF3 (11) [40%] | |||||||||
|
| |||||||||
| -CH3 | -COOH (17) [5%] | -H (18) [5%] | (14) [2%] | (15) [4%] | (16) [1%] | ||||
|
| |||||||||
|
-CH3 | -NO2 (12) [94%] | |||||||
|
| |||||||||
|
H | OH (19) [15%] | |||||||
|
| |||||||||
| -OCH3 (20) [1%] | -OCH3 (21) [1%] | ||||||||
| Br | -NH2 (22) [1%] | -NH2 (23) [2%] | |||||||
The results indicate that most of the compounds are excellent inhibitors of V122I TTR and WT TTR induced cytotoxicity (Fig 2, and supplemental Figs S3–S5). Figure 2A shows cell viability of AC16 cells exposed to V122I TTR pre-incubated with resveratrol (1) and compounds 2–8 which completely prevent V122I TTR-induced cytotoxicity. None of the compounds was cytotoxic to the cells or induced cell proliferation (supplemental Fig S3).
Fig 2. Resveratrol (1) and its analogs prevent V122I TTR and WT TTR-induced cytotoxicity in cardiac AC16 cells.

AC16 cells were treated V122I TTR or WT TTR pre-incubated with the indicated compounds for 24h. Cell viability was measured by resazurin reduction assay (grey bars). The final concentration of TTR was 8 μM; the final concentration of compounds were 8 μM (2A–2C); and 8 μM (grey bars) or 4 μM (white bars) (2D). The first bar in each graph (in black) represents cell viability of the corresponding TTR with no added compound. 2A) Resveratrol (1) and compounds 2–8 prevent V122I TTR-induced cytotoxicity. 2B) Covalent kinetic stabilizers (13–16) and their non-covalent counterparts (17–18) prevent WT TTR-induced cytotoxicity. 2C) Resveratrol and analogs with (1, 3, 5, 7 and 8) or without (19–23) unsaturated linker between the two aromatic rings display similar capacity to prevent WT TTR-induced cardiotoxicity. 2D) Poor inhibitors of acid-mediated TTR aggregation in vitro (9–12) and the positive control resveratrol (1) were tested for their capacity to prevent V122I TTR-induced toxicity to AC16 cells.
Resveratrol analogs that bind covalently to TTR (13–16) and their non-covalent counterparts (17–18) are also very good inhibitors of WT TTR induced cytotoxicity (Fig 2B). Although the former seem to be better kinetic stabilizers of the tetrameric state as observed in the acid-mediated aggregation assay in vitro (Table 1) [13], in tissue culture both classes of compounds are equally good inhibitors of TTR-induced cardiotoxicity.
Some of the compounds (1, 3, 5, 7–8) and their corresponding dihydrostilbene analogs (19–23) were tested side by side for their capacity to prevent WT TTR (Fig 2C) and V122I TTR (supplemental Fig S4) induced cytotoxicity. Both types of compounds, stilbenes and dihydrostilbenes are excellent inhibitors of cytotoxicity and none except compound 23 was toxic to the cells (supplemental Fig S3).
Interestingly, compounds 9–11, which in the acid-mediated aggregation assay in vitro allow as much as 40–61% WT TTR fibril formation (Table 1), were very good inhibitors of TTR-induced toxicity in the cell assay. They have similar inhibitory capacity to that of resveratrol (1) both at 1 and 0.5 molar equivalents with respect to TTR (Fig 2D). Only compound 12 was unable to prevent TTR-induced cytotoxicity. Similar results were obtained when compounds 9–12 and resveratrol (1) were tested in a setting in which the pre-incubation step between compounds and TTR for 24h prior to addition to the cells, was omitted (supplemental Fig S5).
Resveratrol (1) and its analogs have at least a dual mode of preventing TTR-cytotoxicity
It is known that resveratrol (1) and its analogs bind in the TTR T4 binding pocket and kinetically stabilize the native tetrameric state by raising the energetic barrier for TTR dissociation [13; 14]. We tested whether these compounds could prevent TTR toxicity by other mechanisms. We used an engineered monomeric variant of TTR, M-TTR (F87M/L110M TTR) in which the substitution of two residues situated at the dimer-dimer interfaces (Phe87 and Leu110) by Met prevents tetramerization by means of steric hindrance. The resulting variant has identical tertiary structure as wild-type TTR subunits and remains monomeric at the concentrations used in the experiments [17].
Monomeric M-TTR was cytotoxic to AC16 cells. Interestingly, its cytotoxicity can be prevented by resveratrol (1) and the analogs tested (Fig 3A). Thus, the compounds not only stabilize the native tetramer, but they are able to prevent TTR induced cytotoxicity in some other way. We investigated whether these compounds had an effect on the quaternary structure of TTR, i.e. whether they induce the tetramerization of the initial monomer.
Fig 3. Resveratrol (1) and its analogs prevent monomeric M-TTR-induced cytotoxicity in cardiac AC16 cells by modifying M-TTR quaternary structure.

A) AC16 cells were treated with mixtures of monomeric M-TTR (16 μM, i.e. 4 μM tetramer equivalent) and compounds 1–8 (4 μM). Cell viability was measured by resazurin reduction. B) M-TTR and compounds 1–8 were incubated for 1 day under the same conditions as in the cell-based assay. After cross-linking with glutaraldehyde the solutions were analyzed by SDS-PAGE and stained with Coomassie Blue. Left lane: M-TTR, time 0; lane 2: MW standards (defined at the left of the gel); lane 3: M-TTR time 1 day; lanes 4–11: mixtures of M-TTR with compounds, time 1 day. Arrowheads (right) indicate the migration of M, monomer, D, dimer, T, tetramer and A, soluble aggregates.
Solutions of M-TTR and selected compounds were incubated under the same conditions as in the cell assay. The solutions were then analyzed by SDS-PAGE after cross-linking with glutaraldehyde, which prevents dissociation of the quaternary structures in solution (Fig 3B). M-TTR incubated with compounds 2–8 partitions between monomeric and tetrameric states, demonstrating that these compounds can induce tetramerization of the monomer. As a result, the amount of monomeric TTR remaining in solution is not sufficient to produce significant cytotoxic species. Interestingly, resveratrol (1) promoted the oligomerization of the protein as seen by the appearance of a high molecular weight band at the top of the gel.
To confirm this apparent compound-induced structural quaternary conversion, M-TTR solutions (M-TTR alone, with resveratrol (1) and with compounds 5, 7, and 8) were analyzed by sedimentation velocity experiments using analytical ultracentrifugation (AUC) (Table 2). The data show that resveratrol (1) induced the formation of large soluble aggregates whereas its analogs promoted the tetramerization of the monomer.
Table 2.
Quantitation of TTR species in solution by sedimentation velocity analytical ultracentrifugation. Monomeric M-TTR samples (32 μM, i.e. 8 μM tetramer equivalent) were pre-incubated with or without compounds (1, 5, 7 and 8) at 8 μM, for 24 h at 37°C before analysis. Root mean square deviations (rmsd) are shown to demonstrate the goodness of the fit to the experimental data.
| % species in solution
|
Fit rmsd | ||||
|---|---|---|---|---|---|
| monomer | dimer | tetramer | soluble aggregates | ||
| M-TTR | 86 | 13 | 0.0078 | ||
| M-TTR+1 | 54 | 17 | 7 | 22 | 0.0132 |
| M-TTR+5 | 46 | 51 | 2 | 0.0117 | |
| M-TTR+7 | 40 | 51 | 9 | 0.0078 | |
| M-TTR+8 | 33 | 8 | 54 | 5 | 0.0077 |
Finally, M-TTR solutions (M-TTR alone, with resveratrol (1) and with compounds 5, 7, and 8) were analyzed by FPLC using a calibrated Superdex 75 column, which can resolve large soluble aggregates (void volume, 7.8 ml), from tetrameric TTR (9.2 mL) and monomeric TTR (12.2 mL) (Fig 4). Integration of the peak areas in the chromatograms demonstrated that the mass balance was conserved over time (not shown). The data from these analyses confirm those obtained by SDS-PAGE and AUC, i.e. resveratrol (1) promotes the formation of large soluble TTR aggregates (Fig 4B) whereas its analogs, represented by compound 5, promote the tetramerization of the initial monomer (Fig 4C).
Fig 4. Analysis of M-TTR species in solution by gel filtration.

Mixtures of the monomeric M-TTR (16 μM, i.e. 4 μM of tetramer equivalent) and compounds 1 and 5 (4μM) were incubated for 24h at 37°C under the same conditions as in the cell-based assay. Samples were analyzed at time 0 and time 24h by gel filtration (Superdex 75). Chromatograms are shown for A) M-TTR, B) M-TTR + resveratrol (1), and C) M-TTR + 5. Dashed traces: time 0; solid traces: time 24h.
DISCUSSION
While it seems clear that amyloid TTR deposition in peripheral nerves and hearts plays a mechanical role in reducing organ function [29], recent data in cell systems, transgenic mice and human biopsies of peripheral nerves suggest that oligomeric intermediates, which may or may not be on pathway to amyloid fibril formation, produce tissue damage well before tissue deposition can be observed [19; 30; 31]. Furthermore, autopsies from patients carrying the V122I TTR mutation show myocyte atrophy and myocardial replacement by TTR deposits [32; 33] suggesting that cell loss play a role in the disease progression.
Establishing a relevant tissue culture model for the TTR cardiac amyloidoses is important because it is a tool to probe the molecular mechanisms of the disease onset. A tissue culture system is advantageous in that it allows exploring the direct effects of misfolding proteins on a given target without interference in the analysis from neighboring cells or infiltrating non-target cells responding to local damage.
In the present study, we show that the AC16-based human cardiac tissue culture system is sensitive to externally applied amyloidogenic TTR variants related to TTR cardiac amyloidoses, but not to non-amyloidogenic TTR (Fig 1). Moreover, AC16 cells are not sensitive to amyloid fibrils or large soluble aggregates (Fig S2). Stabilization of the native tetrameric state using small molecules, which bind in the TTR T4 binding pocket, prevents cytotoxicity, indicating that the tetramer itself is not toxic to the cells. Thus, as in the previously described FAP model system, monomeric TTR species or small oligomeric forms are responsible for the observed cytotoxicity [19]. The mechanism of amyloidogenic TTR-induced cardiac cell death includes caspase 3/7 activation and superoxide formation (Bourgault and Reixach, in preparation). We have used the cardiac tissue culture model system to screen 22 compounds, analogs of the polyphenol resveratrol (1), for their capacity to prevent TTR-induced cytotoxicity (Fig 2). Resveratrol (1) and its analogs not only stabilize the native tetramer but they are able to modify the quaternary structure of monomeric TTR in solution, by accelerating M-TTR aggregation (resveratrol (1)) or by inducing tetramer formation (analogs 2–8) (Figs 3–4). In both cases, there is a decrease in the concentration of the monomeric TTR in solution, the precursor of the cytotoxic species, resulting in the prevention of cell damage. The observed differential mechanisms of action might be related to how these small molecules interact with TTR. For instance, the crystal structures show that while resveratrol (1) binds to TTR with the p-hydroxyphenyl ring buried in the innermost cavity of the T4 pocket [26], compound 8 can bind in both directions [14]. It is not known how compounds 2–7 bind to TTR but structural studies found the 3,5-dibromo-4-hydroxyphenyl ring in the outermost region of the T4 cavity in 90% of the structures solved [14].
Interestingly, weak inhibitors of acid-mediated TTR fibril formation in vitro (9, 10, 11 and 19) were good inhibitors of TTR-induced cytotoxicity in cell culture, even when they were not pre-incubated with TTR prior to cell treatment (Fig 2C–D and Supplemental Fig S5). This discrepancy was also noted in an FAP model system using analogs of the non-steroidal anti-inflammatory drug diflunisal [34]. Thus it seems that the selection for active compounds in the acid-mediated fibril formation assay is more stringent than in the tissue culture system. It is possible that the conformation that tetrameric TTR adopts under the conditions of the acid-mediated fibril formation assay is different than at physiological pH, and the small molecule binding in the TTR T4 pocket differs under the two sets of conditions. Alternatively, it could be that compounds 9–11 and 19 prevent amyloidogenic TTR cytotoxicity by neutralizing the cellular mechanisms of cell death. The study of the direct effects of small molecules on the target cells is currently under investigation.
The tissue culture system not only presents a more physiologic setting than the TTR acid-mediated fibril formation assay, but it also provides a fast read-out of the intrinsic cytotoxicity of the compounds tested. It has recently been reported that sulfated glycosaminoglycans accelerate TTR amyloidogenesis in vitro [35; 36]. This cardiac tissue culture system should be a useful tool to understand how several components of the extracellular matrix affect TTR cytotoxicity under physiological conditions. Moreover, the tissue culture system has the potential to screen compounds that do not necessarily affect the quaternary structure of TTR but prevent toxicity by direct interaction with the target cells.
In summary, we present a robust tissue culture model system for the TTR cardiac amyloidoses which reflects many aspects of the human clinical syndromes. We have shown that amyloidogenic TTR induces cardiotoxicity, which can be prevented with small molecules that stabilize and promote the formation of tetrameric TTR or accelerate the formation of large non-toxic soluble aggregates. This model should facilitate the investigation of basic aspects of TTR misfolding diseases and their prevention as well as drug discovery.
Supplementary Material
Highlights.
We have established a human cardiac tissue culture model system to study the transthyretin cardiac amyloidoses.
Amyloidogenic transthyretin variants, but not a stable and non-amyloidogenic variant, decrease cardiac cell viability.
Resveratrol analogs prevent amyloidogenic transthyretin-induced cardiotoxicity
Resveratrol analogs stabilize the transthyretin tetrameric native state and modify the quaternary structure of the monomeric state.
Acknowledgments
We thank Dr Mercy Davidson for developing and providing the human cardiac AC16 cells.
FUNDING
This work was supported by the American Heart Association (award 0865061F to N.R.). Additional support from the National Institutes of Health (AG030027 to J.N.B.) is acknowledged. The synthesis of the stilbenes used in this work was supported by the NIH (DK46335 to J.W.K). S.B. is a recipient of a postdoctoral fellowship from the Fonds de la Recherche en Santé du Québec (FRSQ).
Footnotes
The abbreviations used in this manuscript are: AUC, Analytical ultracentrifugation; FAC, Familial Amyloid Cardiomyopathy; FAP, Familial Amyloidotic Polyneuropathy; SSA, Senile Systemic Amyloidosis; TTR, Transthyretin; T4, Thyroxine.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Buxbaum JN. The Amyloidoses. In: Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors. Rheumatology. Elsevier; London: 2008. pp. 1671–1681. [Google Scholar]
- 2.Buxbaum JN. The systemic amyloidoses. Curr Opin Rheumatol. 2004;16:67–75. doi: 10.1097/00002281-200401000-00013. [DOI] [PubMed] [Google Scholar]
- 3.Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. A primer of amyloid nomenclature. Amyloid. 2007;14:179–183. doi: 10.1080/13506120701460923. [DOI] [PubMed] [Google Scholar]
- 4.Baures PW, Peterson SA, Kelly JW. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg Med Chem. 1998;6:1389–1401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 5.Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Abeta toxicity. Proc Natl Acad Sci U S A. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buxbaum JN, Reixach N. Transthyretin: the servant of many masters. Cell Mol Life Sci. 2009;66:3095–3101. doi: 10.1007/s00018-009-0109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 8.Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 9.Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 2003;10:160–184. doi: 10.3109/13506120308998998. [DOI] [PubMed] [Google Scholar]
- 10.Jacobson DR, Pan T, Kyle RA, Buxbaum JN. Transthyretin ILE20, a new variant associated with late-onset cardiac amyloidosis. Hum Mutat. 1997;9:83–85. doi: 10.1002/(SICI)1098-1004(1997)9:1<83::AID-HUMU19>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 11.Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, Buxbaum JN. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Eng J Med. 1997;336:466–473. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 12.Davidson MM, Nesti C, Palenzuela L, Walker WF, Hernandez E, Protas L, Hirano M, Isaac ND. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:133–147. doi: 10.1016/j.yjmcc.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 13.Choi S, Connelly S, Reixach N, Wilson IA, Kelly JW. Chemoselective small molecules that covalently modify one lysine in a non-enzyme protein in plasma. Nat Chem Biol. 2010;6:133–139. doi: 10.1038/nchembio.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi S, Reixach N, Connelly S, Johnson SM, Wilson IA, Kelly JW. A substructure combination strategy to create potent and selective transthyretin kinetic stabilizers that prevent amyloidogenesis and cytotoxicity. J Am Chem Soc. 2010;132:1359–1370. doi: 10.1021/ja908562q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson SM, Connelly S, Wilson IA, Kelly JW. Toward optimization of the linker substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem. 2008;51:6348–6358. doi: 10.1021/jm800435s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White JT, Kelly JW. Support for the multigenic hypothesis of amyloidosis: the binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc Natl Acad Sci U S A. 2001;98:13019–13024. doi: 10.1073/pnas.241406698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang X, Smith CS, Petrassi HM, Hammarstrom P, White JT, Sacchettini JC, Kelly JW. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40:11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- 18.O’Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 19.Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci U S A. 2004;101:2817–2822. doi: 10.1073/pnas.0400062101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikeda S, Nakazato M, Ando Y, Sobue G. Familial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneity. Neurol. 2002;58:1001–1007. doi: 10.1212/wnl.58.7.1001. [DOI] [PubMed] [Google Scholar]
- 21.Ranlov I, Alves IL, Ranlov PJ, Husby G, Costa P, Saraiva MJM. A Danish kindred with familial amyloid cardiomyopathy revisited: identification of a mutant transthyretin-methionine111 variant in serum from patients and carriers. Am J Med. 1992;93:3–8. doi: 10.1016/0002-9343(92)90672-x. [DOI] [PubMed] [Google Scholar]
- 22.Coelho T, Carvalho M, Saraiva M, Alves I, Almeida MR, Costa PP. A strikingly benign evolution of FAP in an individual found to be a compound heterozygote for two TTR mutations: TTR MET 30 and TTR MET 119. J Rheumatol. 1993;20:179. [Google Scholar]
- 23.Hammarstrom P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293:2459–2462. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 24.Lindhagen-Persson M, Vestling M, Reixach N, Olofsson A. Formation of cytotoxic transthyretin is not dependent on inter-molecular disulphide bridges commonly found within the amyloid form. Amyloid. 2008;15:240–245. doi: 10.1080/13506120802524916. [DOI] [PubMed] [Google Scholar]
- 25.Sorgjerd K, Klingstedt T, Lindgren M, Kagedal K, Hammarstrom P. Prefibrillar transthyretin oligomers and cold stored native tetrameric transthyretin are cytotoxic in cell culture. Biochem Biophys Res Commun. 2008;377:1072–1078. doi: 10.1016/j.bbrc.2008.10.121. [DOI] [PubMed] [Google Scholar]
- 26.Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, Sacchettini JC. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 2000;7:312–321. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 27.Purkey HE, Dorrell MI, Kelly JW. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc Natl Acad Sci U S A. 2001;98:5566–5571. doi: 10.1073/pnas.091431798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wenzel E, Somoza V. Metabolism and bioavailability of trans-resveratrol. Mol Nutr Food Res. 2005;49:472–481. doi: 10.1002/mnfr.200500010. [DOI] [PubMed] [Google Scholar]
- 29.Hongo M, Kono J, Yamada H, Misawa T, Tanaka M, Nakatsuka T, Kinoshita O, Okubo S, Sekiguchi M. Doppler echocardiographic assessments of left ventricular diastolic filling in patients with amyloid heart disease. J Cardiol. 1991;21:391–401. [PubMed] [Google Scholar]
- 30.Sousa MM, Cardoso I, Fernandes R, Guimaraes A, Saraiva MJ. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am J Pathol. 2001;159:1993–2000. doi: 10.1016/s0002-9440(10)63050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sousa MM, Fernandes R, Palha JA, Taboada A, Vieira P, Saraiva MJ. Evidence for early cytotoxic aggregates in transgenic mice for human transthyretin Leu55Pro. Am J Pathol. 2002;161:1935–1948. doi: 10.1016/S0002-9440(10)64469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacobson DR, Ittmann M, Buxbaum JN, Wieczorek R, Gorevic PD. Transthyretin Ile 122 and cardiac amyloidosis in African-Americans. 2 case reports. Tex Heart Inst J. 1997;24:45–52. [PMC free article] [PubMed] [Google Scholar]
- 33.Hamour IM, Lachmann HJ, Goodman HJ, Petrou M, Burke MM, Hawkins PN, Banner NR. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant. 2008;8:1056–1059. doi: 10.1111/j.1600-6143.2008.02162.x. [DOI] [PubMed] [Google Scholar]
- 34.Reixach N, Adamski-Werner SL, Kelly JW, Koziol J, Buxbaum JN. Cell based screening of inhibitors of transthyretin aggregation. Biochem Biophys Res Commun. 2006;348:889–897. doi: 10.1016/j.bbrc.2006.07.109. [DOI] [PubMed] [Google Scholar]
- 35.Bourgault S, Solomon JP, Reixach N, Kelly JW. Sulfated glycosaminoglycans accelerate transthyretin amyloidogenesis by quaternary structural conversion. Biochemistry. 2011;50:1001–1015. doi: 10.1021/bi101822y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noborn F, O’Callaghan P, Hermansson E, Zhang X, Ancsin JB, Damas AM, Dacklin I, Presto J, Johansson J, Saraiva MJ, Lundgren E, Kisilevsky R, Westermark P, Li JP. Heparan sulfate/heparin promotes transthyretin fibrillization through selective binding to a basic motif in the protein. Proc Natl Acad Sci U S A. 2011;108:5584–5589. doi: 10.1073/pnas.1101194108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.