

Abstract
Transforming growth factor β (TGF-β) physiologically induces Epstein-Barr virus (EBV) lytic infection by activating the expression of EBV's latent-lytic switch BZLF1 gene. Liang et al. (J. Biol. Chem. 277:23345–23357, 2002) previously identified a Smad-binding element (SBE) within the BZLF1 promoter, Zp; however, it accounts for only 20 to 30% of TGF-β-mediated activation of transcription from Zp. Here, we identified additional factors responsible for the rest of this activation. The incubation of EBV-positive MutuI cells with a TGF-β neutralizing antibody or inhibitors of the TGF-β type I receptor (TβRI) or Smad3 eliminated the TGF-β-induced reactivation of EBV. The coexpression of Smad2, Smad3, and Smad4 together with a constitutively active form of TβRI induced 15- to 25-fold transcription from Zp in gastric carcinoma AGS cells. By electrophoretic mobility shift assays, we identified four additional Smad-binding elements, named SBE2 to SBE5. Substitution mutations in individual SBEs reduced Smad-mediated activation of Zp by 20 to 60%; together, these mutations essentially eliminated it. Chromatin immunoprecipitation assays confirmed that Smad4 newly bound the Zp region of the EBV genome following the incubation of MutuI cells with TGF-β. SBE2 overlaps the ZEB-binding ZV silencing element of Zp. Depending upon posttranslational modifications, Smad4 either competed with ZEB1 for binding or formed a complex with ZEB1 on the Zp ZV element in a cell-free assay system. In transiently transfected cells, exogenously expressed ZEB1 inhibited Smad-mediated transcriptional activation from Zp. We conclude that TGF-β induces EBV lytic reactivation via the canonical Smad pathway by activating BZLF1 gene expression through multiple SBEs acting in concert.
INTRODUCTION
Epstein-Barr virus (EBV) is a human gamma herpesvirus that infects most of the world's population (62). After primary infection, EBV establishes latency in its host, occasionally reactivating into lytic replication, which leads to the production of infectious virus. Thus, reactivation provides a mechanism for EBV to transmit itself to new hosts. The latent form of infection allows EBV to maintain its genome within hosts while evading the immune system.
The switch from latency to lytic replication usually is initiated in EBV-infected cells by activating the expression of the viral immediate-early (IE) BZLF1 gene (38). The BZLF1 (also called Z, Zta, Zebra, and EB1) protein performs several functions, including binding to the viral lytic origin of DNA replication, oriLyt, decreasing the expression of the viral latency promoters, Cp and Wp, and activating the expression of other lytic IE and early (E) genes. Thus, the regulation of BZLF1 gene expression is central to the EBV life cycle.
Transcription from the promoter of the BZLF1 gene, Zp, can be induced by multiple agents, including phorbol esters such as 12-O-tetradecanoyl-phorbol-13-acetate (TPA), calcium ionophores such as ionomycin, sodium butyrate, transforming growth factor-β (TGF-β), and anti-immunoglobulins (anti-Igs) (19, 38). Each of these agents utilizes different cellular signaling pathways to activate BZLF1 gene expression and is dependent upon cell type and EBV latency state. The treatment of B cells with anti-Ig or TGF-β mimics physiologically relevant mechanisms of EBV reactivation into lytic replication.
TGF-β is a cytokine secreted by numerous cell types to regulate cellular processes such as proliferation, differentiation, apoptosis, migration, and immune responses (13, 47, 48). Signaling via the TGF-β pathway alters the expression of more than 500 genes to exert a variety of cellular responses. The pathway is activated by the binding of the TGF-β1 ligand to the TGF-β type II receptor (TβRII), thereby triggering the heterodimerization and transphosphorylation of the TGF-β type I receptor (TβRI). The signal is propagated by TβRI phosphorylating the intracellular proteins Smad2 and Smad3, which then complex with Smad4. This activated complex can shuttle to the nucleus, where it modulates gene expression by binding, along with numerous transcriptional coactivators and corepressors, to Smad-binding elements (SBEs) located within promoters. The consensus binding sequences for Smad proteins are 5′-GTCT-3′ and its complement, 5′-AGAC-3′ (66, 76). Smad binding to DNA is fairly weak. Thus, sufficient DNA-binding affinity for transcriptional activation is achieved by the incorporation of one or more transcriptional cofactors into the activated Smad2/3/4 complex (64, 66).
ZEB1 (also called δEF1, TCF8, AREB6, ZFHEP, NIL-2A, ZFHX1A, and BZP) is one of many transcription factors known to be regulated by TGF-β (4, 5, 58, 59, 67). Our laboratory has identified ZEB1 as a key cellular protein repressing EBV lytic replication in most EBV-positive epithelial and B-cell lines (16, 17, 41, 74, 75). ZEB1 binds via its two zinc finger domains to sequences resembling 5′-CAGGTA-3′ located at nucleotides (nt) −17 to −12 and nt +5 to +10 relative to the transcription initiation site of Zp, referred to as ZV and ZV′, respectively (41, 74, 75). Variants of EBV containing mutations in them spontaneously reactivate out of latency at significantly higher frequencies than does the parental B95.8 strain of EBV (74, 75). Unknown is the mechanism by which the ZEB1 repression of BZLF1 gene expression is alleviated during the induction of EBV lytic replication.
Liang et al. (43) previously reported that TGF-β disrupts EBV latency by activating BZLF1 gene expression through a Smad-binding element, 5′-GTCTG-3′, located at nt −233 to −229 relative to the transcription initiation site of Zp. However, this SBE1 accounts for only 20 to 30% of the total TGF-β-mediated activation of transcription from Zp in transient-transfection assays. Therefore, additional factors likely also significantly contribute to the mechanism by which TGF-β activates transcription from this viral promoter.
Here, we investigated how TGF-β induces the reactivation of EBV out of latency into lytic replication. We show that this activation is mediated through the canonical TGF-β/Smad pathway. We identified four additional Smad-binding elements within the nt −221 to +12 region of Zp, named SBE2 to SBE5. All of these Smad-binding elements, including the previously reported SBE1, contribute in part to the TGF-β-mediated activation of BZLF1 gene expression; together they account for essentially all of it. SBE2 overlaps the ZEB1-binding ZV element of Zp. TGF-β-activated Smad4 forms a complex with ZEB1 on the ZV element. We conclude that TGF-β induces EBV reactivation by activating and alleviating ZEB-mediated repression of Zp through multiple SBEs acting in concert.
MATERIALS AND METHODS
Reagents.
Porcine TGF-β1 and polyclonal anti-TGF-β neutralizing antibody (clone 1D11) were purchased from R&D Systems. Goat-human IgG was purchased from Sigma (I5260). The inhibitors SP600125, Y27632, LY294002, Y27632 (Calbiochem), SB431542, SIS3 (Sigma), and U0126 (Promega) were reconstituted in dimethylsulfoxide (DMSO) and stored at −80°C. Stock solutions of U0126 were used within 1 week.
Cell lines.
EBV-positive Burkitt's lymphoma (BL) Akata cells were obtained from Paul Farrell, EBV-negative T-lymphoblastic Jurkat cells were purchased from the American Type Culture Collection (ATCC), EBV-positive BL MutuI and MutuIII cells were obtained from Alan Rickinson, and EBV-negative gastric carcinoma AGS cells were obtained from Shannon Kenney. Simian virus 40 (SV40)-transformed human embryonic kidney 293T cells were purchased from the ATCC. Akata, MutuI, and MutuIII cells were grown in RPMI medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (HyClone, Thermo Scientific). The 293T cells were grown in RPMI or Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% FBS. AGS cells were maintained in F12 medium supplemented with 10% FBS and 100 U penicillin and streptomycin per ml. All cells were maintained in a 37°C humidified 5% CO2 incubator.
Plasmids.
Plasmid DNAs were purified using Qiagen plasmid maxikits as described by the manufacturer. Expression plasmids encoding Flag-tagged human Smad2 (pCS2-FLAG-Smad2), Flag-tagged human Smad3 (pCS2-FLAG-Smad3), and a constitutively active mutant variant of the TGF-β type I receptor, designated ALK5 CA (pcDNA3-ALK5 T204D), were gifts from F. Michael Hoffmann. A Smad4 expression plasmid, pCMV6-XL5-Smad4, was purchased from Origene. Plasmid pcDNA3.1 served as the empty vector control. The expression plasmid pCi-ZEB1, generously provided by Douglas Dean, encodes full-length human ZEB1. The firefly luciferase reporter pZp-Luc-WT, a gift from Shannon Kenney, contains the nt −496 to +28 region of Zp relative to the transcription initiation site cloned into the vector pGL3-Basic (Promega).
Substitution mutants.
The site-directed mutagenesis of pZp-Luc-WT was performed using 100 ng template, 20 pmol of each primer set, 1× MasterAmp PCR enhancer (Epicentre Biotechnologies), and 2.5 U of Pfu Turbo (Stratagene). The amplifications were performed with a GeneMate Genius PCR machine (ISC BioExpress) for 35 cycles with an initial denaturation for 4 min at 95°C, denaturation for 1 min at 94°C, annealing for 50 s at 50°C, and extension for 7 min at 68°C, followed by a final extension for 15 min at 72°C.
The primers for site-directed mutagenesis (sense strand, mutations indicated by underlining) were the following: nt −241 to −221 (SBE1m), 5′-TGATGAATTTTTGCTGCATGC-3′; nt −211 to −184 (SBE3m), 5′-TTCAACTGGGCTTTTTATTTTTGACACC-3′; nt −185 to −155 (SBE4m), 5′-CCAGCTTATTTTCAACACTTCTGAAAACTGC-3′; nt −114 to −81 (SBE5m), 5′-GCTAATGTACCTCATCAACACACCTAAATTTAGC-3′; nt −33 to −1 (SBE2m), 5′-AAGGGGAGATGTTCAACAGGTAACTCACTAAAC-3′; and nt −26 to +20 (ZV/ZV′dm), 5′-GATGTTAGACAGGCCACTCACTAAACATTGCATTTTGCCGGCCACC-3′. Afterward, the PCRs were incubated with DpnI (New England BioLabs) at 37°C for 2.5 h and transformed into Escherichia coli JM109 cells (Promega) according to the manufacturer's instructions. All plasmids were sequenced to verify that they contained the desired mutations.
Reporter assays.
EBV-negative AGS cells were seeded in 24-well plates. Twenty-four hours later, the indicated plasmid DNAs were cotransfected into the cells using Lipofectamine 2000 reagent (Invitrogen). After incubation for an additional 44 to 48 h, the cells were harvested. Luciferase assays were performed with the Luciferase reporter assay system (Promega) as described by the manufacturer.
Immunoblot analysis.
Cells were harvested and lysed in SUMO buffer followed by sonication as previously described (3). For the detection of the EBV lytic proteins BZLF1 and BMRF1, proteins were loaded onto SDS 4 to 20% gradient polyacrylamide gels (ISC BioExpress), separated by electrophoresis at 100 V for 45 min, and transferred to nitrocellulose membranes (ISC Biosystem). The primary antibodies used for the detection of specific proteins were the following: BZLF1 (1:250; BZ1; Santa Cruz Biotechnology), BMRF1 (1:500; VP-E608; Vector Laboratories), and Smad4 (1:500; H-552; Santa Cruz Biotechnology). The secondary antibody was horseradish peroxidase (HRP)-conjugated mouse (1:1,000; 31430; ThermoScientific) or rabbit (1:1,000; NA934; GE Healthcare) antibody. Blots were developed by enhanced chemiluminescence (ECL) (GE Healthcare). The blots then were stripped by incubation in 100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl (pH 8.2) at 65°C for 1 h and reprobed with antibody specific to β-actin (1:1,000; A2228; Sigma) or α-tubulin (1:1,000; T9026; Sigma) as a loading control.
Fluorescence cytometry.
MutuI cells were fixed by incubation with 1% paraformaldehyde for 10 min in a 37°C humidified 5% CO2 incubator. The cells were washed twice with 1× PBS containing 0.1 mM phenylmethylsulfonyl fluoride (PMSF) and permeabilized by incubation for 5 min at room temperature with 1× PBS containing 0.1 mM PMSF, 0.1% Triton X-100. The cells then were incubated for 1 h with primary antibody specific for BMRF1 (1:50;VP-E608; Vector Laboratories), followed by incubation for 15 min with an Alexa Fluor anti-mouse IgG secondary antibody (1:1,000; A11017; Invitrogen) at room temperature. Cells were analyzed by fluorescence-activated cell sorting (FACS) with a FACSCalibur (BD Bioscience) at an excitation length of 488 nM.
ChIP.
For chromatin immunoprecipitation (ChIP), MutuI cells (5 × 105 cells per IP) were fixed by addition to the cell culture medium of formaldehyde (Sigma) to a final concentration of 1% and incubation for 10 min at 37°C. The cells then were incubated for 1 min with 125 mM glycine to stop the cross-linking reaction. The cells were pelleted by centrifugation at 3,000 rpm for 5 min at 4°C and rinsed with PBS. The pellet was resuspended in nucleus lysis buffer (71) and incubated for 10 min at 0°C. Chromatin was sheared to an average length of 500 bp by three pulses of 15 s each at 4°C with a Model 60 sonic dismembrator (Fisher Scientific) at an output setting of 2. Samples were cleared by incubation at 4°C with rotation for 1 h with ChIP-grade protein G magnetic beads (Cell Signaling). A portion of each precleared chromatin sample was saved as the input. The rest was immunoprecipitated by incubation overnight at 4°C with antibody specific to Smad4 (1:50; no. 9515; Cell Signaling) or IgG (1:500; I5260; Sigma). The immunoprecipitates were incubated for 2 h at 4°C with ChIP-grade protein G magnetic beads. The cross-links were reversed by incubation overnight at 65°C. DNAs were purified with a PCR cleanup kit in Buffer PB (Qiagen). The specific promoter DNAs were detected by PCR with a GeneMate Genius PCR machine (ISC BioExpress) using 50 nM each primer set and Pfu Turbo (Stratagene). The amplifications were performed in the following manner: initial denaturation for 4 min at 95°C, denaturation for 10 s at 95°C, annealing for 30 s at an optimal temperature for each primer set (indicated in parentheses below), and extension for 30 s at 72°C. The primers were the following: BZLF1 (58.2°C) FWD, 5′-TTGACACCAGCTTATTTTAGACAC-3′, and REV, 5′-TTACCTGTCTAACATCTCCCCTTT-3′; and Smad7 (55°C) FWD, 5′-CTGCCTAGGGCATTCATT-3′, and REV, 5′-CTGCCTAGGGCATTCATT-3′ (39). The following primers were used for the EBV sequence located 4.8-kbp upstream of the Zp transcription initiation site (55°C): FWD, 5′-AGAAGGGAGACACATCTGGA-3′, and REV, 5′-AACTTGGACGTTTTTGGGGT-3′ (75). PCR products were analyzed by electrophoresis in 1.5% agarose gels and visualized with UV light after being stained with ethidium bromide.
Smad4 protein.
Purified amino-terminal 6×His-tagged full-length human Smad4 protein produced in E. coli was purchased from ProSpec-Tany TechnoGene (Israel). Amino-terminal glutathione S-transferase (GST)-tagged full-length human Smad4 protein, purified from a wheat germ in vitro translation reaction, was purchased from Abnova (Taiwan). To obtain Smad4 from human cells, we first subcloned full-length human Smad4 cDNA from pCMV6-XL5-Smad4 into pFN21A Halo Tag Flexi vector (Promega) by PCR amplification using as primers 5′-CGAAGCGATCGCCATGGACAATATGTCTATTACGAATACACCAACAAG-3′ and 5′-GTGCGTTTAAACGTCTAAAGGTTGTGGGTCTGCAAT-3′. To produce Halo-tagged Smad4 protein in TGF-β-activated human cells, 9 μg of pFN21A-Smad4 and 3 μg of pcDNA3-ALK5 T204D were cotransfected into 15-cm plates of 293T cells using TransIT-LT1 (Mirus Bio). After incubation for 48 h, the cells were washed with PBS, scraped off the dish, and lysed by sonication in lysis buffer [50 mM HEPES (pH 7.5), 150 mM NaCl, 0.5 mM EDTA, 0.005% IGEPAL CA-630] supplemented with 0.5 mM PMSF and a 1:1,000 dilution of protease inhibitor cocktail III (EMD Biosciences). The purification of the Halo-tagged Smad4 protein was performed as previously described (9), with modifications. In brief, the lysate was cleared by centrifugation at 3,300 rpm for 30 min and incubated with HaloLink resin (Promega) overnight at 4°C on a rotating platform. The resin was washed twice with lysis buffer, incubated with lysis buffer supplemented with 1 M urea for 20 min at room temperature on a rotating platform, and washed twice more with lysis buffer without urea. The HaloLink resin was placed in cleavage buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 0.5 mM EDTA, 0.1 mM dithiothreitol [DTT], 15 μg/ml tobacco etch virus protease), incubated at room temperature for 2 h on a rotating platform, and pelleted by centrifugation at 3,300 rpm for 5 min. The supernatant, containing the Smad4 protein cleaved from its tag, was snap-frozen in liquid nitrogen and stored at −80°C. Protein concentration was determined by a Pierce 660 nm protein assay (Thermo Scientific) with bovine serum albumin (BSA) as a protein standard.
ZEB1 protein.
Extracts containing human ZEB1 protein were prepared by the transfection of 293T cells in 10-cm-diameter dishes with 2 μg of pCi-ZEB1 using TransIT-LT1. Two days later, the cells were harvested and lysed in 200 μl of Freedman's buffer (150 mM NaCl, 50 mM Tris-HCl [pH 7.5], 10% glycerol, 0.1% Nonidet-P40, 0.1 mM NaF, 2 mM EDTA, 0.2 mM PMSF). Lysates were cleared by centrifugation at 10,000 rpm for 5 min at 4°C and stored at −80°C.
EMSAs.
Electrophoretic mobility shift assays (EMSAs) were performed as previously described (46), with modifications. In brief, double-stranded synthetic oligonucleotides (UW-Biotechnology Center) were incubated with T4 polynucleotide kinase (New England BioLabs) and 50 μCi [γ-32P]ATP for 1 h at 37°C. Unincorporated nucleotides were removed with G-50 Sephadex quick spin columns (GE Healthcare). Binding reactions (22-μl total volume) contained 3.5 μl of HGED.0 buffer (20 mM HEPES [pH 7.9], 0.2 mM EDTA, 20% glycerol), 8 μl of HGED.1 buffer (20 mM HEPES [pH 7.9], 0.2 mM EDTA, 20% glycerol, 0.1 M KCl), 1 μg BSA (Thermo Scientific), 1 μg poly(dI-dC)(dI-dC) (Sigma), 0.45 μM ZnCl2, 0.25% Tween 20, 8 ng 32P-labeled double-stranded oligonucleotide probe DNA, and the indicated protein extracts or purified proteins. For the supershift assays, 2 μg of anti-Smad4 antibody (B-8X; Santa Cruz Biotechnology) or anti-ZEB1 antibody (E-20X; Santa Cruz Biotechnology) was incubated with the reaction mixture prior to the addition of the radiolabeled probe. For the DNA competition assays, the indicated amount of the unlabeled double-stranded oligonucleotide was added to the reaction mixture before the addition of the radiolabeled probe. Reaction mixtures were electrophoresed at 232 V for 105 min at 4°C in 4% nondenaturing polyacrylamide gels containing 0.5× Tris-borate-EDTA buffer. Gels were dried and imaged with a Storm PhosphorImager (GE Healthcare).
Double-stranded oligonucleotide DNA sequences for probes and competitors (sense strand shown; mutations are indicated by underlined letters) were the following: consensus SBE, 5′-GGAGTATGTCTAGACTGAGGC-3′; Zp nt −241 to −221 WT (SBE1), 5′-TGATGAATGTCTGCTGCATGC-3′; Zp nt −207 to −187 WT (SBE3), 5′-ACTGGGCTGTCTATTTTTGAC-3′; Zp SBE3 mt −199/-197 (SBE3m), 5′-ACTGGGCTTTTTATTTTTGAC-3′; Zp nt −182 to −162 WT (SBE4), 5′-GCTTATTTTAGACACTTCTGA-3′; Zp SBE4 mt −173/−172 (SBE4m), 5′-GCTTATTTTCAACACTTCTGA-3′; Zp SBE4 dmt −173/−172/−166/−165 (SBE4dm), 5′-GCTTATTTTCAACACTGTTGA-3′; Zp nt −108 to −88 WT (SBE5), 5′-GTACCTCATAGACACACCTAA-3′; Zp SBE5 mt −99/−98 (SBE5m), 5′-GTACCTCATCAACACACCTAA-3′; Zp nt −26 to −6 WT (SBE2), 5′-GATGTTAGACAGGTAACTCAC-3′; Zp SBE2 mt −20/−19 (SBE2m), 5′-GATGTTCAACAGGTAACTCAC-3′; and Zp ZV mt −13/−12 (SBE2-ZVm), 5′-GATGTTAGACAGGCCACTCAC-3′.
Determination of IC50.
The half-maximal inhibitory concentration (IC50) of an SBE was the concentration of unlabeled double-stranded oligonucleotide containing the SBE at which the binding of GST-Smad4 protein to the 32P-radiolabeled consensus SBE was inhibited by 50%. Bands corresponding to the GST-Smad4/DNA complexes in the competition EMSAs were scanned by densitometry, with their intensities being quantified using Image J software (NIH). Dose-response curves were obtained by plotting percent inhibition versus log10 concentration of unlabeled oligonucleotide. IC50s were calculated by nonlinear regression analysis using GraphPad Prism 4.0 software (GraphPad Software, Inc.).
RESULTS
TGF-β reactivation of EBV-positive cells.
EBV-positive MutuI is a representative Burkitt's lymphoma cell line (25) in type I latency, restricted to expressing a minimal set of latency genes, including EBV nuclear antigen 1 (EBNA1), EBV-encoded small RNAs (EBERs), and BamHI-A rightward transcripts (BART) (8). The EBV-positive MutuIII cell line was derived from MutuI following a latency conversion event; it is in type III latency, displaying a fuller set of EBV latency genes (26). While MutuI cells contain an intact, functional TGF-β pathway, MutuIII cells are not responsive to TGF-β, since they do not express the TGF-β type II receptor (30, 31). In the absence of TGF-β, the EBV immediate-early (IE) lytic BZLF1 protein was barely detectable in MutuI cells by immunoblot analysis (Fig. 1 A, lane 4). The incubation of MutuI cells with 100 pM TGF-β1 induced the synthesis of BZLF1 and BMRF1 (also called EA-D) protein (Fig. 1A, lanes 5 and 6). The latter is the product of an EBV early (E) lytic gene. On the other hand, the incubation of MutuIII cells with 100 pM TGF-β1 failed to induce the synthesis of BZLF1 or BMRF1 (Fig. 1A, lanes 2 and 3). Taken together, these findings show that MutuI cells can be used to study the mechanism of TGF-β-induced EBV lytic reactivation out of latency type I.
Fig. 1.
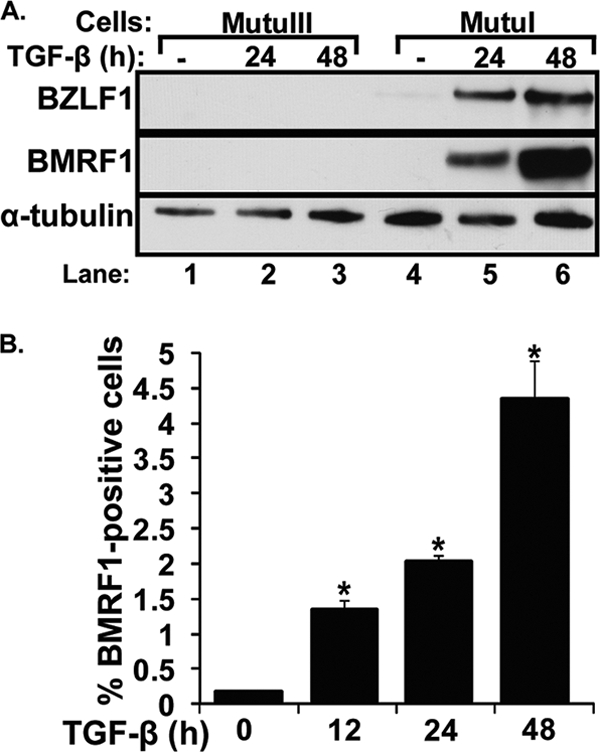
TGF-β1 induces EBV lytic gene expression in MutuI cells. (A) Immunoblots showing IE BZLF1 and E BMRF1 protein levels following treatment with TGF-β1. MutuI and MutuIII cells were incubated for the indicated times with or without (−) 100 pM TGF-β1. Cell lysates were prepared, and relative protein levels of BZLF1, BMRF1, and α-tubulin (as an internal control) were determined by immunoblot analysis. (B) Percentage of MutuI cells containing the EBV BMRF1 protein at various times after the addition of 100 pM TGF-β1. Cells were harvested at the indicated times and analyzed by FACS for the percentage of BMRF1-positive ones. Bars indicate means ± standard errors of the means (SEM) from assays performed in triplicate on three separate occasions. Significant differences (P < 0.05) are indicated with an asterisk.
We used FACS analysis to determine the percentage of the MutuI cells induced into lytic gene expression following treatment with TGF-β1. Approximately 4 to 5% of them contained BMRF1 protein by 48 h after the addition of 100 pM TGF-β1 (Fig. 1B). Thus, we conclude that TGF-β1 induces EBV reactivation, but only in a subset of these MutuI cells, within 2 days.
Reactivation of EBV via T-cell secretion of TGF-β.
Most knowledge regarding the understanding of EBV reactivation out of latency into lytic replication comes from the addition of exogenous factors to EBV-positive cells grown in cell culture by themselves in the absence of other types of cells. Since this is a somewhat artificial situation, we hypothesized that the EBV lytic cycle is naturally induced by immune cells secreting growth factors or cytokines such as TGF-β. To test this possibility, we examined whether EBV-positive MutuI cells can be induced into expressing EBV lytic genes by coculturing them with EBV-negative Jurkat cells, a T-cell line known to secrete TGF-β1 (36). As hypothesized, the coculture of MutuI and Jurkat cells led to the considerable production of BZLF1 protein, almost to the level observed following the direct addition of TGF-β1 to the medium (Fig. 2, lane 7 versus lane 5). The addition of a neutralizing antibody that inhibits the binding of TGF-β ligand to its receptor (11) blocked this induction (Fig. 2, lane 6). As a control, we also examined whether coculturing Jurkat cells can induce BZLF1 gene expression in EBV-positive Akata cells, a BL cell line that does not express TGF-β type II receptor (TβRII) (14, 31, 69). In this case, the presence of the Jurkat cells failed to induce the synthesis of BZLF1 protein (Fig. 2, lane 4). These findings demonstrate that Jurkat cells can specifically induce EBV lytic gene expression in MutuI cells via the TGF-β signaling pathway.
Fig. 2.
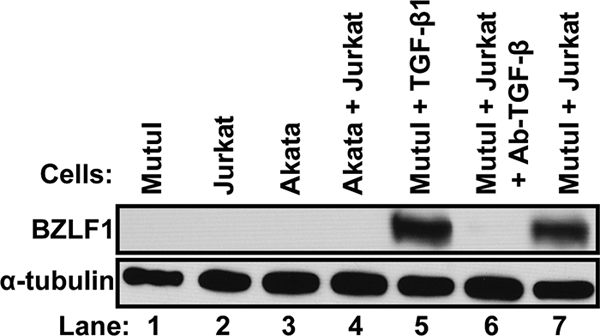
TGF-β1 secreted by T cells reactivates EBV in MutuI cells. Immunoblot showing relative levels of BZLF1 protein after culture of the indicated cells for 48 h under the indicated conditions. Whole-cell extracts were prepared and BZLF1 protein was assayed by immunoblot analysis, with α-tubulin serving as an internal control. Lane 5, cells cultured in the presence of 100 pM TGF-β1; lane 6, cells cultured in the presence of 2.5 μg TGF-β neutralizing antibody; other lanes, cells cultured in medium containing neither added TGF-β nor antibody.
TGF-β1 induces EBV via signaling its canonical Smad pathway.
To begin to determine the mechanism by which TGF-β induces EBV reactivation in MutuI cells, we examined the effects on this induction of kinase inhibitors targeting TβRI, Smad3, Jun NH-terminal kinase (JNK), phosphatidylinositol-3 kinase (PI3K), and mitogen-activated protein kinase (MAPK) kinase/extracellular signal-regulated kinase activator kinase (MEK1) with SB431542, SIS3, SP600125, LY294002, and U0126, respectively. The concentration of each inhibitor added to the cell culture medium was chosen to minimize off-target effects (12, 33, 35). Compared to that of the untreated control, the incubation of MutuI cells with TGF-β1 for 24 h greatly increased the expression of both BZLF1 and BMRF1 (Fig. 3, lane 2 versus lane 1). The addition of either the TβRI inhibitor SB431542 or the Smad3 inhibitor SIS3 completely prevented this induction of lytic gene expression by TGF-β (Fig. 3, lanes 3 and 4, respectively). Thus, TβRI specifically signals the Smad pathway to induce the synthesis of EBV lytic proteins. The addition of either the MEK1/2 inhibitor U0126 or JNK inhibitor SP600125 had little or no effect (Fig. 3, lanes 7 and 5, respectively), while the PI3K inhibitor LY294002 partially prevented this TGF-β induction (Fig. 3, lane 6). The incubation of these cells with the same concentration of U0126 completely inhibited the epidermal growth factor (EGF)-induced phosphorylation of ERK (data not shown), indicating that this drug had retained its activity. Thus, we conclude that TGF-β1 induces EBV lytic gene expression in MutuI cells primarily through the canonical Smad pathway.
Fig. 3.
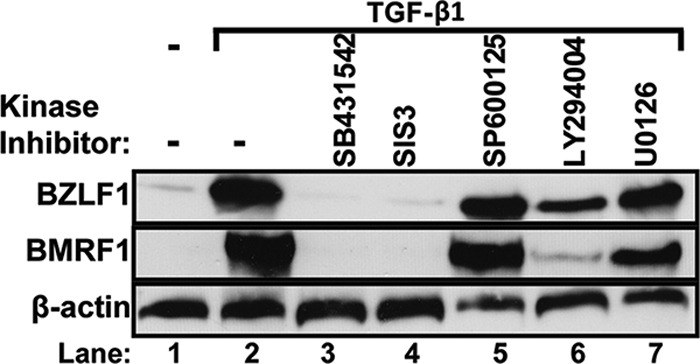
TGF-β1 induces EBV reactivation via Smad proteins. Immunoblots showing relative BZLF1 and BMRF1 protein levels following the incubation of MutuI cells for 24 h with 100 pM TGF-β1 plus the indicated kinase inhibitors. Whole-cell extracts were prepared and proteins were assayed by immunoblot analysis, with β-actin serving as an internal control. Lane 1, no TGF-β1; lane 2, only TGF-β1; lane 3, 1 μM SB431542; lane 4, 1 μM SIS3; lane 5, 10 μM SP600125; lane 6, 1 μM LY294002; and lane 7, 10 μM U0126.
Identification of novel SBEs in Zp.
Liang et al. (43) previously reported the presence of an SBE, located at nt −233 to −229 of Zp relative to the transcription initiation site, referred to here as SBE1, that contributes to the TGF-β induction of BZLF1 gene expression. However, base pair substitution mutations that knock out the binding of Smads to SBE1 result in only a 20 to 30% loss of TGF-β-mediated activation of Zp in transient-transfection assays, a conclusion we confirmed here (see Fig. 8A). Thus, we hypothesized that other functionally important SBEs exist within Zp. To begin to test this possibility, we examined the nt −221 to +12 region of Zp, which is known to be sufficient for the activation of BZLF1 gene expression by inducers such as phorbol esters (22, 34), for putative Smad4-binding elements that might contribute to its induction by TGF-β. We identified four regions containing 5′-GTCT-3′, or its complement, 5′-AGAC-3′, which we named SBE3 (nt −199 to −196), SBE4 (nt −173 to −170), SBE5 (nt −99 to −96), and SBE2 (nt −20 to −17) (Fig. 4 A and B). To determine if Smad4 actually binds any of these putative SBEs, we performed EMSAs using a GST-tagged human Smad4 protein purified from a wheat germ in vitro translation reaction mixture as the protein source and radiolabeled double-stranded 21-mer oligonucleotides spanning the putative Smad-binding elements or mutant variants of them as the probes (Fig. 4C). For a negative control, EMSAs were performed using purified GST protein as the protein source (Fig. 4C, lanes 2, 6, 11, 17, and 22). As a positive control, we performed the assay using as probe an oligonucleotide containing SBE1. As expected, Smad4 bound SBE1 (Fig. 4C, lane 3); the mobilities of the Smad4/SBE1 complexes were retarded when an antibody specific to Smad4 was included in the reaction mixture (Fig. 4C, lane 4). DNA-protein complexes were observed with these same mobilities when the probe DNA included SBE3, SBE4, SBE5, or SBE2 (Fig. 4C, lanes 7, 12, 18, and 23, respectively); the addition of the Smad4-specific antibody resulted in the supershifting of these complexes to the same slower mobilities observed with the SBE1 probe DNA (Fig. 4C, lanes 8, 13, 19, and 24, respectively, versus lane 4). To verify that Smad4 bound these DNAs via their SBE, we also performed EMSAs with variants of these DNAs containing the 2-bp substitution mutations indicated in Fig. 4B. As expected, these mutations eliminated the binding of Smad4 (Fig. 4C, lanes 9, 14, 15, 20, and 25). We also examined the binding of Smad4 to the two SBE-like elements, 5′-TCTG-3′ or its complement, 5′-CAGA-3′, within Zp, previously noted by Liang et al. (43); Smad4 bound only very weakly to them, if at all (data not shown) (Fig. 5). Therefore, we conclude that the nt −221 to +12 region of Zp contains four previously unidentified SBEs.
Fig. 8.

SBEs act cooperatively to enhance transcription from Zp. (A) Effects of mutations in SBEs on the Smad-mediated activation of Zp. AGS cells in 24-well plates were cotransfected with 80 ng of pZp-Luc-WT or the indicated variant of it containing the SBE base pair substitution mutations shown in Fig. 4B (i) and 350 ng of pcDNA3.1 or 100 ng each of pCS2-FLAG-Smad2, pCS2-FLAG-Smad3, pCMV6-XL5 Smad4, and 50 ng of pcDNA3-ALK5 T204D (ii). Two days later, cell lysates were prepared and firefly luciferase activities were measured. Data were internally normalized to the amount of protein in each lysate and externally normalized to the basal level shown in panel B observed with each reporter in the absence of the activated Smads. (B) Effects of mutations in the SBEs on basal transcription from Zp. AGS cells were cotransfected with 80 ng of pZp-Luc-WT or the indicated mutant variant of it along with 350 ng of pcDNA3.1. Data were internally normalized to the amount of protein in each lysate and externally normalized to the level of luciferase activity observed with pZp-Luc-WT. Bars indicate means ± SD of data obtained from assays performed in quadruplicate. The results presented here are typical of data observed in four independent experiments. (C) Effect of activated Smads on expression of the promoterless luciferase reporter, pGL3-Basic. The experiment was performed as described for panels A and B but on a different day.
Fig. 4.

Identification of novel Smad-binding elements within Zp. (A) Schematic diagram indicating cis-acting elements present within the nt −237 to +15 region of Zp relative to the transcription initiation site. Rectangles along Zp indicate approximate locations of regulatory elements with their known trans-acting factors indicated above them and SBEs indicated below. (B) Sequence of the nt −237 to +15 region of Zp. SBEs are indicated within rectangles. The base pair substitution mutations (m) in the SBEs studied here are indicated by italicized letters below the wild-type sequence. Locations of the previously reported cis-acting Z elements are indicated by lines below the sequence. The transcription initiation site is indicated by a rightward arrow. (C) EMSAs showing binding of Smad4 to SBEs. EMSAs were performed with the indicated radiolabeled double-stranded oligonucleotides (8 ng) as probes. Purified GST-Smad4 or GST (120 ng) was used as indicated as the protein source. Immunoshift assays were performed with 2 μg of antibody specific to Smad4. Locations of the protein/DNA complexes are indicated.
Fig. 5.

Competition EMSAs used to determine relative binding affinities of Zp SBEs for Smad4. Purified GST-Smad4 (100 ng) was incubated with 8 ng of radiolabeled double-stranded consensus SBE and various amounts of the indicated unlabeled double-stranded DNAs as competitors. Immunoshift assays were performed with 2 μg of antibody specific to Smad4. Locations of the protein/DNA complexes are indicated. Amounts of unlabeled double-stranded competitors used were the following: 5, 10, 20, and 40 ng for consensus SBE; 40, 100, 250, 625, 1,562, and 3,906 ng for SBE1, SBE2, SBE3, SBE4, and SBE5; and 40, 100, 250, 625, 1,562, 3,906, and 9,765 ng for SBE-like2 and SBE2m. The IC50s shown below the gels were determined from the concentrations of unlabeled oligonucleotide required to inhibit the binding of GST-Smad4 to the radiolabeled consensus SBE probe by 50%.
To determine the relative binding affinities of Smad4 for these SBEs, we performed competition EMSAs with a radiolabeled probe containing a consensus SBE and unlabeled competitor DNAs corresponding to the double-stranded SBE-containing oligonucleotides from Fig. 4. As expected, consensus SBE DNA strongly competed with the probe for binding Smad4; the DNAs containing the other Zp SBEs also competed for binding Smad4 but with somewhat lower affinities (Fig. 5). Quantifying these data, we obtained the following relative Smad4-binding affinities, confirming the lack of binding of Smad4 to the SBE-like2 sequence in Zp: consensus SBE > SBE1 > SBE2 ∼ SBE3 > SBE5 > SBE4 > SBE2m > SBE-like2.
TGF-β1 promotes Smad binding to Zp in vivo.
We performed chromatin immunoprecipitation assays to determine whether Smad4 becomes newly associated with the Zp region of the EBV genome following the treatment of MutuI cells with TGF-β1. We chose to assay for Smad4 since (i) it is expressed in many commonly used Burkitt's lymphoma B-cell lines (31), (ii) it contains a DNA-binding domain (48), and (iii) Smad4 is required for the activated Smad complex to enhance transcription (48). EBV-positive MutuI cells were incubated with 100 pM TGF-β1 for 30 min and then processed for ChIP analysis with an antiserum specific to Smad4. Immunoprecipitated DNA containing the nt −221 to +30 region of Zp was readily detected in the cells that had been incubated with TGF-β1 but not in the untreated cells (Fig. 6, lane 4 versus lane 3). As a positive control, we examined these same ChIP samples for Smad4 binding to a region of the cellular Smad7 promoter shown by others to contain an SBE (52). As negative controls, we probed for an EBV sequence located 4.8 kbp upstream of the transcriptional initiation site of the BZLF1 promoter that does not contain an SBE or immunoprecipitated the chromatin with anti-IgG (Fig. 6, lanes 4 and 5, and data not shown). Thus, we conclude that Smad4 is not bound to the endogenous BZLF1 promoter during latency but becomes bound to Zp within 30 min of the onset of incubating MutuI cells with TGF-β1.
Fig. 6.
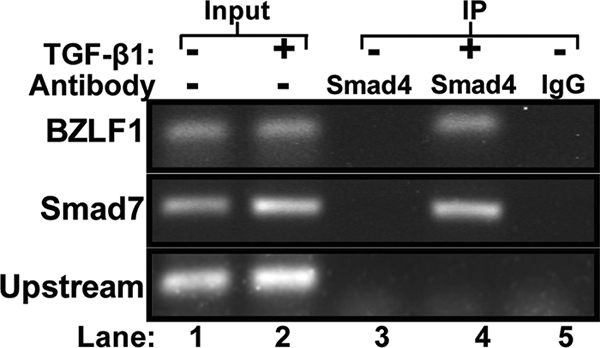
ChIP assay showing TGF-β1 induces Smad4 binding to Zp. MutuI cells were incubated for 30 min in the presence (+) or absence (−) of 100 pM TGF-β1. Cells then were processed for ChIP and immunoprecipitated with a Smad4-specific antibody or IgG as a control. Recovered DNA was amplified by PCR with primers specific to the nt −221 to +30 region of Zp (15 cycles), the promoter region of the cellular Smad7 gene (25 cycles) as a positive control, and an EBV sequence located 4.8 kbp upstream of Zp (15 cycles) as a negative control. The input lanes contained 10% of the amount of DNA included in the IPs.
Activated Smads enhance transcription from Zp.
Promoters of genes targeted by TGF-β usually contain Smad-binding elements for recruiting Smads and their cofactors to DNA (49). The binding of TGF-β ligand to TβRII recruits TβRI (also called Alk5), leading to the phosphorylation and activation of TβRI. The activated TβRI then phosphorylates Smad2 and Smad3, resulting in their heterotrimerization with Smad4. The Smad complexes translocate to the nucleus, where they interact with other transcription factors to regulate gene expression (42, 51).
To test directly whether the presence of activated Smad complexes can enhance transcription from the BZLF1 promoter, we examined the effects of the exogenous addition of Smad2, Smad3, and Smad4 in the presence and absence of a constitutively active mutant variant of the TGF-β type I receptor, Alk5 CA, on transcription from Zp in a transient-transfection assay using a reporter construct, pZp-Luc-WT, which contains the nt −496 to +28 region of Zp. For this experiment, we chose the human gastric carcinoma cell line AGS because (i) it is easily transfected with DNA, (ii) it lacks EBV, and (iii) it does not express the cellular transcription factors ZEB1 and ZEB2 (17, 20), which repress transcription from Zp (17, 41, 74, 75). The addition of Smad2, Smad3, or Smad2/3 together with Smad4 activated the expression of the reporter by 2.5-, 8-, and 11-fold, respectively (Fig. 7). The inclusion of constitutively active Alk5 further enhanced transcription from Zp up to 7.5-, 14-, and 20-fold for Smad2/4, Smad3/4, and Smad2/3/4, respectively. The addition of all three Smads plus Alk5 CA routinely activated the expression of this reporter 15- to 25-fold (Fig. 8 A and C; also see Fig. 10). Thus, we conclude that the presence of activated Smads leads to dramatically increased transcription from the BZLF1 promoter.
Fig. 7.

Effects of expression of Smad2/3/4 and constitutively active TGF-β type I receptor on transcription from Zp. EBV-negative AGS cells in 24-well plates were cotransfected as indicated with 80 ng of the firefly luciferase reporter, pZp-Luc-WT (i), 100 ng of pCS2-FLAG-Smad2 and/or pCS2-FLAG-Smad3 as indicated (ii), 100 ng of pCMV6-XL5 Smad4 (iii), and 50 ng of pcDNA3-ALK5 T204D (iv) where indicated by light gray bars. The empty vector, pcDNA3.1, was added to bring the total DNA to 430 ng per well. Cell lysates were prepared 48 h later, and firefly luciferase activities were measured. Data were internally normalized to the amount of protein in each lysate and externally normalized to the basal activity of pZp-Luc-WT. Bars indicate means ± standard deviations (SD) of data from assays performed in quadruplicate.
Fig. 10.

ZEB1 attenuates TGF-β-mediated activation of transcription from Zp. AGS cells in 24-well plates were cotransfected with 80 ng of pZp-Luc-WT or a mutant variant of it containing the base pair substitution mutations in the ZV and ZV′ elements of Zp indicated in Fig. 4B (i), 350 ng of pcDNA3.1 or 100 ng each of pCS2-FLAG-Smad2, pCS2-FLAG-Smad3, and pCMV6-XL5 Smad4 and 50 ng of pcDNA3-ALK5 T204D (ii), and the indicated amounts of a ZEB1 expression plasmid, pCi-ZEB1, plus pcDNA3.1 for a total of 12 ng DNA (iii). Luciferase activities were measured after incubation for 2 days. Data were internally normalized to the amount of protein in each lysate and externally normalized to basal activity observed for each reporter in the absence of the Smads, Alk5 CA, and ZEB1. Bars indicate means ± SD of data obtained from assays performed in quadruplicate. Results presented here are typical of data obtained on four separate occasions.
SBEs cooperatively contribute to Smad activation of Zp.
To test whether SBE2, SBE3, SBE4, and SBE5 play functional roles in the Smad-mediated activation of transcription from Zp, we constructed 2-bp substitution mutant variants of pZp-Luc-WT containing the same sequence alterations in the SBEs that had been examined in Fig. 4 for Smad4 binding. The mutation in SBE2 was chosen to disrupt Smad binding while leaving intact ZEB binding to the overlapping ZV element (Fig. 9 C). Reporters containing these 2-bp mutations in two, three, four, and all five SBEs also were generated. These DNAs were cotransfected in parallel into AGS cells along with plasmids expressing Smad2, Smad3, Smad4, and Alk5 CA or the empty vector, pcDNA3.1. The inactivation of individual SBEs somewhat reduced the Smad-mediated activation of transcription from Zp (20 to 40% for SBE1, SBE3, and SBE5 and 50 to 60% for SBE4 and SBE2) (Fig. 8A). The concurrent inactivation of multiple SBEs resulted in even greater reduction in Smad-mediated activation of Zp, with the SBE1/2/3/4/5 pentuple mutant exhibiting activation by Smads plus Alk5 CA barely above the 2.5-fold increase observed with the control empty vector, pGL3-Basic (Fig. 8C). Thus, we conclude that all five SBEs contribute to TGF-β-induced BZLF1 gene expression, with SBE2 and SBE4 being of greater importance in AGS cells than the other SBEs despite their having lower affinities than SBE1 for binding by Smad4 in a highly purified cell-free assay (compare Fig. 8A to 5).
Fig. 9.

EMSAs showing Smad4 competing with ZEB1 for binding to Zp or complexing with ZEB1 on the ZV element. (A) Protein competition EMSA with E. coli-expressed Smad4 and ZEB1. EMSAs were performed with 8 ng of radiolabeled double-stranded SBE2-WT probe corresponding to the nt −26 to −6 region of Zp. 6×His-tagged-Smad4 purified from E. coli and an extract from 293T cells that had been transfected with a ZEB1 expression plasmid were used as the protein sources. Lane 1, no protein; lanes 2 and 4, 225 ng of 6×His-Smad4; lane 3, 225 ng of 6×His-Smad4 plus 3 μg of antibody specific to Smad4; lane 5, 2.9 μg of ZEB1-containing extract plus 3 μg of antibody specific to ZEB1; lanes 6 to 9, 2.9 μg of ZEB1-containing extract plus 0, 25, 75, and 225 ng of 6×His-Smad4, respectively. (B) Protein competition EMSA with activated Smad4 and ZEB1. EMSAs were performed with radiolabeled SBE2-WT DNA as the probe, activated, nontagged Smad4 purified from 293T cells cotransfected with expression plasmids for Alk5 CA, and Smad4 and the ZEB1-containing extract used for panel A as the protein sources. Lane 1, no protein; lane 2, 200 ng of activated Smad4 plus 3 μg of antibody specific to Smad4; lane 3, 200 ng of activated Smad4; lane 4, 2.9 μg of ZEB1-containing extract plus 3 μg of antibody specific to ZEB1; lanes 5 to 9, 2.9 μg of ZEB1-containing extract plus 0, 100, 200, 500, and 1,000 ng of activated Smad4, respectively. (C) EMSAs showing the formation of Smad4/ZEB1 complex bound to the ZV element of Zp. EMSAs were performed with 8 ng of radiolabeled SBE2-WT DNA or variants of it containing 2-bp substitution mutations in either the SBE2 or the ZV element. Protein sources were the same ones used for panel B. Lanes 1, 7, and 13, no protein; lanes 2, 10, and 14, 0.5 μg of activated Smad4 plus 3 μg of antibody specific to Smad4; lanes 3, 11, and 15, 0.5 μg of activated Smad4; lanes 4, 8, and 16, 2.9 μg of ZEB1-containing extract plus 3 μg of antibody specific to ZEB1; lanes 5, 9, and 17, 2.9 μg of ZEB1-containing extract; lanes 6, 12, and 18, 0.5 μg of activated Smad4 plus 2.9 μg of ZEB1-containing extract. Locations of the protein/DNA complexes are indicated.
Smad4 can either compete or cooperate with ZEB1 for binding to Zp.
ZEB1 is a key transcriptional repressor of BZLF1 gene expression in many cell lines latently infected with EBV (17); it does so via concurrently binding the ZV and ZV′ elements of Zp (41, 74, 75). SBE2, mapping to nt −20 to −17, appears to overlap with the primary ZEB-binding ZV element, mapping to nt −17 through −12 (Fig. 4B). ZEB1 also can complex with Smads via direct protein-protein interactions (58). Thus, ZEB1 and Smad4 may compete or cooperate for binding to Zp.
To test these possibilities, we performed a series of protein competition EMSAs. In one set of experiments, we used our radiolabeled SBE2 oligonucleotide corresponding to the nt −26 to −6 region of Zp as the probe DNA and purified 6×His-tagged-Smad4 protein that had been expressed in E. coli and whole-cell extract prepared from 293T cells in which ZEB1 had been overexpressed as the sources of the proteins. As expected, Smad4 bound the SBE2 probe DNA; the Smad4/DNA complexes were retarded in mobility when a Smad4-specific antibody was included in the reaction mixture (Fig. 9A, lanes 2, 4, and 3, respectively). ZEB1 also bound this probe DNA; the addition of a ZEB1-specific antibody shifted the position of the ZEB1/DNA complex to the top of the gel (Fig. 9A, lanes 6 and 5, respectively). The addition of E. coli-synthesized Smad4 protein together with the ZEB1 protein led to the appearance of the Smad4/DNA complexes concurrently with some reduction in the abundance of the ZEB1/DNA complexes; slower-migrating complexes consistent with the formation of Smad4/ZEB1/DNA complexes were not observed (Fig. 9A, lanes 7 to 9). Similar results were observed using Smad4 protein that had been partially purified from the DG-75 BL B-cell line, with the abundance of ZEB1/DNA complexes reduced by approximately 50% at the highest level of Smad4 (data not shown). This finding suggests that ZEB1 and nonactivated Smad4 can compete for binding to Zp.
In a second set of experiments, in place of E. coli-synthesized Smad4, which is devoid of posttranslational modifications, we used Smad4 protein purified from 293T cells in which Smad4 and Alk5 CA had been coexpressed to provide the posttranslational modifications that Smad4 may acquire upon TGF-β signaling (15, 54, 63). This activated Smad4 protein by itself only weakly bound the SBE2 probe DNA in the absence of antibody or ZEB1 (Fig. 9B, lane 3, and C, lanes 3 and 11). However, the inclusion of anti-Smad4 antibody in the reaction mixture led to a large increase in the amount of Smad4 observed bound to the probe DNA, with it appearing in the position of the Smad4/Ab/DNA complex (Fig. 9B, lane 2, and C, lanes 2 and 10). Thus, the binding of this antibody to this activated form of Smad4 enhances or stabilizes its binding to DNA. Likewise, the inclusion of activated Smad4 together with ZEB1 led to significant changes in both the abundances and mobilities of the ZEB1/DNA complexes (Fig. 9B, lanes 5 to 9, and C, lane 5 versus lane 6).
One hypothesis consistent with the latter observation is that this slower-migrating band consists of ZEB1/Smad4 protein complexes synergistically bound via both the SBE2 and ZV elements present within this probe DNA. Alternatively, the ZEB1/Smad4 complexes could be bound via only one of these two elements, with their enhanced affinity for the probe resulting from conformational changes in the structures of the proteins that accompany these protein-protein interactions. To distinguish between these two possibilities, EMSAs were performed using these same protein preparations, but with variants of the wild-type SBE2 DNA containing 2-bp substitution mutations in either the ZV element or SBE2 as probes. As expected, Smad4 bound the SBE2-ZVm probe but not the SBE2m one (Fig. 9C, lane 11 versus lane 15), while ZEB1 bound the SBE2m probe but not the SBE2-ZVm one (Fig. 9C, lane 17 versus lane 9). Interestingly, when both activated Smad4 and ZEB1 were present together in the same reaction mixture, the slower-migrating ZEB1/Smad4/DNA complexes were observed with the SBE2-WT and SBE2m probes but not with the SBE2-ZVm probe (Fig. 9C, lanes 6 and 18 versus lane 12). In addition to the Smad4/ZEB1/DNA complex, the binding of Smad4 alone to the SBE probe also was detected in the presence of ZEB1 (Fig. 9B, lane 9, and C, lane 6). Therefore, we conclude that Smad4 can compete with ZEB1 for binding to Zp or can bind primarily via the ZV element as part of protein complexes containing ZEB1.
Smad activation of Zp is attenuated by ZEB1.
The findings described above suggest that Smads activate BZLF1 gene expression by (i) competing with the repressor ZEB1 for promoter occupancy or (ii) complexing with ZEB1, thereby switching ZEB1 from repressor to activator of Zp. To test these possibilities, we cotransfected into ZEB1-negative AGS cells (i) pZp-Luc-WT or pZp-Luc-ZV/ZV′dm, a variant of it containing the 2-bp substitution mutations in the ZEB1-binding ZV and ZV′ elements indicated in Fig. 4B; (ii) the Smad2, Smad3, Smad4, and Alk5 CA expression plasmids or the empty vector, pcDNA3.1; and (iii) various amounts of the ZEB1 expression plasmid, pCi-ZEB1. As expected, in the absence of the activated Smads, the addition of ZEB1 dramatically decreased transcription from the wild-type promoter without significantly affecting transcription from the mutant one (Fig. 10). The addition of Smads and Alk5 CA, but not ZEB1, enhanced transcription 19-fold from both the wild-type and ZV/ZV′ mutant promoters. The latter finding indicates that the Smad-mediated activation of transcription from Zp can occur independently of either ZEB1 protein or the ZEB1-binding elements. The addition of ZEB1 strongly inhibited this Smad-mediated activation of the wild-type promoter while not significantly affecting this activation of the mutant promoter. Thus, we conclude that exogenously expressed ZEB1 primarily functions in AGS cells as a repressor of Zp in transient-transfection reporter assays even when the TGF-β pathway is activated. The level of transcription from Zp likely is determined in part by the molar ratio of activated Smads to ZEB1 present in these transfected cells, i.e., high levels of activated Smad complexes can overcome repression by ZEB1.
DISCUSSION
The goal of this study was to elucidate the mechanism by which TGF-β signaling activates BZLF1 gene expression. First, we demonstrated that the incubation of MutuI cells in type I latency with TGF-β1 provided by either addition to the medium (Fig. 1) or cocultivation with Jurkat T cells (Fig. 2) led to the reactivation of EBV into lytic replication. We then showed that this induction occurred via the canonical TGF-β intracellular Smad pathway (Fig. 3), with Smad4 newly bound to the promoter region of the BZLF1 gene on the EBV genome within 30 min of the addition of TGF-β (Fig. 6). The addition of Smad2, Smad3, and Smad4 along with a constitutively active form of TβRI led to 15- to 25-fold activation of transcription from Zp (Fig. 7, 8, and 10). Four previously unrecognized Smad4-binding elements within Zp, named here SBE2 to SBE5, were identified (Fig. 4 and 5). Each SBE contributed somewhat to the Smad-mediated activation of Zp, with SBE2 and SBE4 being of greatest importance; taken together with SBE1, they accounted for essentially all of the activation (Fig. 8). SBE2 overlaps with the ZEB1-binding ZV element of Zp (Fig. 4B). Smad4 competed with ZEB1 for direct binding to SBE2 or bound as part of a complex with ZEB1 (Fig. 9). The addition of ZEB1 inhibited the transcriptional activation of Zp by activated Smads in a transient reporter assay (Fig. 10). Taking these results together, we conclude that TGF-β induces EBV lytic replication by activating BZLF1 gene expression through Smad-containing complexes binding to multiple SBEs located throughout Zp.
Role of TGF-β in EBV reactivation and survival.
Although the exact site of EBV reactivation within its human host remains unclear, much evidence points to the lymphoid tissue known as the Waldeyer's ring (6, 70). Latently infected memory B cells receive signals from factors present in the Waldeyer's ring, leading to reactivation, lytic replication, and the shedding of virus particles (70). Human tonsillar T cells secrete low levels of TGF-β but increase its production following activation with immune-stimulatory agents (37). Thus, we propose that some types of B and T cells present within certain lymphoid tissues secrete growth factors and cytokines, including TGF-β (37, 56, 65). We showed here that the coculture of the EBV-negative T-cell line Jurkat with the EBV-positive BL cell line MutuI led to viral reactivation (Fig. 2). This reactivation was inhibited by the incubation of the cells with a pan-TGF-β ligand neutralizing antibody (Fig. 2), indicating that the T-cell-induced reactivation of these EBV-infected B cells required TGF-β. These findings suggest that TGF-β is indeed an important player in EBV reactivation in the tonsils in vivo.
TGF-β-induced reactivation of EBV occurs via its canonical pathway.
The reactivation of latent EBV is a complex process that requires the participation and integration of multiple signaling pathways (38). Depending upon the specific cell type, TGF-β can regulate gene expression by activating a variety of different signaling kinases, including the Smads, phosphatidylinositol 3-OH kinase/Akt (PI3K), mitogen-activated protein kinase (MAPK), and extracellular signal-regulated kinases (ERKs) (7, 23, 24, 48, 72, 73). We observed here that the incubation of MutuI cells with either the TβRI inhibitor SB431542 or the Smad3 inhibitor SIS3, but not the Jun NH-terminal kinase (JNK) inhibitor SP600125, completely blocked the TGF-β induction of BZLF1 gene expression (Fig. 3). These findings suggest that the TGF-β canonical signaling pathway is the primary one mediating EBV reactivation, doing so through the activation of Smads. In agreement with Joab and colleagues (19, 55), the incubation of MutuI cells with the Akt inhibitor LY294002 somewhat inhibited TGF-β1-induced BZLF1 gene expression (Fig. 3), suggesting some involvement of Akt. However, whereas we found no effect of the MEK1/2 inhibitor U0126 on TGF-β-mediated EBV reactivation (Fig. 3), Joab and colleagues reported that it did prevent this induction (19, 55). Differences in experimental procedures that might account for this one seemingly contradictory finding include (i) cell lines, (ii) concentration of U0126, and (iii) sensitivity of the assays for detecting the EBV lytic proteins. Regardless, taking these results together, we conclude that TGF-β-mediated EBV lytic reactivation occurs principally through the Smad pathway.
Multiple Smad-binding elements cooperate in TGF-β-mediated activation of Zp.
Liang et al. (43) were the first to identify an SBE, referred to here as SBE1, which plays a role in TGF-β-mediated reactivation. However, Zp reporters containing substitution mutations in SBE1 exhibit only a 20 to 30% reduction in activation by TGF-β in EBV-positive BL P3HR-1 (43) and EBV-negative gastric cancer AGS cells (Fig. 8). While they reported the complete elimination of TGF-β activation by the mutation of SBE1 in EBV-negative BL Ramos cells (43), activation of the WT reporter by the addition of TGF-β was only 2-fold in these cells. Liang et al. suggested that two additional SBE-like elements, located at nt −166 to −163 and nt −59 to −56 of Zp, also contribute to its activation by TGF-β. We noted little, if any, binding of Smad4 to these sites (Fig. 5 and data not shown). However, we did identify four additional SBEs within the nt −221 to +12 region of Zp, named here SBE2 to SBE5 (Fig. 4), with the following relative binding affinities: SBE1 > SBE2 ∼ SBE3 > SBE5 > SBE4 (Fig. 5). Interestingly, the measured IC50s of these SBEs (Fig. 5) did not correlate well with their contributions to TGF-β-mediated activation of Zp (Fig. 8). This apparent discrepancy can be explained by the fact that the binding of Smad4 by itself is weak and does not convey promoter selectivity; rather, Smad4 complexes with other Smads and cofactors to bind promoters and regulate their transcription (32). The concurrent disruption of multiple SBEs led to a nearly complete loss of specific Zp activation by TGF-β (Fig. 8). Thus, we conclude that these five SBEs can cooperatively act for a maximal response to TGF-β; while they may appear to be functionally redundant, they likely provide alternative routes to activate BZLF1 gene expression via forming complexes with a variety of different cofactors that bind nearby elements within Zp.
The nt −221 to +12 region of Zp contains numerous cis-acting elements involved in regulating repression during latency and activation during lytic replication (Fig. 4A) (44, 45). SBE3 is located within the ZIA element overlapping with Sp1/3 and MEF2D binding sites, SBE4 overlaps with the MEF2D-binding site within the ZIB region, and SBE5 is juxtaposed to the Sp1/3-binding site within the ZID region (Fig. 4A). Sp1 can interact with Smad2, Smad3, and Smad4 in response to TGF-β signaling (21, 28, 29, 77). The formation of a Sp1/Smad3/4 complex has been shown to be required for recruiting Smads to SBEs in stimulating transcription from the COL1A2 promoter (29). Therefore, we hypothesize that an activated Smad/Sp1 complex on the SBE3/ZIA and SBE5/ZID regions can lead to the activation of transcription from Zp (Fig. 11 D).
Fig. 11.

Working model for regulation of BZLF1 gene expression by Smads. See Discussion for details.
MEF2D functions as a repressor by recruiting class II histone deacetylases (HDACs) to Zp during EBV latency (27). Smads have been shown to complex with MEF2, enhancing its transcriptional activity in response to TGF-β signaling (60). Thus, MEF2D also may complex with activated Smads following TGF-β signaling, with the Smad/MEF2D complex activating BZLF1 gene expression by binding the SBE3/ZIA, SBE4/ZIB, and, possibly, SBE5/ZID regions. This switch from HDAC/MEF2 to Smad/MEF2 complexes may involve, in part, p38 MAPK-induced posttranslational modifications of MEF2 (10, 27, 60, 78). Alternatively, activated Smads may compete with HDAC/MEF2D complexes for binding to Zp. Additional experiments will be needed to distinguish among these possibilities.
ZEB1 attenuates TGF-β-mediated activation of Zp.
ZEB1 binding to the ZV and ZV′ elements of Zp plays a central role in the maintenance of EBV latency by repressing transcription (17, 40, 41, 74, 75). However, the mechanisms by which this repression is relieved during lytic reactivation remain unknown. Here, a Smad-binding element, SBE2, was identified that overlaps with the primary ZEB-binding ZV element (Fig. 4B). ZEB1 and Smad complexes may compete for binding to Zp (Fig. 11A versus B). Alternatively, Smads may complex with ZEB1 while primarily bound via the ZV element (Fig. 11C). Prior studies showed that ZEB1 complexes and synergizes with activated Smads to activate transcription from the 3TP, p21, p15, and c-jun promoters (53, 58). Nevertheless, the addition of ZEB1 to AGS cells strongly inhibited the Smad-mediated activation of transcription from Zp in a transient reporter assay (Fig. 10). This difference may be due to the fact that ZEB1 binds Zp with high affinity directly over the transcription initiation site. Thus, it may always function as a repressor in this context, needing to be displaced for the activation of Zp. Supporting this possibility is the fact that ZEB1 represses even basal transcription from Zp (Fig. 10A) but not the other promoters mentioned above (58). Possibly, a certain threshold level of activated Smads or ratio of Smads to ZEBs needs to be reached or exceeded within the host cell for TGF-β-mediated reactivation of EBV to occur. Alternatively, our transient assay with reporter plasmids and the addition of ZEB1 may not properly mimic the physiologically relevant state that exists in B cells latently infected with EBV, given that these two systems likely differ with respect to the posttranslationally modified state of ZEB1 and the chromatin structure of Zp. Thus, the data shown in Fig. 10 do not exclude the possibility that TGF-β-mediated activation of Smads leads to the formation of Smad/ZEB1 complexes which function to activate transcription from Zp in the context of endogenous EBV genomes.
Incorporating these findings, we propose a working model for the TGF-β-mediated regulation of BZLF1 gene expression (Fig. 11). ZEB1, MEF2D, and other repressors, such as ZIIR-BP and YY1 (17, 27, 40, 41, 50, 74, 75), recruit corepressors, cooperatively silencing transcription from Zp during latency (Fig. 11A). Upon TGF-β signaling, activated Smad complexes accumulate in the nucleus, synergizing with transcriptional coactivators to bind Zp. Some activated Smad complexes bind SBE2, displacing the ZEBs if present, thereby stimulating transcription from Zp (Fig. 11B) as the homeobox protein Nkx2.5 has been shown to do on the collagen type I alpha 2 chain (col1a2) promoter in vascular smooth-muscle cells (57). However, if activated Smad complexes are (i) present at too low a level to outcompete ZEB1 for binding or (ii) primarily binding via the ZV/ZV′ elements as part of a ZEB1 repressor complex (Fig. 11C), the expression of the BZLF1 gene remains silenced. Activated Smads also can complex with other transcription factors, such as Sp1, stimulating transcription in part by binding the other SBEs located throughout Zp (Fig. 11D and data not shown). If Smad-mediated activation involving multiple SBEs is sufficient to overcome repression by the ZEBs and other repressors, the equilibrium shifts toward BZLF1 gene expression. Alternatively, the binding of Smads switches some of these repressor complexes (e.g., MEF2D and ZEB1) to activator ones.
Other DNA viruses induced by TGF-β.
TGF-β may contribute to the reactivation of other human DNA viruses as well. For example, like EBV, the human neurotropic polyomavirus JC virus sometimes can replicate to high levels in patients immunosuppressed by HIV/AIDS, with high TGF-β levels inducing JC virus-infected cells to produce infectious virus (18, 61, 68). Likewise, BK virus, another human polyomavirus, is frequently reactivated following kidney transplantation and immunosuppressant drug therapy (2). Elevated levels of TGF-β are observed after treatment with immunosuppressive drugs such as cyclosporine. TGF-β induces the expression of the virus-encoded large tumor antigen in renal cells infected with the TU strain of BK virus, doing so via adjacent putative ZEB- and Smad-binding elements present in the promoter region of this gene (1). These findings suggest that many DNA viruses utilize a similar mechanism to regulate reactivation into lytic replication by TGF-β. If true, the development of methods to control the levels of TGF-β and ZEBs may provide ways to control viral loads, especially in immunosuppressed patients for whom viral reactivation can be life threatening.
In summary, TGF-β1 can induce the reactivation of EBV in cells that contain an intact TGF-β pathway. It does so primarily via its canonical Smad signaling pathway, leading to the transcriptional activation of the key latent lytic switch gene, BZLF1, by a mechanism that involves the direct binding of activated Smads to multiple Smad-binding elements in the promoter.
ACKNOWLEDGMENTS
We thank Paul Farrell, Alan Rickinson, Bill Sugden, and Kenzo Takata for cell lines, Michael Hoffmann for pCS2-FLAG-Smad2, pCS2-FLAG-Smad3, and pcDNA3-ALK5 T204D, and Douglas Dean for pCi-ZEB1. We also thank Shannon Kenney, Allen Laughon, Bill Sugden, and members of the Mertz laboratory for reagents, discussions, and critical comments on the manuscript, as well as Joel Puchalski, Erik Puffer, and Kathy Schell of the UWCCC Flow Cytometry Facility for FACS.
This work was supported by U.S. Department of Health and Human Services grants RO1-AI107034, PO1-CA022443, and P30-CA14520 from the National Institutes of Health. S.D. was supported in part by T32-CA09135. T.I. is a Royal Thai Government Scholar with funding from the National Science and Technology Development Agency of Thailand.
Footnotes
Published ahead of print on 18 May 2011.
REFERENCES
- 1. Abend J. R., Imperiale M. J. 2008. Transforming growth factor-beta-mediated regulation of BK virus gene expression. Virology 378:6–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abend J. R., Jiang M., Imperiale M. J. 2009. BK virus and human cancer: innocent until proven guilty. Semin. Cancer Biol. 19:252–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adamson A. L., Kenney S. 2001. Epstein-Barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 75:2388–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aigner K., et al. 2007. The transcription factor ZEB1 (δEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26:6979–6988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aigner K., et al. 2007. The transcription factor ZEB1 (δEF1) represses Plakophilin 3 during human cancer progression. FEBS Lett. 581:1617–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Babcock G. J., Hochberg D., Thorley-Lawson A. D. 2000. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 13:497–506 [DOI] [PubMed] [Google Scholar]
- 7. Bakin A. V., Tomlinson A. K., Bhowmick N. A., Moses H. L., Arteaga C. L. 2000. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 275:36803–36810 [DOI] [PubMed] [Google Scholar]
- 8. Chau C. M., Lieberman P. M. 2004. Dynamic chromatin boundaries delineate a latency control region of Epstein-Barr virus. J. Virol. 78:12308–12319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chumanov R. S., Kuhn P. A., Xu W., Burgess R. R. 2011. Expression and purification of full-length mouse CARM1 from transiently transfected HEK293T cells using HaloTag technology. Protein Expr. Purif. 76:145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corcoran E. E., Means A. R. 2001. Defining Ca2+/calmodulin-dependent protein kinase cascades in transcriptional regulation. J. Biol. Chem. 276:2975–2978 [DOI] [PubMed] [Google Scholar]
- 11. Dasch J. R., Pace D. R., Waegell W., Inenaga D., Ellingsworth L. 1989. Monoclonal antibodies recognizing transforming growth factor-β. Bioactivity neutralization and transforming growth factor β2 affinity purification. J. Immunol. 142:1536–1541 [PubMed] [Google Scholar]
- 12. Davies S. P., Reddy H., Caivano M., Cohen P. 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351:95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Derynck R., Zhang Y. E. 2003. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425:577–584 [DOI] [PubMed] [Google Scholar]
- 14. Di Bartolo D. L., et al. 2008. KSHV LANA inhibits TGF-β signaling through epigenetic silencing of the TGF-β type II receptor. Blood 111:4731–4740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dupont S., et al. 2009. FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell 136:123–135 [DOI] [PubMed] [Google Scholar]
- 16. Ellis-Connell A. L., Iempridee T., Xu I., Mertz J. E. 2010. Cellular microRNAs 200b and 429 regulate the Epstein-Barr virus switch between latency and lytic replication. J. Virol. 84:10329–10343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ellis A. L., Wang Z., Yu X., Mertz J. E. 2010. Either ZEB1 or ZEB2/SIP1 can play a central role in regulating the Epstein-Barr virus latent-lytic switch in a cell-type-specific manner. J. Virol. 84:6139–6152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Enam S., Sweet T. M., Amini S., Khalili K., Del Valle L. 2004. Evidence for involvement of transforming growth factor β1 signaling pathway in activation of JC virus in human immunodeficiency virus 1-associated progressive multifocal leukoencephalopathy. Arch. Pathol. Lab. Med. 128:282–291 [DOI] [PubMed] [Google Scholar]
- 19. Fahmi H., Cochet C., Hmama Z., Opolon P., Joab I. 2000. Transforming growth factor beta 1 stimulates expression of the Epstein-Barr virus BZLF1 immediate-early gene product ZEBRA by an indirect mechanism which requires the MAPK kinase pathway. J. Virol. 74:5810–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feng W. H., et al. 2007. ZEB1 and c-Jun levels contribute to the establishment of highly lytic Epstein-Barr virus infection in gastric AGS cells. J. Virol. 81:10113–10122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feng X. H., Lin X., Derynck R. 2000. Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15Ink4B transcription in response to TGF-β. EMBO J. 19:5178–5193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flemington E., Speck S. H. 1990. Identification of phorbol ester response elements in the promoter of Epstein-Barr virus putative lytic switch gene BZLF1. J. Virol. 64:1217–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gal A., et al. 2008. Sustained TGFβ exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 27:1218–1230 [DOI] [PubMed] [Google Scholar]
- 24. Ghosh Choudhury G., Abboud H. E. 2004. Tyrosine phosphorylation-dependent PI 3 kinase/Akt signal transduction regulates TGFβ-induced fibronectin expression in mesangial cells. Cell Signal. 16:31–41 [DOI] [PubMed] [Google Scholar]
- 25. Gregory C. D., et al. 1988. Isolation of a normal B cell subset with a Burkitt-like phenotype and transformation in vitro with Epstein-Barr virus. Int. J. Cancer 42:213–220 [DOI] [PubMed] [Google Scholar]
- 26. Gregory C. D., et al. 1987. Identification of a subset of normal B cells with a Burkitt's lymphoma (BL)-like phenotype. J. Immunol. 139:313–318 [PubMed] [Google Scholar]
- 27. Gruffat H., Manet E., Sergeant A. 2002. MEF2-mediated recruitment of class II HDAC at the EBV immediate early gene BZLF1 links latency and chromatin remodeling. EMBO Rep. 3:141–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Inagaki Y., et al. 2001. Constitutive phosphorylation and nuclear localization of Smad3 are correlated with increased collagen gene transcription in activated hepatic stellate cells. J. Cell. Physiol. 187:117–123 [DOI] [PubMed] [Google Scholar]
- 29. Inagaki Y., Nemoto T., Nakao A., Dijke Pt, Kobayashi K., Takehara K., Greenwel P. 2001. Interaction between GC box binding factors and Smad proteins modulates cell lineage-specific α2(I) collagen gene transcription. J. Biol. Chem. 276:16573–16579 [DOI] [PubMed] [Google Scholar]
- 30. Inman G. J., Allday M. J. 2000. Apoptosis induced by TGF-β1 in Burkitt's lymphoma cells is caspase 8 dependent but is death receptor independent. J. Immunol. 165:2500–2510 [DOI] [PubMed] [Google Scholar]
- 31. Inman G. J., Allday M. J. 2000. Resistance to TGF-β1 correlates with a reduction of TGF-β type II receptor expression in Burkitt's lymphoma and Epstein-Barr virus-transformed B lymphoblastoid cell lines. J. Gen. Virol. 81:1567–1578 [DOI] [PubMed] [Google Scholar]
- 32. Inman G. J., Hill C. S. 2002. Stoichiometry of active smad-transcription factor complexes on DNA. J. Biol. Chem. 277:51008–51016 [DOI] [PubMed] [Google Scholar]
- 33. Inman G. J., et al. 2002. SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 62:65–74 [DOI] [PubMed] [Google Scholar]
- 34. Israel B. F., Kenney S. C. 2005. EBV lytic infection, p. 571–611 In Robertson E. S. (ed.), Epstein-Barr virus. Caister Academic Press, Norfolk, England [Google Scholar]
- 35. Jinnin M., Ihn H., Tamaki K. 2006. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-β1-induced extracellular matrix expression. Mol. Pharmacol. 69:597–607 [DOI] [PubMed] [Google Scholar]
- 36. Kehrl J. H., et al. 1986. Transforming growth factor β is an important immunomodulatory protein for human B lymphocytes. J. Immunol. 137:3855–3860 [PubMed] [Google Scholar]
- 37. Kehrl J. H., et al. 1986. Production of transforming growth factor β by human T lymphocytes and its potential role in the regulation of T cell growth. J. Exp. Med. 163:1037–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kenney S. C. 2007. Reactivation and lytic replication of EBV. In Arvin A., Campadelli-Fiume G., Mocarski E., Moore P. S., Roizman B., Whitney W., Yamanishi K. (ed.), Human herpesviruses. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 39. Koinuma D., et al. 2009. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor β signaling. Mol. Cell. Biol. 29:172–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kraus R. J., Mirocha S. J., Stephany H. M., Puchalski J. R., Mertz J. E. 2001. Identification of a novel element involved in regulation of the lytic switch BZLF1 gene promoter of Epstein-Barr virus. J. Virol. 75:867–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kraus R. J., Perrigoue J. G., Mertz J. E. 2003. ZEB negatively regulates the lytic-switch BZLF1 gene promoter of Epstein-Barr virus. J. Virol. 77:199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee P. S., Chang C., Liu D., Derynck R. 2003. Sumoylation of Smad4, the common Smad mediator of transforming growth factor-β family signaling. J. Biol. Chem. 278:27853–27863 [DOI] [PubMed] [Google Scholar]
- 43. Liang C. L., Chen J. L., Hsu Y. P., Ou J. T., Chang Y. S. 2002. Epstein-Barr virus BZLF1 gene is activated by transforming growth factor-β through cooperativity of Smads and c-Jun/c-Fos proteins. J. Biol. Chem. 277:23345–23357 [DOI] [PubMed] [Google Scholar]
- 44. Liu S., Borras A. M., Liu P., Suske G., Speck S. H. 1997. Binding of the ubiquitous cellular transcription factors Sp1 and Sp3 to the ZI domains in the Epstein-Barr virus lytic switch BZLF1 gene promoter. Virology 228:11–18 [DOI] [PubMed] [Google Scholar]
- 45. Liu S., Liu P., Borras A., Chatila T., Speck S. H. 1997. Cyclosporin A-sensitive induction of the Epstein-Barr virus lytic switch is mediated via a novel pathway involving a MEF2 family member. EMBO J. 16:143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lo K., Landau N. R., Smale S. T. 1991. LyF-1, a transcriptional regulator that interacts with a novel class of promoters for lymphocyte-specific genes. Mol. Cell. Biol. 11:5229–5243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Massagué J., Gomis R. R. 2006. The logic of TGFβ signaling. FEBS Lett. 580:2811–2820 [DOI] [PubMed] [Google Scholar]
- 48. Massagué J., Seoane J., Wotton D. 2005. Smad transcription factors. Genes Dev. 19:2783–2810 [DOI] [PubMed] [Google Scholar]
- 49. Massagué J., Wotton D. 2000. Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 19:1745–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Montalvo E. A., Cottam M., Hill S., Wang Y. J. 1995. YY1 binds to and regulates cis-acting negative elements in the Epstein-Barr virus BZLF1 promoter. J. Virol. 69:4158–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moustakas A., Heldin C. H. 2009. The regulation of TGFβ signal transduction. Development 136:3699–3714 [DOI] [PubMed] [Google Scholar]
- 52. Nagarajan R. P., Zhang J., Li W., Chen Y. 1999. Regulation of Smad7 promoter by direct association with Smad3 and Smad4. J. Biol. Chem. 274:33412–33418 [DOI] [PubMed] [Google Scholar]
- 53. Nakahata S., Yamazaki S., Nakauchi H., Morishita K. 2010. Downregulation of ZEB1 and overexpression of Smad7 contribute to resistance to TGF-β1-mediated growth suppression in adult T-cell leukemia/lymphoma. Oncogene 29:4157–4169 [DOI] [PubMed] [Google Scholar]
- 54. Ohshima T., Shimotohno K. 2003. Transforming growth factor-β-mediated signaling via the p38 MAP kinase pathway activates Smad-dependent transcription through SUMO-1 modification of Smad4. J. Biol. Chem. 278:50833–50842 [DOI] [PubMed] [Google Scholar]
- 55. Oussaief L., et al. 2009. Phosphatidylinositol 3-kinase/Akt pathway targets acetylation of Smad3 through Smad3/CREB-binding protein interaction: contribution to transforming growth factor β1-induced Epstein-Barr virus reactivation. J. Biol. Chem. 284:23912–23924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ozdemir C., Akdis M., Akdis C. A. 2009. T regulatory cells and their counterparts: masters of immune regulation. Clin. Exp. Allergy 39:626–639 [DOI] [PubMed] [Google Scholar]
- 57. Ponticos M., Partridge T., Black C. M., Abraham D. J., Bou-Gharios G. 2004. Regulation of collagen type I in vascular smooth muscle cells by competition between Nkx2.5 and deltaEF1/ZEB1. Mol. Cell. Biol. 24:6151–6161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Postigo A. A. 2003. Opposing functions of ZEB proteins in the regulation of the TGFβ/BMP signaling pathway. EMBO J. 22:2443–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Postigo A. A., Depp J. L., Taylor J. J., Kroll K. L. 2003. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 22:2453–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Quinn Z. A., Yang C. C., Wrana J. L., McDermott J. C. 2001. Smad proteins function as co-modulators for MEF2 transcriptional regulatory proteins. Nucleic Acids Res. 29:732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Raj G. V., Khalili K. 1994. Identification and characterization of a novel GGA/C-binding protein, GBP-i, that is rapidly inducible by cytokines. Mol. Cell. Biol. 14:7770–7781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Robertson E. S. 2005. Epstein-Barr virus. Caister Academic Press, Wymondham, England [Google Scholar]
- 63. Roelen B. A., et al. 2003. Phosphorylation of threonine 276 in Smad4 is involved in transforming growth factor-β-induced nuclear accumulation. Am. J. Physiol. Cell Physiol. 285:C823–C830 [DOI] [PubMed] [Google Scholar]
- 64. Seoane J., Le H. V., Shen L., Anderson S. A., Massagué J. 2004. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117:211–223 [DOI] [PubMed] [Google Scholar]
- 65. Sesterhenn K., Krueger G. R., Uhlmann C. 1977. Percent distribution of T- and B-cells in tonsils of children, juveniles and adults. Arch. Otorhinolaryngol. 218:37–44 [DOI] [PubMed] [Google Scholar]
- 66. Shi Y., et al. 1998. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-β signaling. Cell 94:585–594 [DOI] [PubMed] [Google Scholar]
- 67. Shirakihara T., Saitoh M., Miyazono K. 2007. Differential regulation of epithelial and mesenchymal markers by δEF1 proteins in epithelial mesenchymal transition induced by TGF-β. Mol. Biol. Cell 18:3533–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stettner M. R., et al. 2009. SMAD proteins of oligodendroglial cells regulate transcription of JC virus early and late genes coordinately with the Tat protein of human immunodeficiency virus type 1. J. Gen. Virol. 90:2005–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Takada K., et al. 1991. An Epstein-Barr virus-producer line Akata: establishment of the cell line and analysis of viral DNA. Virus Genes 5:147–156 [DOI] [PubMed] [Google Scholar]
- 70. Thorley-Lawson D. A., Duca K. A., Shapiro M. 2008. Epstein-Barr virus: a paradigm for persistent infection-for real and in virtual reality. Trends Immunol. 29:195–201 [DOI] [PubMed] [Google Scholar]
- 71. Weinmann A. S., Farnham P. J. 2002. Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods 26:37–47 [DOI] [PubMed] [Google Scholar]
- 72. Wilkes M. C., et al. 2005. Transforming growth factor-β activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. 65:10431–10440 [DOI] [PubMed] [Google Scholar]
- 73. Xie L., et al. 2004. Activation of the Erk pathway is required for TGF-β1-induced EMT in vitro. Neoplasia 6:603–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yu X., et al. 2011. ZIIR element of BZLF1 promoter of Epstein-Barr virus plays central role in establishment and maintenance of viral latency. J. Virol. 85:5081–5090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yu X., Wang Z., Mertz J. E. 2007. ZEB1 regulates the latent-lytic switch in inf ection by Epstein-Barr virus. PLoS Pathog. 3:e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zawel L., et al. 1998. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1:611–617 [DOI] [PubMed] [Google Scholar]
- 77. Zhang W., Ou J., Inagaki Y., Greenwel P., Ramirez F. 2000. Synergistic cooperation between Sp1 and Smad3/Smad4 mediates transforming growth factor β1 stimulation of α2(I)-collagen (COL1A2) transcription. J. Biol. Chem. 275:39237–39245 [DOI] [PubMed] [Google Scholar]
- 78. Zhao M., et al. 1999. Regulation of the MEF2 family of transcription factors by p38. Mol. Cell. Biol. 19:21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]