

Sultams (cyclic sulfonamides) are an important class of molecules that exhibit widespread biological activity against a variety of biological targets.[1] In particular, the benzoxaze-pine-1,1-dioxide motif has exhibited activity towards a variety of targets including: histone deacetylase inhibition (for treatment of cognitive disorders such as Alzheimer’s disease),[2] glucokinase activation,[3] serotonin 5-HT2C activation,[4] modulation of the histamine H3 receptor,[5] inhibition of MDM2-p53,[6] inhibition of sodium–proton exchange, bradykinin B1 receptor antagonism (for treating Alzheimer’s disease),[7] AMPA receptor agonism,[8] and inhibition of metalloproteinase.[9] As a consequence, there is interest in the development of general strategies for the synthesis of organized collections (libraries) of these compounds to serve as probes for biological pathways and as potential therapeutic agents.
Diversity-oriented synthesis (DOS)[10,11] has emerged as an effective strategy to synthesize structurally varied compound collections. The development of strategies that allow the production of versatile core scaffolds in a highly efficient manner is crucial for DOS-based strategies. The advent of rational DOS whereby ‘privileged structures’ are utilized as core scaffolds in DOS approaches has presented opportunities in this regard.[12] The desire to utilize benzofused sultam cores for DOS approaches has prompted the investigation of strategies that would allow facile access to these scaffolds. In this regard α-fluorobenzenesulfonyl chlorides represent a versatile building block synthon. The electron-withdrawing SO2 moiety imparts enhanced electrophilicity at the β-carbon atom, and α-fluorobenzenesulfonyl chlorides may therefore be viewed as bis-electrophiles. Pairing of these bis-electrophilic synthons with amino alcohols (bis-nucleophiles) in a complementary fashion would allow access to benzofused sultams bearing a secondary sulfonamide in a highly efficient manner. The reaction potential of this versatile functionality can be harnessed for functionalization, while placement of halides on the aromatic ring allows either SNAr or metal-catalyzed cross-coupling pathways of diversification; stereochemical and skeletal diversity can be achieved by utilizing a variety of chiral amino alcohols (Figure 1). Additionally, the ability to functionalize around the periphery of the molecule along with access to stereo-chemical diversity allows maximal interactions with macromolecules within biological systems.
Figure 1.

Pairing strategy to benzofused sultams.
The use of an intramolecular SNAr O-arylation route to benzothiazepine-1,1-dioxides containing a secondary sulfonamide was explored. Investigations commenced with the nucleophilic substitution of the sulfonyl chloride (first Click reaction) by a variety of 1,2-amino alcohols by employing modified Schotten–Bauman conditions. Accordingly, addition of α-fluorobenzenesulfonyl chloride to a vigorously stirring CH2Cl2/H2O biphasic system of 1,2-amino alcohols in the presence of NaHCO3 furnished the β-hydroxy α-fluorobenzene sulfonamides in excellent yields. These products were thereafter subjected to microwave irradiation at 140°C for 30 min in DMF in the presence of Cs2CO3 to produce a variety of chiral benzothiaoxazepine-1,1-dioxides in excellent yields (Table 1).
Table 1.
Click–cyclize protocol to diverse benzothiaoxazepine-1,1-dioxides.
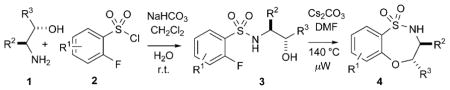 | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Sulfonamide (yield [%])[a] | Sultam (yield [%]) [a] |
| 1 | 4-Br | Ph | H | 3a (93) | 4a (89) |
| 2 | 4-Br | iPr | H | 3b (96) | 4b (87) |
| 3 | 4-Br | iBu | H | 3c (97) | 4c (94) |
| 4 | 4-Br | Bn | H | 3d (89) | 4d (83) |
| 5 | 4-Br | Me | Ph | 3e (92) | 4e (95) |
| 6 | 6-F | iBu | H | 3 f (93) | 4 f (88) |
| 7 | 6-F | Me | Ph | 3g (94) | 4g (92) |
| 8 | 4-F | iBu | H | 3h (95) | 4h (93) |
Yields of isolated product after column chromatography.
With a successful two-step protocol in hand, we next turned our attention to the quantity of final core structure that could be produced. As the building blocks still have to undergo two further diversification steps, it would be necessary to produce these scaffolds on (at least) a 5-gram scale to allow larger library synthesis. The cyclization step worked best (cleanest and highest yielding) when performed under microwave heating while keeping the concentration of 3 at 0.1M. Given the vessel size limitation imposed by the Biotage Initiator Microwave Synthesizer, and the relatively dilute reaction conditions, the only way to obtain the desired quantity of 4 would be to run the reaction repeatedly on small scale in batches and pool the product. Not only is this undesirable, but it dramatically increases the handling and consumables required (e.g., high-pressure reaction tubes) and occupies the microwave synthesizer for prolonged periods (days) of time just to obtain one single scaffold. Movement of the cyclization step (at least) to a flow platform would remove the limitations imposed by scale up. With this goal in mind, we set out to employ a microwave-assisted, continuous flow organic synthesis (MACOS) platform for the scale-up synthesis of sultam 4.
Conducting chemical synthesis in a moveable (flowing) platform offers several significant advantages that make its implementation desirable from a strategic point of view.[13,14] The compartmentalization of starting materials, reagents, and/or catalysts from the site of reaction minimizes their contact with the final product, which, as a result, minimizes side reactions that reduce yield and complicate purification. This means that flowed synthesis is controllable both in terms of space and time. Keeping reactants separate, providing that they are stable in their reservoir (e.g., in a syringe pump or in a larger tank) for extended periods of time, means that the same level of conversion is sustainable, in principle, indefinitely. This means that only a single drop of product mixture is necessary to ascertain how a given set of reaction conditions are going to perform, dramatically reducing the time and waste associated with process optimization. Once a desired set of conditions has been realized, then all that is necessary is to simply collect the effluent from the reaction tube for as long as is necessary to accrue the desired quantity of product. This approach to raising larger amounts of product is termed ‘scaling out’, rather than the conventional ‘scaling up’ process chemistry that has historically dominated the production of fine chemicals. Of course, this is only useful if the reaction proceeds to a high level of conversion; to meet these criteria, microwave irradiation can be applied to the flowing reactant stream to help drive the reaction to completion during the time that any plug of reactant spends flowing through the reactor.
We next sought to optimize the MACOS process for the multi-gram preparation of 4 keeping in mind the brief residence time in the flow tube, while addressing the heterogeneous reaction conditions in Table 1 that are not ideal for flow. To begin, we examined the cyclization of homogeneous mixtures in batches, where the reaction was heated for 60 s to simulate the approximate time that any single plug of moving reaction stream would be receiving microwave irradiation during flow (Table 2).
Table 2.
Optimization of base using batch microwave.[a]
| Entry | Base | Conversion [%] |
|---|---|---|
| 1 | Et3N | decomposition |
| 2 | 2,6-lutidine | no reaction |
| 3 | Cs2CO3 | 80[b] |
| 4 | (CH3)3SiOK | 87 |
| 5 | tBuOK | 100 |
Reaction of 3 to 4 (R1=4-Br, R2=Ph, R3=H) was performed as outlined in Table 1 using DMF at 60°C.
Solvent was DMF–H2O, 3:1.
While amine-derived bases proved ineffective (entries 1 and 2, Table 2),[15] Cs2CO3 solubilized in DMF/water provided reasonable conversion to 4 (entry 3). Potassium silanoxide (entry 4) worked nicely and full conversion was achieved with tBuOK (entry 5); both bases were fully soluble in DMF. This proved that cyclization was a very efficient transformation and the optimized conditions using DMF and tBuOK base could be applied to flow.
Under the conditions given in Table 3, the above solvent and base indeed led to excellent conversion to product using MACOS (entry 1, Table 3). However, while the crude mass was as expected and the crude NMR spectra showed the presence of what appeared to be only product, recovery of 4i was disappointing. With this knowledge, we explored a variety of other solvents (entries 2–4, Table 3) to improve recovery and DMSO provided consistently the best mass balance of product. Moving forward with these conditions, a collection of 19 sultam building blocks was obtained in 5–10 gram quantities each.
Table 3.
Final solvent optimization for MACOS preparation of sultam (4) sublibrary.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R2 | X | Solvent | Time of run (min) | Conversion [%] | Yield [%][a] | Product (grams produced) |
| 1 | Me | 4-Br | DMF | 25 | 100 | 53 | 4i (R) (0.1) |
| 2 | Me | 4-Br | DMA | 25 | 100 | 52 | 4i (R) (0.1) |
| 3 | Me | 4-Br | NMP | 25 | 100 | 49 | 4i (R) (0.09) |
| 4 | Me | 4-Br | DMSO | 25 | 100 | 78 | 4i (R) (0.15) |
| 5 | Me | 4-Br | DMSO | 120 | – | 80 | 4i (R) (6.65) |
| 6 | Me | 4-Br | DMSO | 120 | – | 75 | 4i (S) (4.21) |
| 7 | Ph | 4-Br | DMSO | 120 | – | 73 | 4a (R) (2.07) |
| 8 | iBu | 4-Br | DMSO | 120 | – | 80 | 4c (R) (8.53) |
| 9 | iBu | 4-Br | DMSO | 120 | – | 93 | 4c (S) (9.48) |
| 10 | Bn | 4-Br | DMSO | 120 | – | 86 | 4d (R) (9.83) |
| 11 | Bn | 4-Br | DMSO | 120 | – | 85 | 4d (S) (10.1) |
| 12 | Me | 6-F | DMSO | 120 | – | 84 | 4j (R) (7.72) |
| 13 | Me | 6-F | DMSO | 120 | – | 84 | 4j (S) (7.5) |
| 14 | iBu | 6-F | DMSO | 120 | – | 92 | 4 f (R) (9.9) |
| 15 | iBu | 6-F | DMSO | 120 | – | 89 | 4 f (S) (9.5) |
| 16 | Bn | 6-F | DMSO | 120 | – | 79 | 4k (R) (8.4) |
| 17 | Bn | 6-F | DMSO | 120 | – | 87 | 4k (S) (10.57) |
| 18 | Me | 4-F | DMSO | 120 | – | 79 | 4l (R) (8.45) |
| 19 | Me | 4-F | DMSO | 120 | – | 76 | 4l (S) (7.1) |
| 20 | iBu | 4-F | DMSO | 120 | – | 91 | 4h (R) (10.37) |
| 21 | iBu | 4-F | DMSO | 120 | – | 96 | 4h (S) (10.89) |
| 22 | Bn | 4-F | DMSO | 120 | – | 72 | 4m (R) (6.05) |
| 23 | Bn | 4-F | DMSO | 120 | – | 72 | 4m (S) (7.55) |
Yields of isolated product after column chromatography.
In summary, an effective microwave-assisted flow synthesis protocol for the preparation of multi-gram quantities of benzofused sultams has been developed. The production runs to generate each sultam took approximately 2.5 h. Thus, all compounds were obtained in approximately two weeks, including the time required to optimize the flow process. The preparation of the same number and gram quantity of these compounds using a robotically fed batch microwave would have taken more time and consumables. The bulk sultams thus produced will be elaborated into approximately 1000 compounds using a combination of alkylation and substitution or cross-coupling chemistry. This work is ongoing and will be reported in due course.
Supplementary Material
Acknowledgments
This investigation was generously supported by funds provided by the NIH Center for Chemical Methodologies and Library Development at the University of Kansas (P50 GM069663), and NIGMS Pilot-Scale Libraries Program (NIH P41 GM076302).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201001651.
References
- 1.For an extensive list of biologically active sultams, see: Jiménez-Hopkins M, Hanson PR. Org Lett. 2008;10:2223–2226. doi: 10.1021/ol800649n.Rolfe A, Young K, Volp K, Hanson PR. J Comb Chem. 2009;11:732–738. doi: 10.1021/cc900025e.Jeon K, Rayabarappu DR, Rolfe A, Hanson PR. Tetrahedron. 2009;65:4992–5000. doi: 10.1016/j.tet.2009.03.080.Rolfe A, Samarakoon TB, Hanson PR. Org Lett. 2010;12:1216–1219. doi: 10.1021/ol100035e.Samarakoon TB, Hur MY, Kurtz RD, Hanson PR. Org Lett. 2010;12:2182–2185. doi: 10.1021/ol100495w.
- 2.Rogers K, Patzke H. 0,050,545P. US Patent. 2005
- 3.Campbell L, Pike KG, Suleman A, Waring MJ. 050,101. WOPatent. 2008
- 4.Matsumoto T, Kamo I, Nomura I. 8,007,661. WO. 2008
- 5.Santora VJ, Covel JA, Ibarra JB, Semple G, Smith B, Smith J, Weinhouse MI, Schultz JA. 8,097,261. WO. 2008 doi: 10.1021/jm900857n. [DOI] [PubMed]
- 6.Fotouhi N, Haley GJ, Simonsen KB, Vu BT, Webber SE. 6,097,261. WO. 2006
- 7.Askew BC, Jr, Aya T, Biswas K, Cai G, Chen JJ, Fotsch CH, Han N, Human JB, Li A, Liu Q, Peterkin T, Qian W, Riahi B, Yuan CC, Zhu J. 6,036,664. WO. 2006
- 8.Grove SJA, Zhang M, Shahid M. 2,100,865. WO. 2002
- 9.Duan J, Chen L, Cherney RJ, Decicco CP, Voss ME. 1,994,126. WO. 1988
- 10.a) Schreiber SL. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]; b) Schreiber SL. Nature. 2009;457:153–154. doi: 10.1038/457153a. [DOI] [PubMed] [Google Scholar]
- 11.a) Tempest AP, Armstrong WR. J Am Chem Soc. 1997;119:7607–7608. [Google Scholar]; b) Castellano S, Fiji HDG, Kinderman SS, Watanade M, Leon PD, Tamanoi F, Kwon O. J Am Chem Soc. 2007;129:5843–5845. doi: 10.1021/ja070274n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mao S, Probst D, Werner S, Chen J, Xie X, Brummond KM. J Comb Chem. 2008;10:235–246. doi: 10.1021/cc7001843. [DOI] [PubMed] [Google Scholar]; d) Zhou A, Hanson PR. Org Lett. 2009;11:531–534. doi: 10.1021/ol802467f. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Burke MD, Schreiber SL. Angew Chem. 2004;116:48–60. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:46–58. [Google Scholar]; f) Burke M, Berger EM, Schreiber SL. Science. 2003;302:613–618. doi: 10.1126/science.1089946. [DOI] [PubMed] [Google Scholar]; g) Spring DR. Org Biomol Chem. 2003;1:3867–3870. doi: 10.1039/b310752n. [DOI] [PubMed] [Google Scholar]; h) Tan DS. Nat Chem Biol. 2005;1:74–84. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]
- 12.a) An H, Eum SJ, Koh M, Lee SK, Park SB. J Org Chem. 2008;73:1752–1761. doi: 10.1021/jo702196f. [DOI] [PubMed] [Google Scholar]; b) Ko SK, Jang HJ, Kim E, Park SB. Chem Commun. 2006:2962–2964. doi: 10.1039/b606341a. [DOI] [PubMed] [Google Scholar]
- 13.For the most recent review on flow chemistry, see: Razzaq T, Kappe CO. Chem Asian J. 2010;5:1274–1289. doi: 10.1002/asia.201000010.and references therein
- 14.a) Shore G, Yoo WJ, Li CJ, Organ MG. Chem Eur J. 2010;16:126–133. doi: 10.1002/chem.200902396. [DOI] [PubMed] [Google Scholar]; b) Shore G, Organ MG. Chem Eur J. 2008;14:9641–9646. doi: 10.1002/chem.200801610. [DOI] [PubMed] [Google Scholar]; c) Shore G, Organ MG. Chem Commun. 2008:838–840. doi: 10.1039/b715709f. [DOI] [PubMed] [Google Scholar]; d) Shore G, Morin S, Mallik D, Organ MG. Chem Eur J. 2008;14:1351–1356. doi: 10.1002/chem.200701588. [DOI] [PubMed] [Google Scholar]; e) Bremner S, Organ MG. J Comb Chem. 2007;9:14–16. doi: 10.1021/cc060130p. [DOI] [PubMed] [Google Scholar]; f) Shore G, Morin S, Organ MG. Angew Chem. 2006;118:2827–2832. doi: 10.1002/anie.200503600. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:2761–2766. doi: 10.1002/anie.200503600. [DOI] [PubMed] [Google Scholar]; g) Organ MG, Comer E. Chem Eur J. 2005;11:7223–7227. doi: 10.1002/chem.200500820. [DOI] [PubMed] [Google Scholar]; h) Comer E, Organ MG. J Am Chem Soc. 2005;127:8160–8167. doi: 10.1021/ja0512069. [DOI] [PubMed] [Google Scholar]
- 15.In addition to Et3N and lutidine, the following amine bases also showed no conversion to product at 120°C for 3 min: N,N-diisopropylethylamine, quinuclidine, and 1-methylimidazole. DBU (3 equiv) did show limited promise at 180°C providing 69% conversion.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.