

Non-technical summary
The hippocampus is a neural structure that is critical for some forms of memory function. It performs this function through the ability of its neurons to fire patterns of activity that encode information and the ability of the synaptic connections between neurons to strengthen or weaken. Glutamate, an important synaptic neurotransmitter, can activate different types of receptors, including metabotropic glutamate receptors (mGluRs). mGluRs have been shown to be important for learning and memory. It has also been shown that changes in mGluR type 5 might contribute to mental retardation and autism, suggesting that manipulation of mGluR5 might reduce their symptoms. In this study we examined how mGluR activation can activate neuron membrane channels (SK and TRPC) in hippocampal neurons that regulate their activity. Our findings suggest that mGluR activation of SK and TRPC channels are likely to be important for sculpting patterns of activity that encode information by the hippocampus.
Abstract
Abstract
Group I metabotropic glutamate receptors (mGluRs) play an essential role in cognitive function. Their activation results in a wide array of cellular and molecular responses that are mediated by multiple signalling cascades. In this study, we focused on Group I mGluR activation of IP3R-mediated intracellular Ca2+ waves and their role in activating Ca2+-dependent ion channels in CA1 pyramidal neurons. Using whole-cell patch-clamp recordings and high-speed Ca2+ fluorescence imaging in acute hippocampal brain slices, we show that synaptic and pharmacological stimulation of mGluRs triggers intracellular Ca2+ waves and a biphasic electrical response composed of a transient Ca2+-dependent SK channel-mediated hyperpolarization and a TRPC-mediated sustained depolarization. The generation and magnitude of the SK channel-mediated hyperpolarization depended solely on the rise in intracellular Ca2+ concentration ([Ca2+]i), whereas the TRPC channel-mediated depolarization required both a small rise in [Ca2+]i and mGluR activation. Furthermore, the TRPC-mediated current was suppressed by forskolin-induced rises in cAMP. We also show that SK- and TRPC-mediated currents robustly modulate pyramidal neuron excitability by decreasing and increasing their firing frequency, respectively. These findings provide additional evidence that mGluR-mediated synaptic transmission makes an important contribution to regulating the output of hippocampal neurons through intracellular Ca2+ wave activation of SK and TRPC channels. cAMP provides an additional level of regulation by modulating TRPC-mediated sustained depolarization that we propose to be important for stabilizing periods of sustained firing.
Introduction
Group I metabotropic glutamate receptors (mGluRs) are Gq protein-coupled receptors that play an important role in fundamental neural processes, from development to memory (Huber et al. 2000; Kleppisch et al. 2001; Zho et al. 2002; Hayashi et al. 2007; Niswender & Conn, 2010). Not surprisingly, disruption of mGluRs has been associated with cognitive dysfunction and some neuropathological conditions, including epilepsy, mental retardation, schizophrenia, autism and Alzheimer's disease (Merlin & Wong, 1997; Chuang et al. 2001; Lee et al. 2002; Rutecki et al. 2002; Thuault et al. 2002; Dolen et al. 2007). The diversity of mGluR-associated neuronal responses reflects a vast array of cellular and molecular events triggered by multiple signalling cascades.
mGluR activation of Gq proteins leads to the activation of phospholipase C (PLC), which in turn cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) into soluble inositol 1,4,5-triphosphate (IP3) and membrane-bound diacyl glycerol (DAG). The ability of PLC to act as a biochemical manifold can lead to at least three distinct consequences in neurons: (1) membrane depolarization through deactivation of constitutively active PIP2-dependent K+ channels (GIRK and KCNQ) (Suh et al. 2004; Falkenburger et al. 2010), (2) DAG-dependent stimulation of protein kinase C (PKC)-dependent signalling cascades, and (3) IP3 receptor (IP3R)-mediated internal Ca2+ release and subsequent activation of a variety of Ca2+-dependent K+ channels and second messengers (Berridge, 1998).
We have recently shown that mGluR-mediated Ca2+ waves evoke a Ca2+-activated small-conductance K+ (SK) channel-mediated transient hyperpolarization and a Ca2+-dependent non-specific cationic (CAN) channel-mediated sustained depolarization in pyramidal neurons of the medial prefrontal cortex (mPFC) (Hagenston et al. 2008). We and others have proposed that the sustained depolarization mediated by the CAN current (ICAN) contributes to stabilizing the persistent activity that encodes working memory function (Marder et al. 1996; Camperi & Wang, 1998; Wang, 2001; Wyart et al. 2005; Hagenston et al. 2008; Yoshida et al. 2008). Given the important role that ICAN is likely to play in PFC cognitive function, and in light of evidence for a functional significance of mGluR activation and consequent internal Ca2+ release in the hippocampus (Yeckel et al. 1999; Raymond et al. 2000), we examined whether mGluR activation and IP3R-mediated Ca2+ release in general, and ICAN in particular, may play a part in regulating the excitability of hippocampal pyramidal neurons. More specifically, we used whole-cell patch-clamp recordings combined with high-speed Ca2+ fluorescence imaging in brain slices to test whether synaptic and pharmacological stimulation of mGluRs and subsequent IP3R-mediated intracellular Ca2+ waves lead to a hyperpolarization and depolarization via activation of SK and CAN channels, respectively. Consistent with previous findings, the hyperpolarization results from activation of SK channels (Stutzmann et al. 2003; Hagenston et al. 2008). We show that the depolarization is mediated by activation of transient receptor potential C (TRPC) 1, 4 or 5 channel isoforms, but not TRPC3. Furthermore, the TRPC-mediated response is suppressed by rises in intracellular cAMP concentration. These findings support the hypothesis that mGluRs are an important regulator of neuronal excitability in hippocampal pyramidal neurons and suggest a vital role for SK and TRPC channels in glutamatergic synaptic transmission.
Methods
Ethical approval of hippocampal slice preparation
Acute hippocampal slices (350 μm) were extracted from 3- to 8-week-old Sprague–Dawley rats using procedures described elsewhere (Gipson & Yeckel, 2007; Fitzpatrick et al. 2009). All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee at the Yale University School of Medicine.
Recordings and solutions
Whole-cell patch-clamp recordings were made from visualized CA1 pyramidal neurons (n = 101). Slices were continuously perfused with oxygenated artificial cerebral spinal fluid (31–33°C) containing (in mm): 124 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 10 dextrose and viewed through a 40× water-immersion objective using differential interference contrast optics. Recording electrodes (3–5 MΩ) were typically filled with (in mm): 134 KMeSO4, 3.0 KCl, 10 Hepes, 1.0 MgCl2, 4.0 Mg-ATP, 0.5 Na-GTP, 5 K2-phosphocreatine, 5.0 Na2-phosphocreatine, 50 units ml−1 creatine phosphokinase (pH 7.53, ∼ 288 mosmol l−1). In experiments examining current–voltage relationships, electrodes were filled with (in mm): 130 CsOH, 5.0 CsCl, 130 gluconic acid, 4.0 MgATP, 10 phosphocreatine, 0.3 NaGTP, 10 Hepes (pH 7.45, ∼ 290 mosmol l−1). In most experiments, a low-affinity Ca2+ indicator dye was included in the recording pipette (fura-2FF, 200 μm). Where indicated, higher affinity dyes were used (bis-fura-2 or fluo-4, both at 100 μm). Also included in the recording pipette was an inert fluorescent dye (Alexa 488 or Alexa 564, 5–15 μm) for better visualization of neuronal processes. Recordings were made in bridge or discontinuous voltage-clamp mode with an SEC 05L amplifier (npi electronic, Tamm, Germany), digitized and analysed with custom software developed with IGOR Pro (WaveMetrics; Portland, OR, USA). Average resting membrane potential was −62 ± 3 mV (uncorrected for liquid junction potential of ∼11 mV); neurons with a resting membrane potential greater than −50 mV were discarded. Average input resistance was 88.3 ± 5.6 MΩ and whole-cell series resistance was between 10 and 30 MΩ. Unless otherwise stated, cells were held at approximately −65 mV.
Ca2+ fluorescence imaging
Dye fluorescence was imaged using a cooled CCD camera (Quantix 57 or Cascade 57B; Photometrics, Tucson, AZ, USA). Images were collected at 25 or 50 Hz with 5 × 5 or 4 × 4 pixel binning. Dark noise and autofluorescence were subtracted and relative changes in [Ca2+]i were quantified as changes in ΔF/F, where F represents baseline fluorescence intensity before stimulation and ΔF represents the magnitude of fluorescence change following stimulation (see Hagenston et al. 2008). We did not correct for bleaching, which under our recording conditions was typically <3%ΔF/F over 5–10 s of light exposure.
Stimulation of intracellular Ca2+ waves
Intracellular Ca2+ waves were elicited by one of the following methods: (1) electrical stimulation (30–100 pulses at 100 Hz; 0.1 ms duration pulse) of Shaffer collaterals with a glass, ACSF-filled pipette (5–10 μm tip diameter) with a fine tungsten rod glued to its side, (2) brief pressure application (puff; 50–100 ms; 10–20 psi = 67.9–137.9 kPa) of an mGluR agonist DHPG or ACPD (400 μm) applied less than 3 μm from the primary apical dendrite through a 2–5 MΩ patch pipette, or (3) photolysis of 1-(2-nitro-phenyl)ethyl (NPE)-caged IP3 (97 μm) with flashes of UV light (50–500 ms duration) produced by a 100 W mercury lamp (Carl Zeiss, Inc.; Thornwood, NY, USA). The photolysis beam (∼20 μm in diameter) was directed at different locations along the apical dendrite or in the soma using a custom-made fibre optic spot illumination system fitted to the aperture stop port in the epi-illumination pathway of an Olympus BX51WI microscope (Rapp OptoElectronic GmbH, Hamburg, Germany).
Pharmacology/antibodies
The following drugs were obtained from Tocris Bioscience: DHPG, ACPD, CGP55845, cyclopiazonic acid (CPA), MPEP and LY367385. Apamin, atropine, tetrodotoxin (TTX), forskolin, flufenamate (FFA) and SKF96365 were obtained from Sigma Aldrich. Antibodies to TRPC isoforms were acquired from Alomone laboratories. Inactivation of the antibodies was achieved by incubating the antibodies at 90°C for 10 min.
Statistics
All data are presented as mean ± SEM. Statistical significance (P≤ 0.05) was tested using unpaired Student's t tests (t test) assuming unequal variance, or using a one-way ANOVA with Fisher's post hoc analysis (ANOVA), as appropriate.
Results
mGluR-mediated intracellular Ca2+ waves trigger biphasic membrane potential changes
The basic characteristics of intracellular Ca2+ waves in hippocampal pyramidal neurons have been well described (Nakamura et al. 1999; Kapur et al. 2001; Watanabe et al. 2006; Fitzpatrick et al. 2009). To date, however, there have been few studies investigating the consequences of mGluR-mediated intracellular Ca2+ waves on neuronal function. To investigate the functional consequences of Ca2+ waves in CA1 pyramidal neurons, we performed patch-clamp recordings in acute hippocampal slices from neurons filled with either a low-affinity or high-affinity Ca2+-sensitive dye (200 μm fura-2FF or 100 μm fura-2, respectively). Ca2+ waves were elicited by brief trains of electrical stimulation delivered to Schaffer collaterals (30–100 pulses; 100 Hz). Synaptic stimulation elicited rises in cytosolic Ca2+ concentration ([Ca2+]i) that typically exhibited two readily distinguishable phases. First, there was a relatively small, transient rise in [Ca2+]i that occurred simultaneously in the soma and apical dendrites. This was followed by a longer [Ca2+]i rise that propagated as a wave in the primary apical dendrite (Fig. 1A). As demonstrated previously (Nakamura et al. 1999; Kapur et al. 2001; Hong & Ross, 2007; Fitzpatrick et al. 2009), the initial rise in [Ca2+]i is due to Ca2+ influx through voltage-gated calcium channels (VGCCs) made active during stimulation-evoked action potentials and the delayed rise in [Ca2+]i is due to mGluR-mediated mobilization of IP3 and consequent IP3R-dependent release of Ca2+ stored in the endoplasmic reticulum (ER). Another feature of mGluR-mediated intracellular Ca2+ waves is that they typically propagate through ‘hot spots’ of release located at branch points where type 1 IP3Rs cluster on the ER (Hertle & Yeckel, 2007; Fitzpatrick et al. 2009) (Fig. 1A). An important feature of this mechanism of wave propagation is that when Ca2+ waves eventually stop, they almost always fail at cold spots of internal Ca2+ release, thus endowing cold spots with the passive ability to regulate wave propagation.
Figure 1. Synaptically evoked intracellular Ca2+ waves and an associated biphasic membrane potential change in CA1 pyramidal neurons.
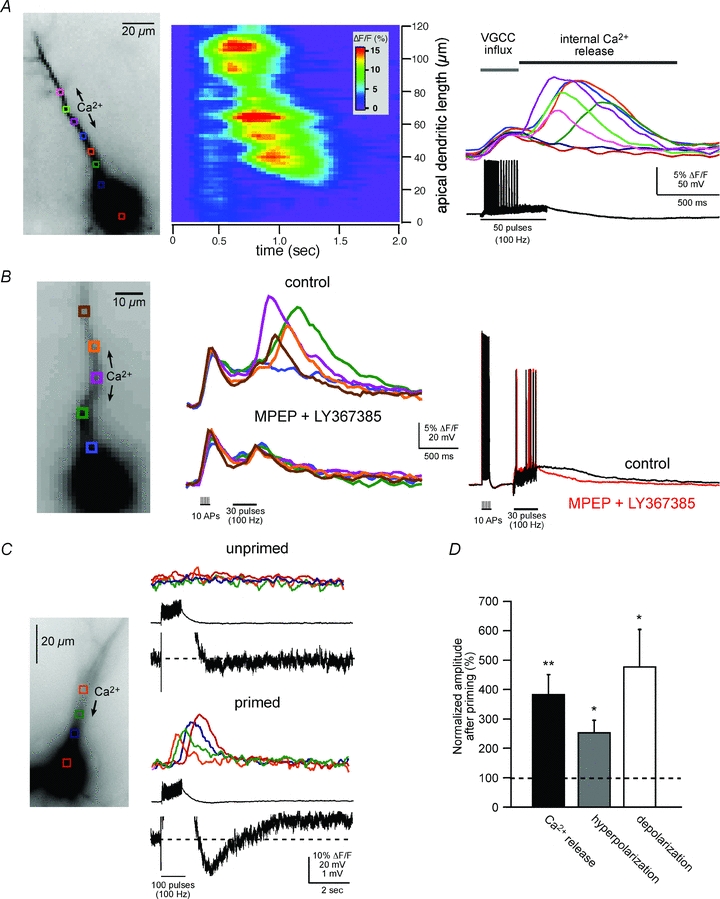
A, left panel, fluorescence image of a fura-2FF-filled neuron. Middle and right panels, two representations of the same imaging data elicited by synaptic stimulation (50 pulses at 100 Hz; see electrical response in right panel). The middle panel is a pseudo-linescan showing that intracellular Ca2+ waves propagate through hot and cold spots of release. The right panel shows coloured waveforms corresponding to Ca2+ rises occurring at the colour-coded boxes (regions of interest, ROIs) over the cell in the left panel. Note the difference in magnitude and timing of the VGCC-mediated Ca2+ rise occurring during the synaptically elicited action potentials and the delayed intracellular Ca2+ wave. B, Group I mGluR blockers selectively blocked intracellular Ca2+ waves, but not VGCC-mediated rises in [Ca2+]i. Rises in [Ca2+]i were first initiated by activation of VGCCs with current injection-evoked spikes (2 ms, 2 nA, 10 spikes at 100 Hz) followed by synaptic stimulation (30 pulses at 100 Hz). Middle panel, mGluR antagonists, MPEP (10 μm) and LY367385 (100 μm), blocked synaptically elicited internal Ca2+ release. Right panel, the evoked electrical waveforms recorded during the Ca2+ responses shown in the middle panel. Note the suppression of the membrane depolarization following bath application of the mGluR antagonists. C, synaptically elicited intracellular Ca2+ waves correlate with a biphasic membrane potential change. Synaptic stimulation, subthreshold for eliciting a rise in [Ca2+]i (100 pulses at 100 Hz), failed to elicit a biphasic membrane potential change. 30 s after ‘priming’ the cell with a train of current injection-evoked spikes (2 ms, 2 nA current injection; 100 spikes at 100 Hz) and consequent VGCC-mediated Ca2+ influx (not shown), the previously subthreshold synaptic stimulation elicited internal Ca2+ release and a hyperpolarization and depolarization. Note that the fast EPSPs evoked by electrical stimulation were not affected by priming. In this example, mAChRs and GABABRs were blocked (1 μm atropine and 1 μm CGP55845, respectively). D, summary data showing priming-induced facilitation of synaptically elicited internal Ca2+ release and associated membrane potential changes (normalized to pre-priming response averages; n = 7; *P < 0.01, **P < 0.001; ANOVA).
Consistent with our previous findings from layer V mPFC neurons (Hagenston et al. 2008), we observed that synaptically elicited intracellular Ca2+ waves were usually associated with a transient hyperpolarization and a sustained depolarization (Fig. 1C). The hyperpolarization was time-locked to internal Ca2+ release and was observed in 83% of cells exhibiting synaptic stimulation-evoked internal Ca2+ release (n = 15/18; 1.81 ± 1.04 mV). The depolarization, which was elicited in 61% of the neurons tested (n = 11/18; 0.28 ± 0.15 mV), had a longer latency-to-onset time than the hyperpolarization and lasted much longer than the rise in [Ca2+]i (up to 60 s, the longest collection period that was used). In 50% of the synaptically stimulated cells, both a hyperpolarization and a depolarization were observed (n = 9/18). As shown previously in cortical pyramidal neurons and cerebellar Purkinje neurons (Finch & Augustine, 1998; Hagenston et al. 2008), internal Ca2+ release and the associated membrane potential changes were blocked by the addition of Group I mGluR1 and mGluR5 antagonists (100 μm LY367385 and 10 μm MPEP, respectively; n = 4/4; Fig. 1B). Blocking muscarinic acetylcholine receptors (mAChRs), another Gq coupled receptor that has been shown to trigger Ca2+ waves in hippocampal neurons (Power & Sah, 2002), with atropine (1 μm) did not affect synaptically elicited internal Ca2+ release under our experimental conditions (n = 6/6). In a few cases (n = 3), when the stimulation electrode was positioned close to the soma/proximal apical dendrite, a rapid membrane hyperpolarization was evoked that did not correlate with internal Ca2+ release. We attribute this to GABAA receptor activation. GABAB receptor activation did not appear to contribute significantly to the transient hyperpolarization under our experimental conditions based on the inability of the GABAB receptor antagonist CGP55845 (1 μm) to block the hyperpolarizing response (n = 6/6).
Further support for the hypothesis that the biphasic electrical response was causally linked to internal Ca2+ release was obtained by selectively enhancing Ca2+ release and observing a concomitant enhancement of both the hyperpolarization and depolarization magnitude. Internal Ca2+ release was enhanced by ‘priming’ the neurons with a train of brief, large depolarizing current injections (2 ms, 2 nA; 50–100 injections at 100 Hz) 20–60 s before synaptically eliciting internal Ca2+ release. Priming is generally thought to result from an increase in ER Ca2+ concentration following influx of Ca2+ into the cytosol through VGCCs during current injection (Jaffe & Brown, 1994; Berridge et al. 1998; Nakamura et al. 1999; Yeckel et al. 1999; Watanabe et al. 2006; Hong & Ross, 2007; Hagenston et al. 2008; Fitzpatrick et al. 2009). By the time internal Ca2+ release is elicited, cytosolic [Ca2+] has returned to baseline levels (within 1 s of the spike train). Additionally, there were no obvious priming-related effects on synaptic transmission or the intrinsic membrane properties of neurons (data not shown). Besides showing that the previous history of cellular activity is an important determinant of internal Ca2+ release, this technique provides a useful tool for selectively manipulating internal Ca2+ release. Following priming there was a 380 ± 71% enhancement of internal Ca2+ release amplitude (Fig. 1C and D). Moreover, Ca2+ waves were more readily evoked when priming was performed prior to synaptic stimulation, and the amplitude of [Ca2+]i rises decreased approximately linearly as the time between priming and synaptic stimulation increased (R2 = 0.92 ± 0.07, n = 6; data not shown). Correlated with the enhancement of internal Ca2+ release were an increase in hyperpolarization amplitude (253 ± 44%) and an increase in depolarization amplitude (479 ± 125%; Fig. 1C). Consistent with the hypothesis that these membrane potential changes depend on intracellular Ca2+ release, we found that their magnitudes decreased in a linear fashion as the time following priming increased (hyperpolarization, R2 = 0.93 ± 0.03, n = 9; depolarization, R2 = 0.89 ± 0.06, n = 6).
ISK and ICAN– the ionic bases of mGluR-mediated, Ca2+ release-dependent excitability changes
To investigate the properties of membrane potential changes elicited by mGluR-mediated internal Ca2+ release, we bypassed glutamatergic synaptic transmission by applying an mGluR agonist (400 μm ACPD or 400 μm DHPG) to various sites along the apical primary dendrite (50–150 μm from the soma). Brief pressure application of agonist (50–100 ms puffs) directly onto the primary apical dendrite triggered intracellular Ca2+ waves in every cell tested (n = 37/37). Similar to synaptically elicited release, we observed a transient hyperpolarization (2.88 ± 0.96 mV) that correlated with the rise in [Ca2+]i followed by a sustained (2 to 60 s) depolarization (0.93 ± 0.10 mV) in 70% of the cells (n = 15/22), that was capable of eliciting persistent spiking when neurons were held at membrane potentials slightly subthreshold for triggering action potentials (Fig. 2). In another series of experiments we voltage-clamped neurons (−63 to −65 mV) during application of ACPD. Of the cells in which internal Ca2+ release was observed, 11/14 exhibited a transient outward current (28.75 ± 2.37 pA) and 13/14 exhibited a sustained inward current (20.79 ± 3.54 pA) (Fig. 2C). In every cell in which a hyperpolarizing or depolarizing current was observed, Ca2+ waves propagated from the release initiation site on the apical dendrite (<70 μm from the soma) into the soma. In the three cells in which there was no hyperpolarizing current, Ca2+ waves failed to propagate into the proximal dendrite and soma region. The depolarization was observed in two of the three cells in which waves failed to propagate into the soma. These observations are consistent with our previous findings showing that in layer V mPFC pyramidal neurons the hyperpolarizing current depends on Ca2+ wave propagation into the proximal apical dendrite and soma region, whereas the depolarizing current correlates with total Ca2+ wave propagation distance in the proximal apical dendrite (Hagenston et al. 2008).
Figure 2. mGluR agonists elicit a Ca2+ wave-dependent hyperpolarization and depolarization.
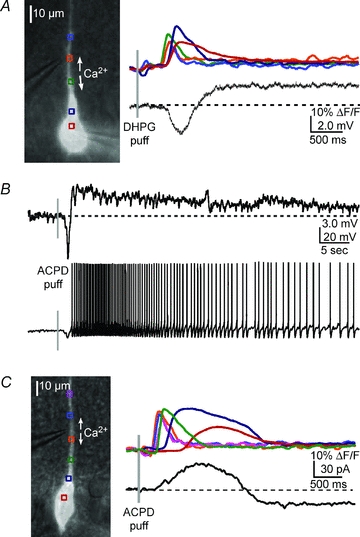
A, left, overlay of a DIC image and fluorescence neuron image showing the position of the pressure application pipette near the primary apical dendrite of the recorded cell. Right, a DHPG puff (400 μm, 50 ms) onto the primary apical dendrite triggered a bidirectionally propagating Ca2+ wave and associated hyperpolarization and depolarization of a CA1 pyramidal neuron. B, upper panel, in a different neuron, an ACPD puff (400 μm, 50 ms) elicited a Ca2+ wave (not shown) and a transient hyperpolarization and a sustained depolarization. Lower panel, when the neuron was held at a membrane potential slightly subthreshold for spiking (∼−53 mV), ACPD elicited a transient hyperpolarization and a sustained train of action potentials. C, in voltage clamp, an ACPD puff elicited an outward current and inward current (see Results for summary data).
We further tested the ionic bases of the biphasic electrical responses. We determined that the reversal potential for the hyperpolarization was −85 ± 1 mV (n = 5), close to the reversal potential of K+, and that it was blocked by the SK antagonist apamin (100 nm) (Fig. 3A and B). These findings are consistent with previous reports showing that the hyperpolarization was due to activation of Ca2+-dependent K+ channels (Jaffe & Brown, 1994; Morikawa et al. 2000; Stutzmann et al. 2003; Yamada et al. 2004; Gulledge & Kawaguchi, 2007; Hagenston et al. 2008). We have shown previously in mPFC pyramidal neurons that a sustained depolarization similar to that described here has electrophysiological properties consistent with ICAN. To further examine this possibility, we determined the reversal potential of the CA1 pyramidal cell depolarization. These experiments were conducted in voltage-clamp mode using a cesium-based internal solution to block K+ channels (5 mm CsCl), and in the presence of Na+ channel and GABABR blockers (1 μm TTX and 1 μm CGP55845, respectively). Our results showed that the current–voltage relation of the depolarizing current was roughly linear and reversed at 12 ± 3 mV (n = 5) (Fig. 3C) consistent with the involvement of cationic channels (Crepel et al. 1994; Congar et al. 1997; Gee et al. 2003). Our previous efforts using pharmacological agents (FFA) or cation substitution (N-methylglucamine for Na+) to characterize the internal Ca2+ release-mediated depolarization were problematic because they also blocked internal Ca2+ release (Hagenston et al. 2008). In subsequent experiments in this study we sought to more definitively identify the origin of the sustained depolarization.
Figure 3. The hyperpolarization and depolarization are due to SK channels and CAN channels, respectively.
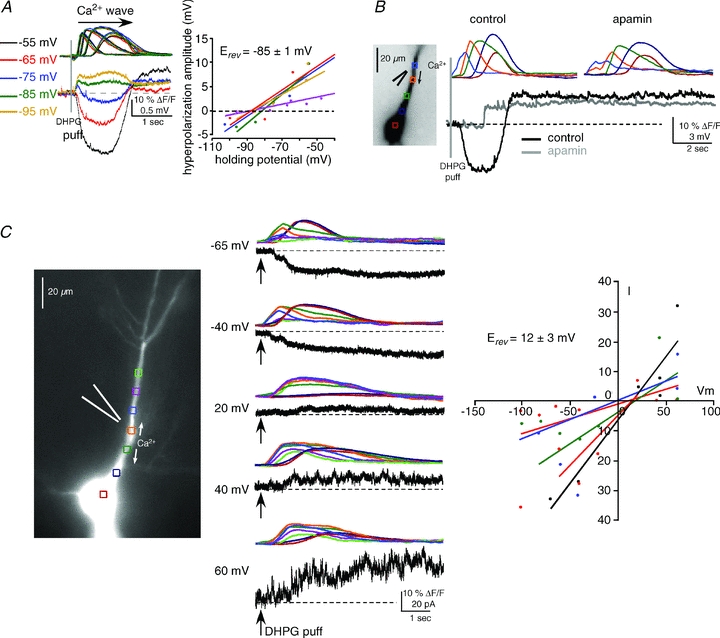
A, left, the reversal potential (Erev) of the hyperpolarizing potential was determined to be ∼−85 mV by applying DHPG puffs (400 μm, 50 ms) at different holding potentials in current clamp and measuring membrane potential changes. Intracellular Ca2+ waves were similar at all holding potentials (waves are colour-coded by holding potential). Right panel, summary graph showing the reversal potential for all cells tested (n = 5; each cell is represented by a different colour). B, consistent with a mechanism involving SK channels, apamin (100 nm) blocked the hyperpolarization. This treatment unveiled the isolated depolarizing potential and revealed its delayed onset. C, left and middle panels, the mGluR-mediated, Ca2+-dependent depolarizing current was isolated in voltage clamp and its Erev was determined to be ∼12 mV. DHPG puffs (400 μm, 50 ms) were delivered to the primary apical dendrite in the presence of voltage-gated K+ channel and Na+ channel blockers, and GABABR blockers (see Results). Right, summary I–V graph for all cells tested (n = 5; each cell is represented by a different colour) shows data consistent with activation of CAN channels.
The sustained depolarization is mGluR- and Ca2+-dependent
Earlier work examining Ca2+ wave-dependent changes in membrane potentital of mPFC pyramidal neurons showed that the sustained depolarization required concomitant activation of mGluRs and intracellular Ca2+ waves (Hagenston et al. 2008). We found this also to be true in CA1 pyramidal neurons. Our findings also corroborate studies showing that an mGluR-mediated ICAN depends on a rise in [Ca2+]i in hippocamapal pyramidal neurons (Crepel et al. 1994; Congar et al. 1997; Partridge & Valenzuela, 2000; Gee et al. 2003). One previous study, however, showed an mGluR-mediated ICAN that occurred independently of G protein activation (Gee et al. 2003). To test whether the sustained depolarization we observed was downstream from IP3 mobilization, we bypassed mGluR activation by directly raising the intracellular concentration of IP3 through photolysis of caged NPE-IP3 (97 μm; 50–500 ms UV exposure) on the proximal apical dendrite (Fig. 4A). In only one cell (n = 1/14) was a small depolarization elicited (<0.7 mV). In contrast, a hyperpolarization was evoked in all of the cells examined (n = 14/14; 5.03 ± 0.84 mV). In another series of experiments we elicited intracellular Ca2+ waves without activating either mGluRs or IP3Rs, but rather by pharmacologically activating ryanodine receptors (RyRs). RyRs are another ER receptor channel that when activated can lead to internal Ca2+ release. Although RyRs have not been shown to play a role in the generation and propagation of Ca2+ waves in wild-type animals (Nakamura et al. 1999; Kapur et al. 2001), they have been implicated in Alzheimer's disease (Kelliher et al. 1999; Stutzmann, 2005). Under some experimental conditions, directly applying caffeine to pyramidal cell dendrites can lead to intracellular Ca2+ release (Sandler & Barbara, 1999; Hagenston et al. 2009). We found that direct application of caffeine to CA1 pyramidal neuron dendrites (50 mm; 30–70 ms puffs) elicited internal Ca2+ release and a hyperpolarization (2.5 ± 0.33 mV) in every cell tested, but failed to elicit a depolarization in any of the cells (n = 6/6) (Fig. 4B).
Figure 4. The sustained depolarization requires both group I mGluR receptor activation and a rise in [Ca2+]i.
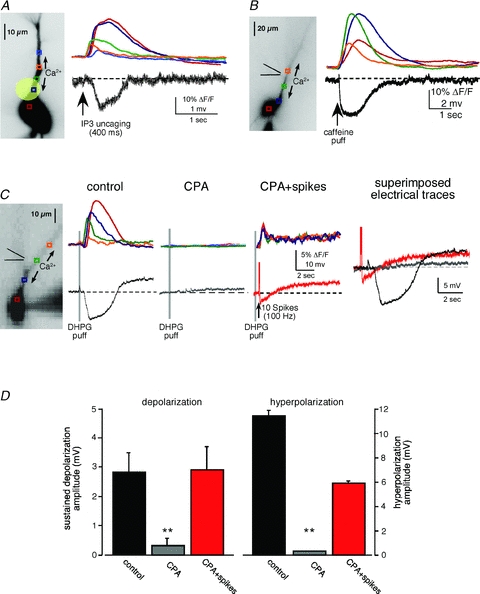
A, fluorescence image of a neuron filled with fluo-4 (100 μm) and NPE-caged IP3 (97 μm). Coloured boxes indicate the regions of interest ROIs in apical dendrites corresponding to the optical traces showing internal Ca2+ release on the right. UV flashes directed at the proximal primary apical dendrite (20 μm diameter, 400 ms duration; represented by yellow circle) elicited internal Ca2+ release and a hyperpolarizing potential, but not a depolarization. B, a caffeine puff (50 mm, 50 ms) onto the proximal apical dendrite elicited intracellular Ca2+ waves and a transient hyperpolarization, but no depolarization. C, the sustained depolarization depended on mGluR activation and a rise in [Ca2+]. Depleting Ca2+ stores with CPA (50 μm) prevented mGluR-mediated internal Ca2+ release and membrane potential changes. In the absence of internal Ca2+ release, pairing mGluR activation with VGCC-mediated Ca2+ influx during spikes evoked by current injection (2 ms, 2 nA current injections; 10–50 spikes at 100 Hz) elicited a similar sustained depolarization. Under these conditions, spiking elicited a VGCC-mediated hyperpolarization that was not affected by agonist application (data not shown). D, summary data showing rescue of membrane potential changes when internal stores are depleted (n = 5, **P < 0.001, ANOVA).
The results described above demonstrate that internal Ca2+ release alone was not sufficient for eliciting the sustained depolarization. To test whether mGluR activation was sufficient for eliciting the sustained depolarization, we depleted the ER of Ca2+ with the Ca2+-ATPase pump blocker CPA (50 μm; 30–40 min bath perfusion) (Seidler et al. 1989). CPA application led to a loss of DHPG-mediated internal Ca2+ release and to the absence of membrane potential changes (n = 4/4). To test whether the ICAN depended exclusively on intracellular Ca2+ waves, we paired DHPG puffs with current-injected spiking to trigger influx of Ca2+ through VGCCs (10 spikes elicited with 2 ms, 2 nA current injections at 100 Hz). Under conditions where ER Ca2+ release was depleted, the sustained depolarization was rescued by VGCC-mediated rises in [Ca2+]i (control, 2.72 ± 0.65 mV; post-CPA plus spikes, 2.86 ± 0.78 mV; n = 5, P = 0.89, t test). These data demonstrate that the ICAN-mediated sustained depolarization we observe requires both Group I mGluR activation and rises in [Ca2+]i, but appears to be insensitive to the source of Ca2+.
The sustained depolarization is mediated by TRPC channels
Several studies suggest that ICAN results from cation flux through TRPC channels in neurons (Strubing et al. 2001; Kim et al. 2003; Faber et al. 2006; Yan et al. 2009). To test the prediction that the sustained depolarization we observe in CA1 pyramidal neurons is due to TRPC channel activation, we bath-applied drugs reported to suppress ITRPC or introduced TRPC antibodies into neurons through our patch pipette. We first tested FFA (100 μm) and SKF96365 (30 μm), in combination or individually. Both drugs have been reported to suppress some TRPC channel isoforms (Boulay et al. 1997; Lee et al. 2003; Faber et al. 2006; Zhang et al. 2008). In our preliminary studies, however, we found that bath application of the drugs suppressed internal Ca2+ release (n = 4), similar to our findings for application of FFA to layer V mPFC neurons (Hagenston et al. 2008). To eliminate confounds associated with suppressing internal Ca2+ release, we again depleted ER Ca2+ stores with bath application of CPA (50 μm) and examined whether putative TRPC antagonists blocked the depolarization elicited by DHPG puffs in combination with VGCC-mediated rises in [Ca2+]i (50 spikes at 100 Hz). Under these conditions we found that combining FFA and SKF96345 significantly suppressed both the depolarization and hyperpolarization (depolarization, 0.5 ± 0.12 mV; hyperpolarization, 4.6 ± 0.93 mV) compared to CPA alone (depolarization, 2.32 ± 0.21 mV; hyperpolarization, 6.92 ± 0.88 mV; n = 5, P < 0.01, ANOVA) (Fig. 5A). Further analysis of these compounds showed that FFA alone (n = 3; Fig. 5B) completely blocked the depolarization and had little effect on the hyperpolarization (95% of control), whereas SKF96365 (n = 5) did not affect either the depolarization or the hyperpolarization (compared to CPA alone, 102 ± 4.7% and 109 ± 15%, respectively) (Fig. 5C).
Figure 5. Pharmacological characterization of the sustained depolarization—non-specific blockers of ICAN/ITRPC suppressed the sustained depolarization.
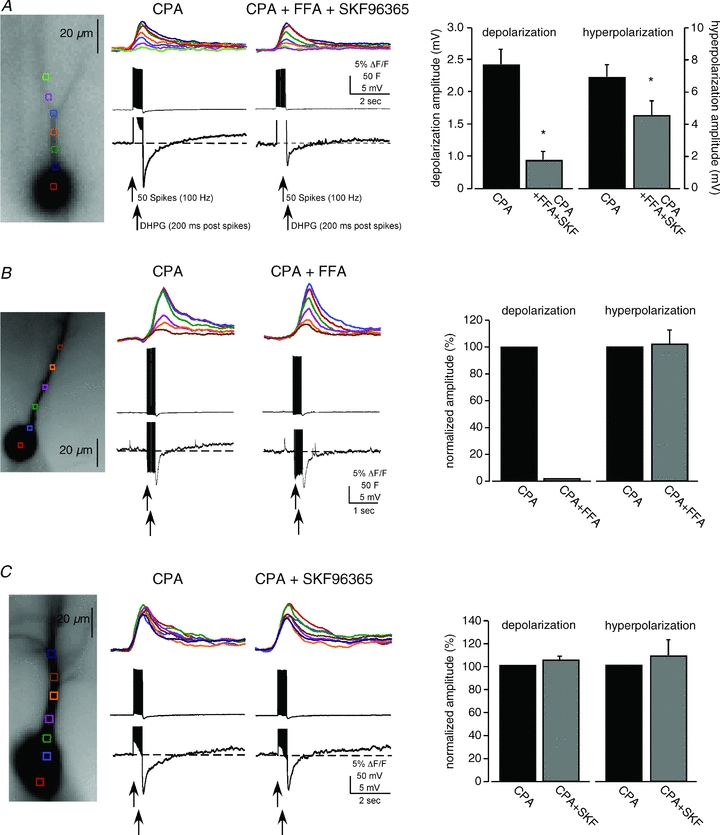
A, internal Ca2+ stores were first depleted with CPA (50 μm). VGCC-mediated rises in [Ca2+]i were elicited with current-injected spikes and paired with puffs of DHPG. Addition of both flufenamate (FFA; 100 μm) and SKF96365 (30 μm) suppressed both the depolarization and the hyperpolarization (n = 5; P < 0.01, t test). B, addition of FFA (100 μm) alone blocked the depolarization (n = 3). C, addition of SKF96365 (30–100 μm) alone had no effect on either the depolarization or the hyperpolarization under these conditions (n = 5).
Due to the lack of specificity of the pharmacological TRPC antagonists, we used a strategy successfully used by others in which antibodies directed against TRPC isoforms are introduced into cells (Faber et al. 2006; Amaral & Pozzo-Miller, 2007b). In this series of experiments, we examined the depolarization over time in neurons dialysed with either anti-TRPC1 (n = 5), anti-TRPC3 (n = 3), anti-TRPC4 (n = 5) or anti-TRPC5 (n = 5) (1:100 dilution; rabbit). In an effort to determine whether the antibodies were having non-specific effects on recorded neurons we dialysed neurons with heat-inactivated TRPC antibodies (n = 5; 90°C for 10 min) or dialysed neurons with control IgG antibodies (n = 7; 1:100 dilution; rabbit). TRPC1, 3, 4 and 5 are known to be highly expressed in CA1 pyramidal neurons (Strubing et al. 2001; Chung et al. 2006; Fowler et al. 2007). The depolarizing amplitude was normalized to the starting amplitude measured in the first 5 min after going into whole-cell recording configuration. We found that including anti-TRPC1, anti-TRPC4 or anti-TRPC5 in our recording pipettes selectively and significantly (P < 0.01, t test) suppressed the sustained depolarization within 20 min of going into whole-cell mode (anti-TRPC1, 32.1 ± 6%; anti-TRPC4, 38 ± 13%; anti-TRPC5, 20 ± 2%) compared to heat-inactivated antibody (inactive anti-TRPC1, 80.1 ± 7%; inactive anti-TRPC5, 82.1 ± 4%) or the IgG control antibody (94%± 3%) (Fig. 6). Neither the amplitude of internal Ca2+ release nor the hyperpolarization amplitude were affected (P > 0.5 for all treatments). Anti-TRPC3, however, had little effect on either the depolarization or hyperpolarization (96 ± 8% and 87 ± 9%, respectively). Taken together, we conclude that the mGluR-mediated, Ca2+ wave-dependent sustained depolarization is due to the opening of multiple TRPC channel isoforms.
Figure 6. TRPC1, TRPC4 and TRPC5 antibodies block the mGluR-mediated and intracellular Ca2+ wave-dependent depolarization.
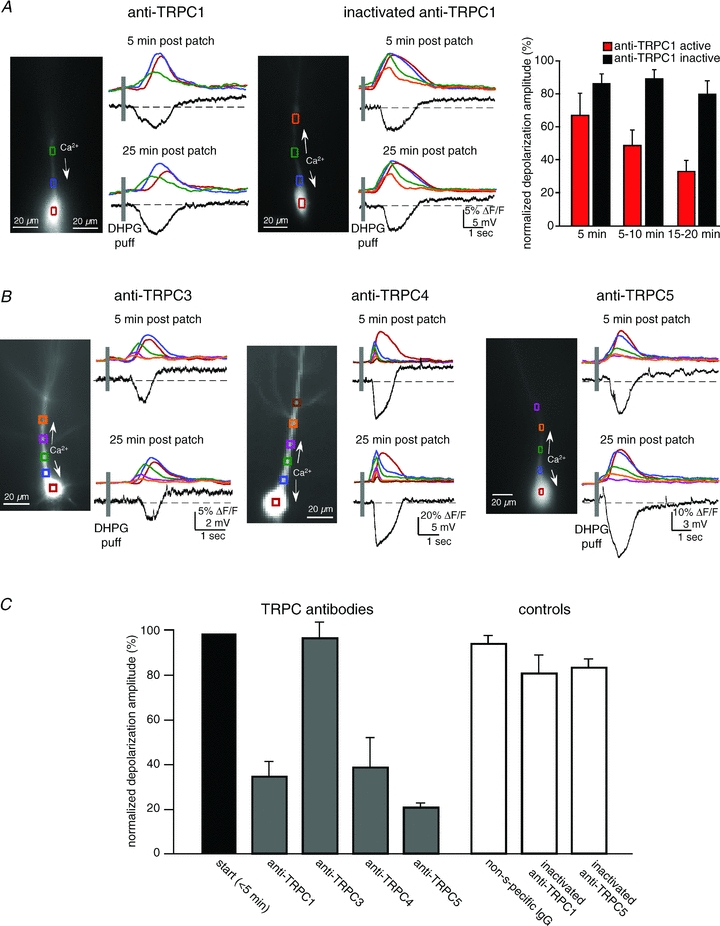
Antibodies to TRPC were loaded into patch recording pipettes (1:100 dilution). In some cases antibodies were heat inactivated. Responses recorded ∼5 min after breaking into the cell were compared to responses recorded ∼20 min after breaking in. A, an example of data collected from a CA1 pyramidal neuron loaded with anti-TRPC1 and an example of a neuron loaded with heat-inactivated anti-TRPC1. Anti-TRPC1 selectively blocked the sustained depolarization. B, examples of neurons loaded with anti-TRPC3, anti-TRPC4 or anti-TRPC5. TRPC3 did not affect the depolarization (n = 3, P > 0.1, t test); TRPC4 (n = 5), like TRPC1 (n = 5) and TRPC5 (n = 5; data not shown), suppressed the mGluR/IP3R evoked-depolarization (P < 0.01 for each antibody, t test). C, summary data for anti-TRPCs, and the controls, IgG (n = 7) or inactivated anti-TRPC1 (n = 5) or TRPC5 (n = 5).
Modulation of TRPC by rises in [cAMP]i
Based on a previous report showing that rises in [cAMP]i suppress a CAN-like current in Helix burster neurons (Partridge et al. 1990), we tested the hypothesis that cAMP modulates the mGluR-mediated depolarization in CA1 pyramidal neurons by administering forskolin to brain slices. We first observed, however, that forskolin application (5–10 μm) suppressed internal Ca2+ release over time (see Fig. 7A). Therefore, we elicited the ITRPC by pairing VGCC-mediated rises in [Ca2+]i (10 spikes at 100 Hz) with puffs of DHPG. As predicted, we found that forskolin completely blocked the sustained depolarization (control, 1.16 ± 0.24 mV; forskolin, 0.12 ± 0.1 mV; n = 9; P < 0.01, t test) (Fig. 7B). The SK-mediated hyperpolarization was also slightly decreased, but this appeared to be due to the decrease in the rise of [Ca2+]i. This observation is consistent with some reports showing a reduction of a slow IAHP by forskolin or cAMP analogues (Madison & Nicoll, 1986; Pedarzani & Storm, 1993; Khawaja et al. 2007; Vatanparast et al. 2007), but is inconsistent with other reports showing a cAMP enhancement of ISK (Blumenthal & Kaczmarek, 1992, 1994).
Figure 7. Rises in intracellular cAMP differentially suppressed the mGluR-mediated depolarization and hyperpolarization.
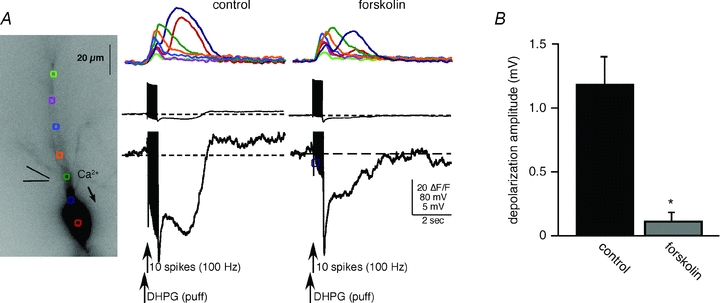
A, DHPG application paired with current injection-evoked action potentials elicited robust rises in [Ca2+]i and associated membrane potential changes. Bath application of forskolin (5–10 μm) totally blocked the TRPC channel-mediated depolarization. Forskolin also partially suppressed the SK channel-mediated AHP, but not the fast Ca2+-dependent AHP. Forskolin also suppressed internal Ca2+ release in this cell. B, summary data showing suppression of the depolarization (n = 9; *P < 0.01, t test).
SK and TRPC channels modulate CA1 pyramidal neuron excitability
To determine whether intracellular Ca2+ waves are capable of regulating the activity of CA1 pyramidal neurons through SK and TRPC channels, we stimulated mGluRs while neurons were depolarized to a membrane potential that triggered spontaneous-like spike activity (∼−45 mV). Under these conditions, application of DHPG to the primary dendrite of spiking pyramidal neurons elicited Ca2+ waves and a hyperpolarization-mediated pause in spiking followed by a 1.8-fold increase in firing frequency (Fig. 8A and D; pre-wave, 3.55 ± 0.7 Hz; post-wave, 6.45 ± 0.7; n = 7; P < 0.05, t test). These results are consistent with previous findings showing that IP3R-mediated internal Ca2+ release can modulate firing patterns in cortical pyramidal neurons (Morikawa et al. 2000; Stutzmann et al. 2003; Hagenston et al. 2008). Puff application of caffeine and consequent RyR-mediated internal Ca2+ release triggered an SK-mediated pause in firing, but did not elicit an increase in firing rate (Fig. 8D; P > 0.1; n = 5).
Figure 8. TRPC1, 4 and 5 antibodies suppress mGluR-mediated increases in spike frequency.
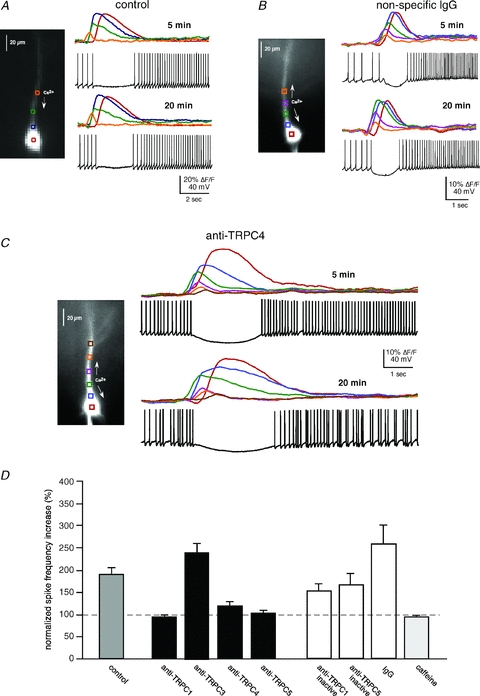
A, mGluR activation of intracellular Ca2+ waves modulates the firing pattern of CA1 pyramidal neurons. Puffing DHPG (50 ms) onto the apical dendrite of a spiking pyramidal neuron held at ∼−45 mV in current clamp suppressed and then increased the firing frequency of this representative pyramidal neuron. B, an example of a neuron loaded with IgG. mGluR regulation of firing frequency was not affected by IgG (or inactivated anti-TRPCs; not shown). C, an example of a neuron loaded with anti-TRPC4. Addition of anti-TRPC1, 4 or 5 (1:100 dilution) to the patch pipette suppressed the increase in firing frequency 20 min after breaking into the cell. D, summary data showing that anti-TRPC1, 4 and 5 suppress mGluR-mediated increases in firing frequency (cumulatively P < 0.01, t test). TRPC3 (n = 3) did not alter mGluR-mediated regulation of firing (P > 0.1, t test). Inactivated anti-TRPCs (n = 4) and IgG (n = 7) did not have a significant effect on mGluR-mediated increases in firing rate (P > 0.1, t test). Caffeine puffs (50 mm; n = 5), which activate an RyR-mediated ISK but not an ITRPC, did not elicit an increase in firing frequency (P > 0.1, t test).
Based on our finding showing that TRPC antibodies suppress the sustained membrane depolarization (Fig. 7), we tested whether they might also block the mGluR-mediated increase in firing frequency. We found that including anti-TRPC1 (n = 3), anti-TRPC4 (n = 5) or anti-TRPC5 (n = 3) in the recording pipette prevented the mGluR-mediated increase in spiking frequency (n = 11, P > 0.1) without affecting the transient pause in spiking which occurred during the evoked Ca2+ wave (Fig. 8C and D). In contrast, addition of anti-TRPC3 (n = 3), heat-inactivated TRPCs (n = 4) or IgG (n = 7) to the recording pipette did not affect increases in spike frequencies that were normally elicited with mGluR application (P < 0.01 for each condition; see Fig. 8). It should be noted that addition of anti-TRPC1, 4 or 5 to recording pipettes typically resulted in an increase in basal firing frequency when compared to inactivated anti-TRPC1 or 5 (data not shown; anti-TRPC1/4/5: pre-puff, 8.64 ± 0.6 Hz; post-puff, 9.11 ± 0.4 Hz, n = 11 vs. inactivated anti-TRPC1/5: pre-puff, 4.41 ± 1.31 Hz, post-puff, 6.75 ± 1.85 Hz; n = 4, P < 0.01, ANOVA), and when compared to controls (control: pre-puff, 3.55 ± 0.7 Hz, post- puff, 6.45 ± 0.7 Hz; n = 7; P < 0.001, ANOVA). However, we found no significant changes in the basal firing frequency between controls and inactivated anti-TRPC antibodies neurons (P > 0.5, ANOVA). We attribute this to an increase in input resistance (Ri) that we hypothesize to be caused by the closing of constitutively active TRPC channels, as has been described previously (Zhou et al. 2008) (control, Ri start: 71.35 ± 8.63 MΩvs. 25–30 min post-patch Ri = 77.5 ± 8.8 MΩ, n = 7, P = 0.63, t test; anti-TRPC active: Ri start: 91.83 ± 11.75 MΩvs. 25–30 min post-patch Ri: 128.4 ± 18 n = 9, P < 0.05, t test; inactive anti-TRPC, Ri start: 101.47 ± 22 MΩvs. 25–30 min post-patch Ri: 110.51 ± 25 MΩ, n = 5, P = 0.8, t test).
Taken together, these data show that mGluR- and IP3R-mediated intracellular Ca2+ waves are capable of robustly regulating activity patterns in CA1 pyramidal neurons. In particular, stimulation of mGluRs and subsequent internal Ca2+ release can sequentially decrease and increase firing frequency via the activation of SK and TRPC1/4/5 channels, respectively.
Discussion
We show that glutamatergic synaptic transmission or focal pharmacological stimulation of Group I mGluRs on CA1 pyramidal neurons triggers IP3R-dependent intracellular Ca2+ waves. These Ca2+ waves, in turn, contribute to an SK channel-mediated transient hyperpolarization followed by a TRPC1/4/5 channel-mediated sustained depolarization. We also found that the TRPC-mediated depolarization was suppressed by forskolin-induced rises in intracellular cAMP. Lastly, we show that activation of SK and TRPC channels can robustly affect neuronal firing by suppressing and enhancing, respectively, the firing frequency of CA1 pyramidal neurons. Consistent with our previous findings in layer V mPFC (Hagenston et al. 2008), the present results show that mGluR- and IP3R-mediated intracellular Ca2+ waves provide an adjunct means by which glutamatergic synaptic transmission regulates pyramidal neuron activity.
SK channels have been well characterized in many neuron types: their activation depends on rises in [Ca2+]i, their reversal potential is near that of the Nernst potential for K+ and they are sensitive to apamin. These studies have mostly focused on extracellular Ca2+ influx through VGCCs. Consistent with previous findings, however, SK channel-mediated hyperpolarization can also be elicited by internal Ca2+ release (Jaffe & Brown, 1994; Morikawa et al. 2000; Stutzmann et al. 2003; Gulledge & Stuart, 2005; Hagenston et al. 2008). The internal Ca2+ release-evoked hyperpolarization requires activation of Gq-coupled proteins by mGluRs or mAChRs and propagation of Ca2+ waves from the initiation site to the proximal apical dendrite where SK type 2 channels are located (Sailer et al. 2002; Gulledge & Stuart, 2005; Hagenston et al. 2008).
In contrast to the transient hyperpolarization, which solely required a rise in [Ca2+]i, the sustained depolarization required both a rise in [Ca2+]i and mGluR stimulation. The hyperpolarization and depolarization also differed in their dependency on Ca2+. The hyperpolarization amplitude and kinetics correlated with the amplitude and kinetics of the [Ca2+]i rise, consistent with direct activation of SK channels; the depolarization, on the other hand, lasted far longer than the rise in [Ca2+]i, suggesting that Ca2+ was involved in the initiation of a signalling cascade that contributes to sustaining the response (Blair et al. 2009). We determined that this CAN-like depolarization resulted from TRPC channel activation based on its reversal potential close to 0 mV (Strubing et al. 2001; Kim et al. 2003; Yan et al. 2009), its suppression with non-specific pharmacological agents that antagonize ITRPC and ICAN (e.g. FFA) (Haj-Dahmane & Andrade, 1999), and its sensitivity to antibodies targeted to brain-specific TRPC isoforms (Faber et al. 2006; Amaral & Pozzo-Miller, 2007b). The ability of TRPC1, 4 and 5 antibodies to suppress the sustained depolarization is consistent with immunohistochemistry data showing their expression in the CA1 region (Strubing et al. 2001; Chung et al. 2006). Partial suppression by an individual antibody type suggests that the different TRPC isoforms are homomers and when one subtype is blocked the others remain operational. More simply, the antibodies are not totally effective at blocking their targeted isoform or at blocking potential TRPC heteromers (Goel et al. 2002; Hofmann et al. 2002). The inability of anti-TRPC3 to block the mGluR-mediated depolarization is interesting because it has been shown previously to block a longer duration (minutes) BDNF-mediated inward current (Amaral & Pozzo-Miller, 2007a,b;). Unlike the mGluR-mediated ITRPC, IBDNF was reported to be insensitive to anti-TRPC5 and sensitive to SKF96396 (Amaral & Pozzo-Miller, 2007a,b;), suggesting an important distinction for the role of TRPC3 and TRPC5 in neuronal function.
We also observed a suppression of the mGluR-mediated depolarization in response to bath application of forskolin and consequent activation of the adenylyl cyclase–cAMP signalling cascade. This finding is consistent with a study showing that forskolin suppresses ICAN in Helix burster neurons (Partridge et al. 1990), but inconsistent with another study reporting the negative finding that mGluR-mediated ICAN in hippocampal neurons is unaffected by forskolin (Congar et al. 1997). This apparent discrepancy is best explained by procedural differences: we applied puffs of DHPG and continuously bath-applied forskolin, whereas Congar and colleagues bath-applied ACPD and applied forskolin discontinuously (i.e. immediately before, during and after application of ACPD), suggesting that their [cAMP]i rises were not sufficient to affect ICAN. It has also been suggested that PKC can suppress a DHPG-elicited TRPC-mediated current (Fowler et al. 2007). This study, however, did not examine whether raising [PKC] with the PKC activator PdBU also suppressed internal Ca2+ release, as suggested by studies showing that PKC can inhibit PLC function (Ryu et al. 1990; Yue et al. 2000).
TRPC channels have been proposed to be activated when ER Ca2+ stores decline, leading to extracellular Ca2+ influx, and subsequent replenishing of ER stores through SERCA pumps. Whether they are associated with so-called store-operated channels is still open for debate (Parekh & Putney, 2005; Ramsey et al. 2006). Although our experiments were not designed to address this issue, we have never observed any changes in neuronal function during pharmacological depletion of ER Ca2+ stores that might indicate activation of store-operated channels. Additionally, we did not observe a rise in Ca2+ fluorescence during the sustained depolarization, as might be predicted based on data reporting Ca2+ influx through TRPC channels in culture cell preparations (Rychkov & Barritt, 2007). Although it is possible that our imaging apparatus is not sufficiently sensitive to detect small rises in [Ca2+]i that might occur during the depolarization, we favour the conclusion that TRPC1/4/5 channels do not gate Ca2+ as efficiently as other TRP channel families (Ramsey et al. 2006). More generally, it is not surprising that neurons might not depend on store-operated channel function, given their ability to activate voltage- and ligand-gated channels that are permeable to Ca2+.
Our investigation primarily focused on mGluR-mediated changes in membrane potential that depended on IP3R-dependent internal Ca2+ release. We also show that both the hyperpolarization and depolarization can occur without internal Ca2+ release if there is extracellular Ca2+ influx through VGCCs. This raises the question of whether internal Ca2+ release might be a vestigial mechanism of developing neurons for raising [Ca2+]i before they express voltage-gated or ligand-gated Ca2+ channels, or whether it might represent a redundant mechanism that is engaged under non-physiological conditions. Given the robustness of internal Ca2+ release compared to VGCC-mediated rises (see Fig. 1A) and the ease with which it can be elicited (as few as 2–3 stimulation pulses can evoke intracellular Ca2+ waves; Nakamura et al. 1999; Kapur et al. 2001; Yeckel et al. 2008), we believe that Ca2+ release is likely to play an important role in neuronal function. Furthermore, because internal Ca2+ release can occur without VGCC activation, particularly after priming (see Fig. 1C), it does not appear to be redundant. This priming process highlights the capability for internal Ca2+ release to transduce VGCC-mediated rises in [Ca2+]i occurring during a spike train into robust membrane potential changes, and suggests that IP3R-mediated Ca2+ release from ER stores can encode information related to patterns of neuronal activity.
An important characteristic of TRPC channel activation is their requirement for concomitant activation of Gq-coupled receptors and rises in [Ca2+]i (Blair et al. 2009). Activation of mGluR1/5, and consequent activation of IP3R-mediated internal Ca2+ release, is capable of fulfilling both requirements. TRPC channels can also function as integrators through their ability to integrate glutamatergic synaptic input and activity-dependent influx of Ca2+ through VGCCs. These distinct mechanisms for raising [Ca2+]i suggest the possibility that TRPC channels are engaged under different physiological conditions. In contrast to considerable evidence showing that rises in [Ca2+]i contribute to TRPC channel activation, very little is known about other downstream molecules triggered by Gq-coupled protein activation. The Gq/PLC cascade can lead to numerous molecules that might be involved in activating TRPC channels, none of which have been conclusively linked to TRPCs. For example, it has been reported that activation of TRPC3, TRPC6 and the heteromer TRPC1/TRPC3 depend on DAG, but activation of TRPC4 and TRPC5 do not require DAG (Hofmann et al. 1999; Lintschinger et al. 2000). It has also been reported that PIP2 might suppress TRP channels in Drosophila and that cleavage of PIP2 by PLC leads to activation of TRP channels (Hardie, 2003). Lastly, it has been suggested that production of phosphoinositide metabolites such as IP3 and IP4 might directly trigger TRPC channel opening (Okada et al. 1998). At least with respect to IP3, this suggestion is inconsistent with our findings showing that uncaging IP3 does not elicit ITRPC (see Fig. 4A; Hagenston et al. 2008). An understanding of the signalling cascade contributing to TRPC channel function is likely to provide the basis for understanding how cAMP modulates ITRPC.
mGluR function in the hippocampus has been shown to be important for learning and spatial memory function (Naie & Manahan-Vaughan, 2004; Hayashi et al. 2007; Gil-Sanz et al. 2008). Recently, it has been shown that a reduction of mGluR5 signalling can reverse the fragile X phenotype in mice (Dolen et al. 2007), suggesting that manipulation of mGluR5 might ameliorate some of the symptoms of fragile X mental retardation, as well as some of the symptoms of autism. To date, mGluR research at the cellular level has predominantly focused on regulation of long-term changes in synaptic transmission in the hippocampus (e.g. LTD and LTP) (Conquet et al. 1994; Yeckel et al. 1999; Dolen et al. 2007; Dolen & Bear, 2008; Anwyl, 2009). Along with their role in LTP and LTD, our findings suggest that mGluR activation of SK and TRPC channels are also likely to be important for sculpting patterns of activity that encode information by the hippocampus. For example, reduction in SK channel-like activity has been shown to contribute to increases in CA1 pyramidal cell firing rate that precedes the acquisition of the nictitating membrane conditioned response (Berger et al. 1976; Disterhoft et al. 1986). Likewise, the sustained depolarization we observe might serve a similar role in the transient storage of information as proposed for sustained depolarizations observed in mPFC and entorhinal cortical pyramidal cells (Egorov et al. 2002; Hagenston et al. 2008). A possible role of mGluR regulation of neuronal excitability might also be provided by the observation that bursts of presynaptic activity optimally activate mGluRs (Nakamura et al. 1999; Kapur et al. 2001), presumably due to their distribution on the periphery of the postsynaptic density (Baude et al. 1993; Lujan et al. 1996). Synchronized bursting activity in hippocampal pyramidal neurons that occurs during sharp waves (Buzsaki et al. 1992; Csicsvari et al. 2000), and which correlates with different forms of hippocampus-dependent behaviour (O'Neill et al. 2006; Csicsvari et al. 2007), is a promising candidate for providing this pattern of input to CA1 pyramidal neurons. Sharp wave-evoked activation of ITRPC, and consequent enhancement of CA1 spiking activity, might contribute to reverbatory network activity through the hippocampal formation such as has been observed in vivo (Chrobak & Buzsaki, 1994), and proposed to be critical for memory consolidation (Axmacher et al. 2008).
Acknowledgments
We wish to thank Dr Babak Tahvildari and Dr Yury Morozov for assistance with miscellaneous reagents. Funded by the Whitehall Foundation, the Kavli Foundation, the Dart Foundation, NIMH RO1-MH067830 and P50-MH068789 and NIAAA 1RL1AA017536-01 (M.F.Y.).
Glossary
Abbreviations
- CAN
Ca2+-dependent non-specific cationic
- cAMP
cyclic adenosine monophosphate
- CPA
cyclopiazonic acid
- DAG
diacyl glycerol
- ER
endoplasmic reticulum
- FFA
flufenamate
- IP3
inositol 1,4,5-triphosphate
- mGluR
metabotropic glutamate receptor
- mPFC
medial prefrontal cortex
- NPE
1-(2-phenyl)ethyl
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PKC
protein kinase C
- PLC
phospholipase C
- RyR
ryanodine receptor
- SK
Ca2+-dependent, small conductance K+
- TRPC
transient receptor potential classical
- TTX
tetrodotoxin
- VGCC
voltage-gated Ca2+ channel
Author contributions
1. Conception and design of the experiments: L.El-H. and M.F.Y. 2. Collection, analysis and interpretation of data: L.El-H., A.H., L.B.D'A. and M.F.Y. 3. Drafting the article or revising it critically for important intellectual content: L.El-H. and M.F.Y. All authors approved the final version.
Author's present address
A. M. Hagenston: Institute of Neurobiology, Ruprecht- Karls University of Heidelberg, Germany.
References
- Amaral MD, Pozzo-Miller L. BDNF induces calcium elevations associated with IBDNF, a nonselective cationic current mediated by TRPC channels. J Neurophysiol. 2007a;98:2476–2482. doi: 10.1152/jn.00797.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral MD, Pozzo-Miller L. TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J Neurosci. 2007b;27:5179–5189. doi: 10.1523/JNEUROSCI.5499-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology. 2009;56:735–740. doi: 10.1016/j.neuropharm.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Axmacher N, Elger CE, Fell J. Ripples in the medial temporal lobe are relevant for human memory consolidation. Brain. 2008;131:1806–1817. doi: 10.1093/brain/awn103. [DOI] [PubMed] [Google Scholar]
- Baude A, Nusser Z, Roberts JD, Mulvihill E, McIlhinney RA, Somogyi P. The metabotropic glutamate receptor (mGluR1α) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron. 1993;11:771–787. doi: 10.1016/0896-6273(93)90086-7. [DOI] [PubMed] [Google Scholar]
- Berger TW, Alger B, Thompson RF. Neuronal substrate of classical conditioning in the hippocampus. Science. 1976;192:483–485. doi: 10.1126/science.1257783. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signalling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Lipp P. Calcium – a life and death signal. Nature. 1998;395:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- Blair NT, Kaczmarek JS, Clapham DE. Intracellular calcium strongly potentiates agonist-activated TRPC5 channels. J Gen Physiol. 2009;133:525–546. doi: 10.1085/jgp.200810153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal EM, Kaczmarek LK. Modulation by cAMP of a slowly activating potassium channel expressed in Xenopus oocytes. J Neurosci. 1992;12:290–296. doi: 10.1523/JNEUROSCI.12-01-00290.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal EM, Kaczmarek LK. The minK potassium channel exists in functional and nonfunctional forms when expressed in the plasma membrane of Xenopus oocytes. J Neurosci. 1994;14:3097–3105. doi: 10.1523/JNEUROSCI.14-05-03097.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Zhu X, Peyton M, Jiang M, Hurst R, Stefani E, Birnbaumer L. Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J Biol Chem. 1997;272:29672–29680. doi: 10.1074/jbc.272.47.29672. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Horvath Z, Urioste R, Hetke J, Wise K. High-frequency network oscillation in the hippocampus. Science. 1992;256:1025–1027. doi: 10.1126/science.1589772. [DOI] [PubMed] [Google Scholar]
- Camperi M, Wang XJ. A model of visuospatial working memory in prefrontal cortex: recurrent network and cellular bistability. J Comput Neurosci. 1998;5:383–405. doi: 10.1023/a:1008837311948. [DOI] [PubMed] [Google Scholar]
- Chrobak JJ, Buzsaki G. Selective activation of deep layer (V-VI) retrohippocampal cortical neurons during hippocampal sharp waves in the behaving rat. J Neurosci. 1994;14:6160–6170. doi: 10.1523/JNEUROSCI.14-10-06160.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang SC, Bianchi R, Kim D, Shin HS, Wong RK. Group I metabotropic glutamate receptors elicit epileptiform discharges in the hippocampus through PLCβ1 signalling. J Neurosci. 2001;21:6387–6394. doi: 10.1523/JNEUROSCI.21-16-06387.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YH, Sun Ahn H, Kim D, Hoon Shin D, Su Kim S, Yong Kim K, Bok Lee W, Ik Cha C. Immunohistochemical study on the distribution of TRPC channels in the rat hippocampus. Brain Res. 2006;1085:132–137. doi: 10.1016/j.brainres.2006.02.087. [DOI] [PubMed] [Google Scholar]
- Congar P, Leinekugel X, Ben-Ari Y, Crepel V. A long-lasting calcium-activated nonselective cationic current is generated by synaptic stimulation or exogenous activation of group I metabotropic glutamate receptors in CA1 pyramidal neurons. J Neurosci. 1997;17:5366–5379. doi: 10.1523/JNEUROSCI.17-14-05366.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, et al. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- Crepel V, Aniksztejn L, Ben-Ari Y, Hammond C. Glutamate metabotropic receptors increase a Ca2+-activated nonspecific cationic current in CA1 hippocampal neurons. J Neurophysiol. 1994;72:1561–1569. doi: 10.1152/jn.1994.72.4.1561. [DOI] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Mamiya A, Buzsaki G. Ensemble patterns of hippocampal CA3-CA1 neurons during sharp wave-associated population events. Neuron. 2000;28:585–594. doi: 10.1016/s0896-6273(00)00135-5. [DOI] [PubMed] [Google Scholar]
- Csicsvari J, O'Neill J, Allen K, Senior T. Place-selective firing contributes to the reverse-order reactivation of CA1 pyramidal cells during sharp waves in open-field exploration. Eur J Neurosci. 2007;26:704–716. doi: 10.1111/j.1460-9568.2007.05684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disterhoft JF, Coulter DA, Alkon DL. Conditioning-specific membrane changes of rabbit hippocampal neurons measured in vitro. Proc Natl Acad Sci U S A. 1986;83:2733–2737. doi: 10.1073/pnas.83.8.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolen G, Bear MF. Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J Physiol. 2008;586:1503–1508. doi: 10.1113/jphysiol.2008.150722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransen E, Hasselmo ME, Alonso AA. Graded persistent activity in entorhinal cortex neurons. Nature. 2002;420:173–178. doi: 10.1038/nature01171. [DOI] [PubMed] [Google Scholar]
- Faber ES, Sedlak P, Vidovic M, Sah P. Synaptic activation of transient receptor potential channels by metabotropic glutamate receptors in the lateral amygdala. Neuroscience. 2006;137:781–794. doi: 10.1016/j.neuroscience.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Falkenburger BH, Jensen JB, Hille B. Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J Gen Physiol. 2010;135:99–114. doi: 10.1085/jgp.200910345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ. Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick JS, Hagenston AM, Hertle DN, Gipson KE, Bertetto-D'Angelo L, Yeckel MF. Inositol-1,4,5-trisphosphate receptor-mediated Ca2+ waves in pyramidal neuron dendrites propagate through hot spots and cold spots. J Physiol. 2009;587:1439–1459. doi: 10.1113/jphysiol.2009.168930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler MA, Sidiropoulou K, Ozkan ED, Phillips CW, Cooper DC. Corticolimbic expression of TRPC4 and TRPC5 channels in the rodent brain. PLoS One. 2007;2:e573. doi: 10.1371/journal.pone.0000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee CE, Benquet P, Gerber U. Group I metabotropic glutamate receptors activate a calcium-sensitive transient receptor potential-like conductance in rat hippocampus. J Physiol. 2003;546:655–664. doi: 10.1113/jphysiol.2002.032961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Sanz C, Delgado-Garcia JM, Fairen A, Gruart A. Involvement of the mGluR1 receptor in hippocampal synaptic plasticity and associative learning in behaving mice. Cereb Cortex. 2008;18:1653–1663. doi: 10.1093/cercor/bhm193. [DOI] [PubMed] [Google Scholar]
- Gipson KE, Yeckel MF. Coincident glutamatergic and cholinergic inputs transiently depress glutamate release at rat schaffer collateral synapses. J Neurophysiol. 2007;97:4108–4119. doi: 10.1152/jn.01051.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel M, Sinkins WG, Schilling WP. Selective association of TRPC channel subunits in rat brain synaptosomes. J Biol Chem. 2002;277:48303–48310. doi: 10.1074/jbc.M207882200. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Kawaguchi Y. Phasic cholinergic signalling in the hippocampus: functional homology with the neocortex? Hippocampus. 2007;17:327–332. doi: 10.1002/hipo.20279. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci. 2005;25:10308–10320. doi: 10.1523/JNEUROSCI.2697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenston AM, Fitzpatrick JS, Yeckel MF. MGluR-mediated calcium waves that invade the soma regulate firing in layer V medial prefrontal cortical pyramidal neurons. Cereb Cortex. 2008;18:407–423. doi: 10.1093/cercor/bhm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenston AM, Rudnick ND, Boone CE, Yeckel MF. 2-Aminoethoxydiphenyl-borate (2-APB) increases excitability in pyramidal neurons. Cell Calcium. 2009;45:310–317. doi: 10.1016/j.ceca.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R. Muscarinic receptors regulate two different calcium-dependent non-selective cation currents in rat prefrontal cortex. Eur J Neurosci. 1999;11:1973–1980. doi: 10.1046/j.1460-9568.1999.00612.x. [DOI] [PubMed] [Google Scholar]
- Hardie RC. Regulation of TRP channels via lipid second messengers. Annu Rev Physiol. 2003;65:735–759. doi: 10.1146/annurev.physiol.65.092101.142505. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Yoshihara T, Ichitani Y. Involvement of hippocampal metabotropic glutamate receptors in radial maze performance. Neuroreport. 2007;18:719–723. doi: 10.1097/WNR.0b013e3280d9e880. [DOI] [PubMed] [Google Scholar]
- Hertle DN, Yeckel MF. Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience. 2007;150:625–638. doi: 10.1016/j.neuroscience.2007.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Ross WN. Priming of intracellular calcium stores in rat CA1 pyramidal neurons. J Physiol. 2007;584:75–87. doi: 10.1113/jphysiol.2007.137661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Jaffe DB, Brown TH. Metabotropic glutamate receptor activation induces calcium waves within hippocampal dendrites. J Neurophysiol. 1994;72:471–474. doi: 10.1152/jn.1994.72.1.471. [DOI] [PubMed] [Google Scholar]
- Kapur A, Yeckel M, Johnston D. Hippocampal mossy fibre activity evokes Ca2+ release in CA3 pyramidal neurons via a metabotropic glutamate receptor pathway. Neuroscience. 2001;107:59–69. doi: 10.1016/s0306-4522(01)00293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelliher M, Fastbom J, Cowburn RF, Bonkale W, Ohm TG, Ravid R, Sorrentino V, O'Neill C. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer's disease neurofibrillary and β–amyloid pathologies. Neuroscience. 1999;92:499–513. doi: 10.1016/s0306-4522(99)00042-1. [DOI] [PubMed] [Google Scholar]
- Khawaja FA, Alonso AA, Bourque CW. Ca2+-dependent K+ currents and spike-frequency adaptation in medial entorhinal cortex layer II stellate cells. Hippocampus. 2007;17:1143–1148. doi: 10.1002/hipo.20365. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ. Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature. 2003;426:285–291. doi: 10.1038/nature02162. [DOI] [PubMed] [Google Scholar]
- Kleppisch T, Voigt V, Allmann R, Offermanns S. Gαq-deficient mice lack metabotropic glutamate receptor-dependent long-term depression but show normal long-term potentiation in the hippocampal CA1 region. J Neurosci. 2001;21:4943–4948. doi: 10.1523/JNEUROSCI.21-14-04943.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AC, Wong RK, Chuang SC, Shin HS, Bianchi R. Role of synaptic metabotropic glutamate receptors in epileptiform discharges in hippocampal slices. J Neurophysiol. 2002;88:1625–1633. doi: 10.1152/jn.2002.88.4.1625. [DOI] [PubMed] [Google Scholar]
- Lee YM, Kim BJ, Kim HJ, Yang DK, Zhu MH, Lee KP, So I, Kim KW. TRPC5 as a candidate for the nonselective cation channel activated by muscarinic stimulation in murine stomach. Am J Physiol Gastrointest Liver Physiol. 2003;284:G604–G616. doi: 10.1152/ajpgi.00069.2002. [DOI] [PubMed] [Google Scholar]
- Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier WF, Romanin C, Zhu MX, Groschner K. Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J Biol Chem. 2000;275:27799–27805. doi: 10.1074/jbc.M002705200. [DOI] [PubMed] [Google Scholar]
- Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Actions of noradrenaline recorded intracellularly in rat hippocampal CA1 pyramidal neurones, in vitro. J Physiol. 1986;372:221–244. doi: 10.1113/jphysiol.1986.sp016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Abbott LF, Turrigiano GG, Liu Z, Golowasch J. Memory from the dynamics of intrinsic membrane currents. Proc Natl Acad Sci U S A. 1996;93:13481–13486. doi: 10.1073/pnas.93.24.13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin LR, Wong RK. Role of group I metabotropic glutamate receptors in the patterning of epileptiform activities in vitro. J Neurophysiol. 1997;78:539–544. doi: 10.1152/jn.1997.78.1.539. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Imani F, Khodakhah K, Williams JT. Inositol 1,4,5-triphosphate-evoked responses in midbrain dopamine neurons. J Neurosci. 2000;20:RC103. doi: 10.1523/JNEUROSCI.20-20-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naie K, Manahan-Vaughan D. Regulation by metabotropic glutamate receptor 5 of LTP in the dentate gyrus of freely moving rats: relevance for learning and memory formation. Cereb Cortex. 2004;14:189–198. doi: 10.1093/cercor/bhg118. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Fujiyama R, Miyamoto T, Sato T. Inositol 1,4,5-trisphosphate activates non-selective cation conductance via intracellular Ca2+ increase in isolated frog taste cells. Eur J Neurosci. 1998;10:1376–1382. doi: 10.1046/j.1460-9568.1998.00151.x. [DOI] [PubMed] [Google Scholar]
- O'Neill J, Senior T, Csicsvari J. Place-selective firing of CA1 pyramidal cells during sharp wave/ripple network patterns in exploratory behaviour. Neuron. 2006;49:143–155. doi: 10.1016/j.neuron.2005.10.037. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Partridge LD, Swandulla D, Muller TH. Modulation of calcium-activated non-specific cation currents by cyclic AMP-dependent phosphorylation in neurones of Helix. J Physiol. 1990;429:131–145. doi: 10.1113/jphysiol.1990.sp018248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge LD, Valenzuela CF. Block of hippocampal CAN channels by flufenamate. Brain Res. 2000;867:143–148. doi: 10.1016/s0006-8993(00)02275-7. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- Power JM, Sah P. Nuclear calcium signalling evoked by cholinergic stimulation in hippocampal CA1 pyramidal neurons. J Neurosci. 2002;22:3454–3462. doi: 10.1523/JNEUROSCI.22-09-03454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Thompson VL, Tate WP, Abraham WC. Metabotropic glutamate receptors trigger homosynaptic protein synthesis to prolong long-term potentiation. J Neurosci. 2000;20:969–976. doi: 10.1523/JNEUROSCI.20-03-00969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutecki PA, Sayin U, Yang Y, Hadar E. Determinants of ictal epileptiform patterns in the hippocampal slice. Epilepsia. 2002;43:179–183. doi: 10.1046/j.1528-1157.43.s.5.34.x. [DOI] [PubMed] [Google Scholar]
- Rychkov G, Barritt GJ. Handbook of Experimental Pharmacology. part I. Vol. 179. Springer; 2007. TRPC1 Ca2+-permeable channels in animal cells; pp. 23–52. [DOI] [PubMed] [Google Scholar]
- Ryu SH, Kim UH, Wahl MI, Brown AB, Carpenter G, Huang KP, Rhee SG. Feedback regulation of phospholipase C-β by protein kinase C. J Biol Chem. 1990;265:17941–17945. [PubMed] [Google Scholar]
- Sailer CA, Hu H, Kaufmann WA, Trieb M, Schwarzer C, Storm JF, Knaus HG. Regional differences in distribution and functional expression of small-conductance Ca2+-activated K+ channels in rat brain. J Neurosci. 2002;22:9698–9707. doi: 10.1523/JNEUROSCI.22-22-09698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler VM, Barbara JG. Calcium-induced calcium release contributes to action potential-evoked calcium transients in hippocampal CA1 pyramidal neurons. J Neurosci. 1999;19:4325–4336. doi: 10.1523/JNEUROSCI.19-11-04325.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem. 1989;264:17816–17823. [PubMed] [Google Scholar]
- Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE. Calcium dysregulation, IP3 signalling, and Alzheimer's disease. Neuroscientist. 2005;11:110–115. doi: 10.1177/1073858404270899. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, LaFerla FM, Parker I. Ca2+ signalling in mouse cortical neurons studied by two-photon imaging and photoreleased inositol triphosphate. J Neurosci. 2003;23:758–765. doi: 10.1523/JNEUROSCI.23-03-00758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Horowitz LF, Hirdes W, Mackie K, Hille B. Regulation of KCNQ2/KCNQ3 current by G protein cycling: the kinetics of receptor-mediated signalling by Gq. J Gen Physiol. 2004;123:663–683. doi: 10.1085/jgp.200409029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuault SJ, Davies CH, Randall AD, Collingridge GL. Group I mGluRs modulate the pattern of non-synaptic epileptiform activity in the hippocampus. Neuropharmacology. 2002;43:141–146. doi: 10.1016/s0028-3908(02)00095-3. [DOI] [PubMed] [Google Scholar]
- Vatanparast J, Janahmadi M, Asgari AR. Forskolin potentiates the paraoxon-induced hyperexcitability in snail neurons by blocking afterhyperpolarization. Neurotoxicology. 2007;28:1178–1183. doi: 10.1016/j.neuro.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Wang XJ. Synaptic reverberation underlying mnemonic persistent activity. Trends Neurosci. 2001;24:455–463. doi: 10.1016/s0166-2236(00)01868-3. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Hong M, Lasser-Ross N, Ross WN. Modulation of calcium wave propagation in the dendrites and to the soma of rat hippocampal pyramidal neurons. J Physiol. 2006;575:455–468. doi: 10.1113/jphysiol.2006.114231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyart C, Cocco S, Bourdieu L, Leger JF, Herr C, Chatenay D. Dynamics of excitatory synaptic components in sustained firing at low rates. J Neurophysiol. 2005;93:3370–3380. doi: 10.1152/jn.00530.2004. [DOI] [PubMed] [Google Scholar]
- Yamada S, Takechi H, Kanchiku I, Kita T, Kato N. Small-conductance Ca2+-dependent K+ channels are the target of spike-induced Ca2+ release in a feedback regulation of pyramidal cell excitability. J Neurophysiol. 2004;91:2322–2329. doi: 10.1152/jn.01049.2003. [DOI] [PubMed] [Google Scholar]
- Yan HD, Villalobos C, Andrade R. TRPC channels mediate a muscarinic receptor-induced afterdepolarization in cerebral cortex. J Neurosci. 2009;29:10038–10046. doi: 10.1523/JNEUROSCI.1042-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeckel MF, Kapur A, Johnston D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci. 1999;2:625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeckel MF, Sleeper AA, Fitzpatrick JS, Hertle D, Hagenston AM, Garner RT. Intracellular calcium waves transmit synaptic information to the nucleus in hippocampal pyramidal neurons. In: Dudek SM, editor. Transcirptional Regulation By Neuronal Activity. Springer Science Publishing; 2008. pp. 73–89. [Google Scholar]
- Yoshida M, Fransen E, Hasselmo ME. mGluR-dependent persistent firing in entorhinal cortex layer III neurons. Eur J Neurosci. 2008;28:1116–1126. doi: 10.1111/j.1460-9568.2008.06409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue C, Ku CY, Liu M, Simon MI, Sanborn BM. Molecular mechanism of the inhibition of phospholipase C β3 by protein kinase C. J Biol Chem. 2000;275:30220–30225. doi: 10.1074/jbc.M004276200. [DOI] [PubMed] [Google Scholar]
- Zhang C, Roepke TA, Kelly MJ, Ronnekleiv OK. Kisspeptin depolarizes gonadotropin-releasing hormone neurons through activation of TRPC-like cationic channels. J Neurosci. 2008;28:4423–4434. doi: 10.1523/JNEUROSCI.5352-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zho WM, You JL, Huang CC, Hsu KS. The group I metabotropic glutamate receptor agonist (S)-3,5-dihydroxyphenylglycine induces a novel form of depotentiation in the CA1 region of the hippocampus. J Neurosci. 2002;22:8838–8849. doi: 10.1523/JNEUROSCI.22-20-08838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FW, Matta SG, Zhou FM. Constitutively active TRPC3 channels regulate basal ganglia output neurons. J Neurosci. 2008;28:473–482. doi: 10.1523/JNEUROSCI.3978-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]