

Abstract
This paper takes a close look at balanced permutations, a recently developed sample reuse method with applications in bioinformatics. It turns out that balanced permutation reference distributions do not have the correct null behavior, which can be traced to their lack of a group structure. We find that they can give p-values that are too permissive to varying degrees. In particular the observed test statistic can be larger than that of all B balanced permutations of a data set with a probability much higher than 1/(B + 1), even under the null hypothesis.
Key words: functional genomics, gene expression, genomics, statistics
1. Introduction
Sample reuse methods such as permutation testing and the bootstrap are invaluable tools in high-throughput genomic settings, such as microarray analyses. They are used to test hypotheses and compute p-values without making strong parametric assumptions about the data, and they adapt readily to complicated test statistics.
Permutation tests, described in more detail below, work by permuting the treatment labels of the data and comparing the resulting values of a test statistic to the original one. Suppose for example, that there are n observations 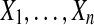 in the treatment group and also n observations
in the treatment group and also n observations 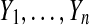 in the control group. We might measure the treatment effect via
in the control group. We might measure the treatment effect via 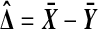 where
where 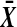 and
and 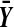 are averages of the treatment and control observations, respectively. There are
are averages of the treatment and control observations, respectively. There are 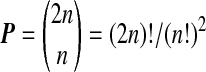 ways to redo the assignment of treatment versus control labels, and they each give a value for the treatment effect. If the actual treatment effect is larger than that from all of the other permutations, then we may claim a p-value of 1/P. More generally, if the observed effect beats (is larger than) exactly b of these values we can claim p = 1 − b/P. The smallest available p value is 1/P because the actual treatment allocation is always included in the reference set.
ways to redo the assignment of treatment versus control labels, and they each give a value for the treatment effect. If the actual treatment effect is larger than that from all of the other permutations, then we may claim a p-value of 1/P. More generally, if the observed effect beats (is larger than) exactly b of these values we can claim p = 1 − b/P. The smallest available p value is 1/P because the actual treatment allocation is always included in the reference set.
One reason why permutation tests work and are intuitively reasonable is their symmetry. If 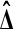 is the actual difference and
is the actual difference and 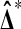 is the difference for any reassignment of labels, chosen without looking at the X and Y values, then
is the difference for any reassignment of labels, chosen without looking at the X and Y values, then
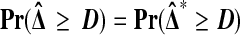 |
(1) |
holds for all D, under the null hypothesis 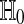 that Xi and Yi are all independent and identically distributed. That is,
that Xi and Yi are all independent and identically distributed. That is, 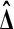 and
and 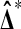 have the same distribution.
have the same distribution.
Recently, a special form of permutation analysis, called balanced permutation, has been employed. In a balanced permutation, we make sure that after relabeling, the new treatment group has exactly n/2 members that came from the original treatment group and n/2 from the original control group. The number of balanced permutations is 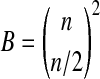 which can be much smaller than the total number P of permutations. Balanced permutations require n to be even, but nearly balanced permutations are possible for odd n.
which can be much smaller than the total number P of permutations. Balanced permutations require n to be even, but nearly balanced permutations are possible for odd n.
These balanced permutations are mentioned in remark E on page 1159 of Efron et al. (2001). This is the earliest mention we have found. Since then, they have been applied numerous times in the literature on statistical analysis of microarrays. As of 2008, the National Cancer Institute describes balanced permutations in their page on statistical tests at http://discover.nci.nih.gov/microarrayAnalysis/Statistical.Tests.jsp. They say that more extreme p-values typically result, but they remark on a granularity problem. For fixed n, 1/B is bigger than 1/P, and so balanced permutations require larger sample sizes n than ordinary ones do, if one is to attain very small p-values.
Balanced permutations are a subset of all possible permutations, and so equation (1) holds for them too. Intuitively, a balanced permutation analysis should be even better than a permutation analysis. If 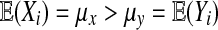 then
then 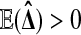 while
while 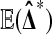 remains at 0 due to cancellation. By contrast, for ordinary permutations some of the
remains at 0 due to cancellation. By contrast, for ordinary permutations some of the 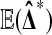 are positive and some are negative.
are positive and some are negative.
Equation (1) is not enough to make balanced permutations give properly calibrated p-values. The theory of permutation tests also requires a group structure for the set of permutations used, and balanced permutations do not satisfy this condition.
Though they fail a sufficient condition for giving exact p values, that does not in itself mean that they give bad p values. The goal of this paper is to investigate the p-values produced via balanced permutations. What we find is that those p values can be much smaller than they should be. This is most extreme in the case where the original test statistic beats all of the balanced permutations.
The outline of this paper is as follows. Section 2 describes permutation based inferences including balanced permutations and it distinguishes balanced from stratified permutations. Section 3 shows through theory and simulations that the reference distribution provided by balanced permutations can lead to permissive p-values, sometimes by astonishingly large factors.
The major use of balanced permutations is in estimating a pooled p-value distribution for N test statistics assumed to have the same distribution, and for estimating the false discovery rate (FDR) of a procedure. Section 4 looks at balanced permutations for pooled reference distributions. It is well known that including non-null cases makes a permutation analysis conservative. We find that including non-null cases in the balanced permutation scheme has the opposite effect, making it overly permissive. Even purely null cases can give rise to a permissive test, though this problem diminishes in large scale problems, when the null cases are independent of the one being tested. Section 5 summarizes our conclusions on balanced permutations, recommending against their use.
2. Background
To be concrete, suppose that we are comparing gene expression for two groups of subjects, treatment and control. Gene expression is a proxy measurement for the amount of product, either protein or RNA, made by a gene. In the past decade, various high-throughput technologies, generally termed microarrays, have made it possible to measure the expression activity of all genes in a genome with a single assay. We will later look at the issues that arise when there are many gene expression measurements to consider. For simplicity, we begin with the expression of a single gene.
We have n ≥ 2 subjects in each group. For the treatment subjects we measure 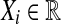 for
for 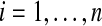 and for the control group we measure
and for the control group we measure 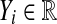 for
for 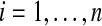 . We assume an unpaired framework, so there is no connection between Xi and Yi. The Xi are modeled as n independent samples from a distribution F and the Yi are similarly sampled from G. We will always assume that F and G both have finite means. This is reasonable for the applications we have in mind and it rules out uninteresting corner cases.
. We assume an unpaired framework, so there is no connection between Xi and Yi. The Xi are modeled as n independent samples from a distribution F and the Yi are similarly sampled from G. We will always assume that F and G both have finite means. This is reasonable for the applications we have in mind and it rules out uninteresting corner cases.
We are usually interested in 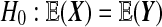 . We also consider the stricter hypothesis
. We also consider the stricter hypothesis 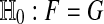 . There is no exact nonparametric test for a one-sample mean (Bahadur and Savage 1956) and so we cannot expect one for H0 either. One often makes do with exact tests for
. There is no exact nonparametric test for a one-sample mean (Bahadur and Savage 1956) and so we cannot expect one for H0 either. One often makes do with exact tests for 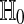 .
.
The customary test for H0 is Student's t test. Let 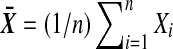 and
and 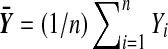 . Then under H0, the statistic
. Then under H0, the statistic
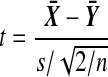 |
(2) |
where
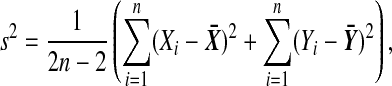 |
has the t(2n−2) distribution. The t-test rejects H0 if 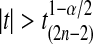 . There are also one tailed versions.
. There are also one tailed versions.
The t test is designed for the case where Xi and Yi are normally distributed with common variance and possibly different means. The test holds up fairly well for non-normal data. In this particular setting with equal sample sizes of n, it is also reasonably accurate when Xi and Yi have unequal variances. For a discussion of the properties of t tests when assumptions may be violated, see Miller (1986).
2.1. Permutation tests
Permutation tests are very simple and intuitive. The original treatment assignment is one of 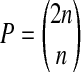 possible assignments. Under the null hypothesis, they all have an equal chance to be the most extreme. The explanation is not based on the P test statistics being independent and identically distributed. In fact, they are not independent. The explanation comes from a symmetry argument which in turn stems from the group structure of the set of permutations.
possible assignments. Under the null hypothesis, they all have an equal chance to be the most extreme. The explanation is not based on the P test statistics being independent and identically distributed. In fact, they are not independent. The explanation comes from a symmetry argument which in turn stems from the group structure of the set of permutations.
Here we make a formal presentation of permutation tests of 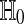 , so that we may later highlight their differences with balanced permutations. Balanced permutations also seem intuitively reasonable and even seem intuitively better than ordinary permutations. Readers who primarily want to apply permutation methods may prefer to skip this subsection.
, so that we may later highlight their differences with balanced permutations. Balanced permutations also seem intuitively reasonable and even seem intuitively better than ordinary permutations. Readers who primarily want to apply permutation methods may prefer to skip this subsection.
We suppose as before that Xi ∼ F and Yi ∼ G are all independent, for 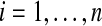 . The sample mean difference is
. The sample mean difference is 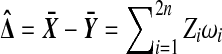 , where
, where
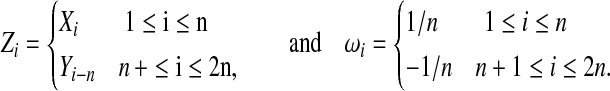 |
We use 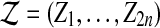 to represent both samples combined.
to represent both samples combined.
Suppose that we relabel the data, changing which subjects are treatment and which are control. Any relabeling of the data can be obtained via a permutation of indices as follows. Let 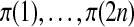 be a permutation of the integers
be a permutation of the integers 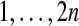 . We shuffle the components of
. We shuffle the components of 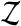 obtaining the vector
obtaining the vector 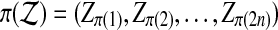 . We use the symbol π for two meanings, a permutation of indices and the resulting rearrangement of a vector, but this should cause no confusion.
. We use the symbol π for two meanings, a permutation of indices and the resulting rearrangement of a vector, but this should cause no confusion.
Now let
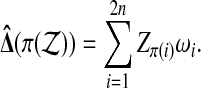 |
(3) |
Under 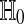 the distribution of
the distribution of 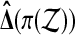 is the same as that of
is the same as that of 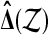 , for all permutations π.
, for all permutations π.
The set of all permutations of 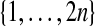 comprises a group, called
comprises a group, called 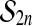 . The group operation is composition. For two permutations
. The group operation is composition. For two permutations 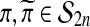 their composition, denoted by
their composition, denoted by 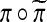 , is the permutation one gets by applying
, is the permutation one gets by applying 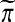 first and then applying π to the result.
first and then applying π to the result.
Although there are M = (2n)! permutations of 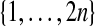 there are only
there are only 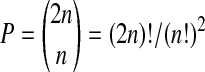 distinct cases for
distinct cases for 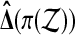 determined by which n observations get π(i) ≤ n and which get π(i) > n. The ratio M/P = (n!)2 reflects the fact that there are n! ways to permute the observations with π(i) ≤ n, (and similarly the ones with π(i) > n) without affecting
determined by which n observations get π(i) ≤ n and which get π(i) > n. The ratio M/P = (n!)2 reflects the fact that there are n! ways to permute the observations with π(i) ≤ n, (and similarly the ones with π(i) > n) without affecting 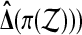 .
.
A permutation test uses the uniform distribution on all M possible sample values of 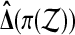 as a reference distribution for
as a reference distribution for 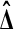 , to test
, to test 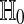 . In practice we accomplish the permutation test by the shortcut of computing only the P distinct cases. Those cases all have multiplicity M/P and so we can still use a uniform distribution on them. When even P is too large for enumeration to be feasible, then one commonly substitutes a very large random sample of permutations for the complete enumeration.
. In practice we accomplish the permutation test by the shortcut of computing only the P distinct cases. Those cases all have multiplicity M/P and so we can still use a uniform distribution on them. When even P is too large for enumeration to be feasible, then one commonly substitutes a very large random sample of permutations for the complete enumeration.
Here we construct permutation tests of 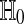 at level
at level 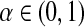 based on Chapter 15.2 of Lehmann and Romano (2005). We will work with all M relabelings, instead of reducing to the P distinct ones, in order to use the simple group structure of
based on Chapter 15.2 of Lehmann and Romano (2005). We will work with all M relabelings, instead of reducing to the P distinct ones, in order to use the simple group structure of 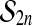 .
.
To test 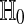 , let T be a real valued function such that
, let T be a real valued function such that 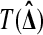 takes larger values in response to stronger evidence against
takes larger values in response to stronger evidence against 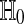 . For example T (Δ) = |Δ| for a usual two tailed test and T (Δ) = Δ or −Δ for one tailed tests. To get a test with level α we reject
. For example T (Δ) = |Δ| for a usual two tailed test and T (Δ) = Δ or −Δ for one tailed tests. To get a test with level α we reject 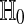 if the observed
if the observed 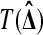 is larger than Mα of the permuted values. We sort the M values of the permuted test statistics
is larger than Mα of the permuted values. We sort the M values of the permuted test statistics 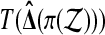 , as π ranges through
, as π ranges through 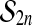 , getting
, getting
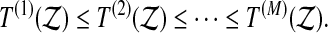 |
Let 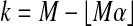 . Roughly speaking, the test will reject
. Roughly speaking, the test will reject  if
if 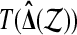 is larger than
is larger than 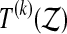 but the recipe below takes care of ties, the number of which can in principle depend on the data
but the recipe below takes care of ties, the number of which can in principle depend on the data 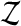 . We let
. We let 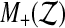 be the number of
be the number of 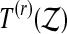 values strictly larger than
values strictly larger than 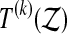 and
and 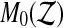 be the number of
be the number of 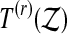 values exactly equal to T(k). Then define the tie breaker quantity
values exactly equal to T(k). Then define the tie breaker quantity 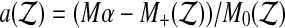 and let
and let
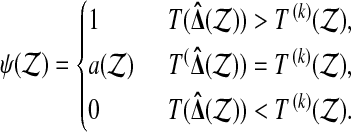 |
By construction
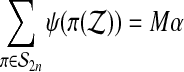 |
(4) |
holds for any  .
.
The interpretation of ψ is as follows. If ψ = 1 then we reject 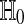 . If ψ = 0 then we don't reject it. If 0 < ψ < 1 then we reject
. If ψ = 0 then we don't reject it. If 0 < ψ < 1 then we reject 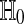 with probability ψ. It is best to have all the ψ values equal to 0 or 1 to avoid randomized decisions. We can usually do this for special choices of α. For continuously distributed data all P possible sample differences are distinct with probability one. Then taking α = A/M for a positive integer A < M yields
with probability ψ. It is best to have all the ψ values equal to 0 or 1 to avoid randomized decisions. We can usually do this for special choices of α. For continuously distributed data all P possible sample differences are distinct with probability one. Then taking α = A/M for a positive integer A < M yields 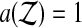 and so eliminates randomized decisions.
and so eliminates randomized decisions.
Theorem 1
Suppose that Xi ∼ F and Yi ∼ G for 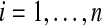 are all independent and that
are all independent and that 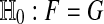 holds. For a test statistic T (z) and the randomization test quantity ψ constructed above from the permutation group
holds. For a test statistic T (z) and the randomization test quantity ψ constructed above from the permutation group 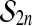 ,
, 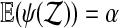 .
.
Remark
Theorem 1 follows from Theorem 15.2.1 of Lehmann and Romano (2005) of which it is a special case. We recap their proof, for later use.
Proof
Using 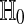 and then equation (4), we get
and then equation (4), we get
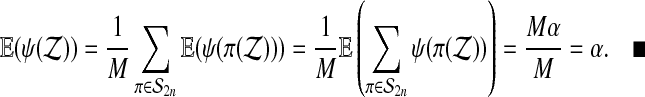 |
When we have arranged for ψ to be either 0 or 1, then 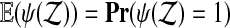 . Therefore, the test rejects
. Therefore, the test rejects 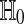 with probability α. The p value is the smallest α rejected (among the non-randomized choices).
with probability α. The p value is the smallest α rejected (among the non-randomized choices).
2.2. Stratified permutations and balanced permutations
Two different ideas have been called “balanced permutation” in the literature. In one kind of balanced permutation, the subjects can be split into two groups in two different ways. The split of primary interest is treatment versus control. The second split has levels that are variously called strata or blocks. It may already be known to be quite important, but it is not of primary interest. For example, test and control might correspond to young and old mice while the strata might correspond to two different strains. We might want a test for age that adjusts for known differences between strains.
Suppose that we have n test subjects, where n is even for simplicity, with n/2 from stratum A and n/2 from stratum B. Similarly, there are n/2 control subjects from each stratum. This study design has balanced the treatment versus control dichotomy with respect to the strata, and it is reasonable to enforce such a balance on all the permutations. To get a permutation that is balanced with respect to the stratification, we may select n/2 of the subjects from stratum A and n/2 of the subjects from stratum B to be relabeled as test subjects. The remaining n subjects are relabeled as controls, and of course are also balanced. Stratified permutation tests of this type can be generalized to stratification factors with more than 2 levels and also to settings with more than 1 factor.
Tusher et al. (2001) employed this form of stratification to compare effects of radiation on genes, adjusting for cell lines. Their subjects had a third dichotomy, corresponding to aliquots, that need not concern us here.
The other method called balanced permutations is the one described in the introduction. These balanced permutations are not the same as stratified permutations. The difference is that the former stratifies on a covariate dichotomy, while the latter stratifies on the dichotomy of interest. Stratification on a covariate is sometimes studied as a restricted randomization and has a long history in nonparametric methods; for example, see Good (2000) and Edgington and Onghena (2007).
We will use the term “balanced permutation” to describe only the new method of stratifying on the variable of interest. As described in the introduction, there is reason to be optimistic about the performance of balanced permutations. On the other hand, an ordinary unbalanced permutation test applied to 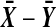 , is asymptotically equivalent to Student's t-test. That test is the appropriate one for Gaussian data, and is asymptotically well behaved otherwise, under mild moment conditions. We might therefore expect that there is little room for improvement from balanced permutations, at least for
, is asymptotically equivalent to Student's t-test. That test is the appropriate one for Gaussian data, and is asymptotically well behaved otherwise, under mild moment conditions. We might therefore expect that there is little room for improvement from balanced permutations, at least for 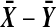 and moderately large n.
and moderately large n.
To justify using balanced permutations to construct a reference distribution, we need to apply a result like Theorem 1. The key ingredient is the group structure. Stratification has a group structure. In the setting above we may employ permutations πA and πB within strata A and B respectively. The permutation of the whole data set may be represented by the pair (πA, πB). These pairs have a group structure. Formally, it is the direct sum of the groups to which πA and πB belong. Practically, the permutations for strata A and B are just handled separately and independently. The group structure can extend to three or more strata.
The set 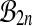 of balanced permutations is not a group. To begin with, the original data
of balanced permutations is not a group. To begin with, the original data 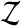 are the identity permutation of the data, but the identity permutation is not a balanced permutation. We might simply include the identity permutation, with the same multiplicity as each balanced permutation into
are the identity permutation of the data, but the identity permutation is not a balanced permutation. We might simply include the identity permutation, with the same multiplicity as each balanced permutation into 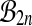 , getting the set
, getting the set 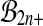 . Then our putative p value is the proportion of permutations
. Then our putative p value is the proportion of permutations 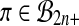 for which
for which 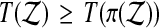 holds.
holds.
The problem with this approach is that the set 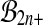 is not a group either, under composition. A balanced permutation of a balanced permutation is not necessarily another balanced permutation. Therefore,
is not a group either, under composition. A balanced permutation of a balanced permutation is not necessarily another balanced permutation. Therefore, 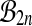 and
and 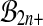 both fail the closure requirement of a group. An example is illustrated in Figure 1.
both fail the closure requirement of a group. An example is illustrated in Figure 1.
FIG. 1.

This figure shows that the result of applying two balanced permutations in succession need not be another balanced permutation. The first column shows treatment and control groups, T and C, of four observations each. The middle column shows the results of a balanced permutation that swapped two of the treatment subjects for two of the controls as indicated by line segments. The third column shows the result of a second such swap. The final treatment group has three members of the original treatment group and one member of the original control group, and so it is unbalanced.
It is not obvious at first sight where the proof of Theorem 1 makes use of the group property of 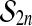 . But if we replace the group
. But if we replace the group 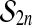 by
by 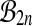 (or
(or 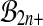 ), and follow the steps, the proof fails at the third equality sign. We can still pick k so that
), and follow the steps, the proof fails at the third equality sign. We can still pick k so that 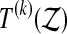 is beaten by a fraction α of
is beaten by a fraction α of 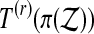 taken over balanced permutations π. For a group of permutations, that means T(k) will be beaten by a fraction α of all
taken over balanced permutations π. For a group of permutations, that means T(k) will be beaten by a fraction α of all 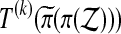 as
as 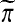 ranges over the group and π is any fixed group member. This happens for the group
ranges over the group and π is any fixed group member. This happens for the group 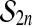 , because
, because 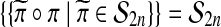 . But no such result holds for
. But no such result holds for 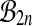 or
or 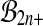 . For balanced permutations, the set of ordered values of
. For balanced permutations, the set of ordered values of 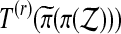 as
as 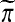 varies for fixed π is not constant. That set depends on π and so its 1 − α quantile will in general depend on π.
varies for fixed π is not constant. That set depends on π and so its 1 − α quantile will in general depend on π.
3. Coverage Properties Of Balanced Permutation
Because balanced permutations do not satisfy Theorem 1, we cannot be sure that they provide a suitable p value for 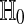 . We look at some theoretical and empirical results to see how they do.
. We look at some theoretical and empirical results to see how they do.
In microarray experiments, n is often small, because arrays are costly. For example, Tusher et al. (2001) compare two groups of n = 4 arrays. Our main interest is therefore in values of n between 4 and 20. We also look briefly at n = 2 because a mathematical answer is possible there and because it illustrates the issues.
3.1. Theory for n = 2
For n = 2 there are only B = 4 distinct balanced permutations, and so we can easily look at all of them. Suppose that 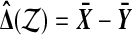 . Then we can write out results from all of the balanced permutations as a vector
. Then we can write out results from all of the balanced permutations as a vector
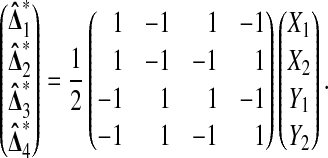 |
Theorem 2
Suppose that X1, X2, Y1, and Y2 are independent random variables with the same continuous distribution F. Let 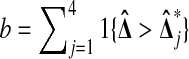 be the number of balanced permutations that the original permutation beats. Then
be the number of balanced permutations that the original permutation beats. Then 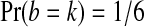 for
for 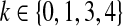 and
and 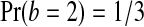 .
.
Proof
We easily find that 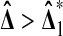 if and only if X2 > Y1. From the other three cases, we find that
if and only if X2 > Y1. From the other three cases, we find that 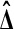 beats all 4 balanced permutations if and only if both Xi are larger than both Yi. This has probability 1/6 by symmetry. A similar argument shows that
beats all 4 balanced permutations if and only if both Xi are larger than both Yi. This has probability 1/6 by symmetry. A similar argument shows that 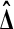 has probability 1/6 of beating none of the
has probability 1/6 of beating none of the 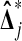 . For
. For 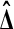 to beat exactly 3 of the
to beat exactly 3 of the 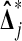 , the X's must rank first and third among the four data points. Thus Pr(b = 3) = 1/6 as well. Similarly Pr(b = 1) = 1/6, and by subtracting we find Pr(b = 2) = 1/3. ▪
, the X's must rank first and third among the four data points. Thus Pr(b = 3) = 1/6 as well. Similarly Pr(b = 1) = 1/6, and by subtracting we find Pr(b = 2) = 1/3. ▪
Thus, for n = 2, the balanced permutation histogram for comparing means is not uniform. It is slightly conservative. When the original permutation beats all 4 balanced permutations, we would naively claim a p value of 1/5. But this event only has probability 1/6 of happening. Balanced permutations are not likely to be applied when n = 2, except perhaps when results from multiple genes are to be pooled as described in Section 4. Unfortunately, when n gets larger, the non-uniformity does not disappear, and the method switches from conservative to permissive.
It is interesting to note that the number of permutations beaten has a double high spike in the middle at 2. If we insert a pseudo value 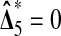 , then by symmetry we split that spike into 2 equal pieces and obtain a uniform histogram for
, then by symmetry we split that spike into 2 equal pieces and obtain a uniform histogram for 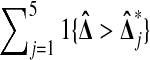 . This spike at the median appears in all of the balanced permutation histograms for treatment effects that we have investigated. It is not a simple consequence of the histogram being symmetric. For instance, the histogram for the full permutation set is symmetric but it does not have a double high spike in the middle.
. This spike at the median appears in all of the balanced permutation histograms for treatment effects that we have investigated. It is not a simple consequence of the histogram being symmetric. For instance, the histogram for the full permutation set is symmetric but it does not have a double high spike in the middle.
3.2. Numerical results for small n>2
The method used to prove Theorem 2 does not give us an answer for n > 2. When n = 4 the argument there shows that the observed difference will beat all 36 balanced permutations if and only if the smallest n/2 = 2 of the Xi have a higher sum than the largest 2 of the Yi. The probability of this event depends on the underlying distribution of Xi and Yi. We proceed numerically, using the N(0, 1) distribution for illustration.
For n = 4 observations per group there are now 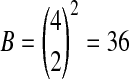 balanced permutations. The observed
balanced permutations. The observed 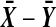 will beat some number in
will beat some number in 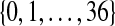 of them. Ideally, the number of balanced permutations giving
of them. Ideally, the number of balanced permutations giving 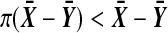 should have the uniform distribution on those 37 levels. The top plot in Figure 2 shows the actual distribution, from 10,000 simulations of the case where
should have the uniform distribution on those 37 levels. The top plot in Figure 2 shows the actual distribution, from 10,000 simulations of the case where 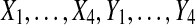 are independent N(0, 1) random variables. The histogram is nearly uniform except that the bars at 0, 18, and 36 are about twice as high as desired. There is also a slight tendency for small and large multiples of 3 to have higher bars than their neighbors.
are independent N(0, 1) random variables. The histogram is nearly uniform except that the bars at 0, 18, and 36 are about twice as high as desired. There is also a slight tendency for small and large multiples of 3 to have higher bars than their neighbors.
FIG. 2.

These figures plot the probability mass function of the number of balanced permutations of X − Y beaten by the original mean difference. The scale is chosen so that the heights of the bars average to 1 in each figure. The top figure is for n = 4 N(0,1) random variables contributing to each of 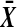 and
and 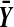 . With n = 4 there are 36 balanced permutations. The bottom figure is for n = 6 where there are 400 balanced permutations. Both figures differ from the ideal in which all bars have height 1.
. With n = 4 there are 36 balanced permutations. The bottom figure is for n = 6 where there are 400 balanced permutations. Both figures differ from the ideal in which all bars have height 1.
The bottom plot in Figure 2 considers the case of n = 6. Now there are B = 400 balanced permutations. The bar at 200 is about twice the desired height, while the ones at 0 and 400 are about 6.8 times as high as they should be.
Histograms for n = 8 and 10 (not shown) are also nearly flat except for extra height in the middle and much greater height at the ends. The heights at the extreme ends are described in Section 3.3, for values of n from 2 to 20.
The p values quoted above are one-tailed. For two-tailed p-values one might simply double both the nominal and observed quantities. The true two-tailed p value for a mean that beats all 400 balanced permutations would be about 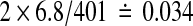 instead of
instead of 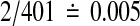 . In principle we might be better off putting both
. In principle we might be better off putting both 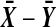 and
and 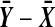 into the reference group. Then the two-tailed p-value would be about 2 × 6.8/402, but changing the denominators from 401 to 402 is a much smaller effect than the others we're studying, so we don't consider it further.
into the reference group. Then the two-tailed p-value would be about 2 × 6.8/402, but changing the denominators from 401 to 402 is a much smaller effect than the others we're studying, so we don't consider it further.
For n = 2 there is enough symmetry to determine the histogram of p values. For larger n and Gaussian data it may be possible to apply computational geometry. For example, when the treatment difference beats all balanced permutations the combined vector Z lies in an intersection of 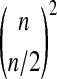 half spaces in
half spaces in 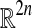 . But as remarked above, the answer is not distribution free. To confirm this distribution effect, we compared results for exponential data to those for normal data for n = 4.
. But as remarked above, the answer is not distribution free. To confirm this distribution effect, we compared results for exponential data to those for normal data for n = 4.
Results of a simulation of 100,000 cases for n = 4 are reported in Table 1. The simulations were run with independent N (0, 1) data for all Xi and Yi. We counted how many times the observed 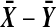 beat all 36 balanced permutation values. Then the simulation was rerun changing the N(0, 1) values to exponential (mean 1) values. The second simulations were coupled to the first by the probability integral transformation,
beat all 36 balanced permutation values. Then the simulation was rerun changing the N(0, 1) values to exponential (mean 1) values. The second simulations were coupled to the first by the probability integral transformation, 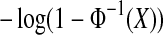 which turns a N(0, 1) random variable X into an exponential one. There were 370 cases where the Gaussian simulation beat all 36 of its balanced permutations while the exponential did not. There were 801 reverse cases. This matched pair difference is statistically significant by McNemar's test. It is equivalent to more than 12 standard deviations using equation 10.4 of Agresti (2002). The size of the effect however is not extremely large. For Gaussian data, the observed treatment effect beats all 36 balanced permutations about 1.843 times as often as it should, while for exponential data the ratio is about 2.003.
which turns a N(0, 1) random variable X into an exponential one. There were 370 cases where the Gaussian simulation beat all 36 of its balanced permutations while the exponential did not. There were 801 reverse cases. This matched pair difference is statistically significant by McNemar's test. It is equivalent to more than 12 standard deviations using equation 10.4 of Agresti (2002). The size of the effect however is not extremely large. For Gaussian data, the observed treatment effect beats all 36 balanced permutations about 1.843 times as often as it should, while for exponential data the ratio is about 2.003.
Table 1.
Results of 100,000 Simulations of a Balanced Permutation Test for the Difference of Means with n = 4 per Group
| Exp. < 36 | Exp. = 36 | |
|---|---|---|
| Gaussian < 36 | 94217 | 801 |
| Gaussian = 36 | 370 | 4612 |
The simulation was run with Gaussian values (all observations in both groups were independent N (0, 1)). Then it was rerun with values from the exponential distribution of mean 1.The simulations were coupled. The table shows counts of the number of times that the original permutation beat all 36 balanced permutations.
3.3. Results for larger n
The most interesting feature of the balanced permutation histogram is the probability that 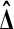 is larger than all
is larger than all 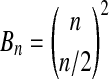 balanced permutations. It is fairly easy to investigate this probability in the Gaussian setting because of the following lemma.
balanced permutations. It is fairly easy to investigate this probability in the Gaussian setting because of the following lemma.
Lemma 1
Let n ≥ 2 be an even integer, and suppose that 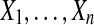 and
and 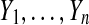 are independent N (μ, σ2) random variables for some σ > 0. Then the observed treatment effect
are independent N (μ, σ2) random variables for some σ > 0. Then the observed treatment effect 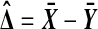 is statistically independent of the whole collection of balanced permutation treatment effects
is statistically independent of the whole collection of balanced permutation treatment effects 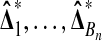 .
.
Proof
Under the hypothesis 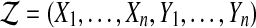 has a multivariate normal distribution. Therefore, so does
has a multivariate normal distribution. Therefore, so does 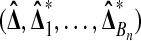 . Because of the balance, the correlation between
. Because of the balance, the correlation between 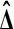 and
and 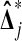 is zero. Therefore,
is zero. Therefore, 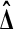 is independent of
is independent of 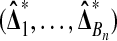 . ▪
. ▪
Lemma 1 lets us estimate the probability that 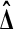 beats all Bn balanced permutations by sampling Xi and Yi for
beats all Bn balanced permutations by sampling Xi and Yi for 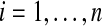 many times, computing
many times, computing 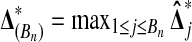 each time, and then averaging the values of
each time, and then averaging the values of
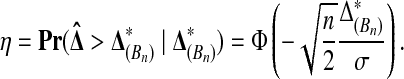 |
The results of this computation are shown in Table 2. For each even n from 2 to 20 inclusive, 100,000 data sets were investigated. The probability pn of beating all balanced permutations grows much faster than 1/(Bn + 1). By n = 20 the p values are off by roughly 400,000. A least squares fit of 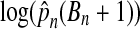 versus n has intercept −2.335, slope 0.745 and an R2 of 0.995. While the fit is good over the range 2 to 20, there is also positive curvature suggesting that extrapolating this model will underestimate the ratio.
versus n has intercept −2.335, slope 0.745 and an R2 of 0.995. While the fit is good over the range 2 to 20, there is also positive curvature suggesting that extrapolating this model will underestimate the ratio.
Table 2.
Rounded Numerical Estimates of the Probability that an Estimated Treatment Effect from n Observations per Group Beats All 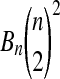 of its Balanced Permutation Treatment Effects
of its Balanced Permutation Treatment Effects
| n | 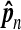 |
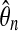 |
2.57CVn |
|---|---|---|---|
| 2 | 0.166000 | 0.83 | 0.006 |
| 4 | 0.051000 | 1.89 | 0.008 |
| 6 | 0.016700 | 6.71 | 0.011 |
| 8 | 0.005700 | 27.90 | 0.014 |
| 10 | 0.001980 | 125.00 | 0.017 |
| 12 | 0.000699 | 596.00 | 0.021 |
| 14 | 0.000248 | 2,920.00 | 0.026 |
| 16 | 0.000089 | 14,800.00 | 0.030 |
| 18 | 0.000032 | 75,500.00 | 0.037 |
| 20 | 0.000012 | 401,000.00 | 0.046 |
The estimate 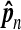 is in the second column. The normalized estimate
is in the second column. The normalized estimate 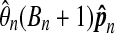 is in the third colunn. Ideally this should be near 1. The fourth column has Φ−1 (0.995) = 2.57 times the sampling coefficient of variation of
is in the third colunn. Ideally this should be near 1. The fourth column has Φ−1 (0.995) = 2.57 times the sampling coefficient of variation of 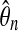 . For example, the approximate 99% confidence interval for θ8 is 1.4% higher or lower than the estimate 27.9.
. For example, the approximate 99% confidence interval for θ8 is 1.4% higher or lower than the estimate 27.9.
3.4. Balanced permutation tests based on t statistics
In high-throughput settings, permutation tests are often run on the t-statistic of equation (2), instead of the raw treatment effect 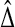 . Let t* be the value of t computed after a reassignment of the treatment versus control labels.
. Let t* be the value of t computed after a reassignment of the treatment versus control labels.
For a single test statistic this may seem redundant, because the permutation test on 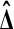 already behaves similarly to the t statistic. But in high throughput settings we often have many test statistics, for instance one per probe, whose underlying data have extremely different variances. The
already behaves similarly to the t statistic. But in high throughput settings we often have many test statistics, for instance one per probe, whose underlying data have extremely different variances. The 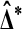 from different probes are not comparable. The t* values are more comparable because they normalize
from different probes are not comparable. The t* values are more comparable because they normalize 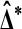 by a standard error. Permutations of t* from one probe may thus be useful in forming a test for another probe.
by a standard error. Permutations of t* from one probe may thus be useful in forming a test for another probe.
The theorem below shows that balanced permutations of t statistics have the same calibration problem that balanced permutations of treatment differences do. As a result, the values in Table 2 apply to t tests too.
Theorem 3
Let n ≥ 2 be an even number. Let 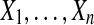 ,
, 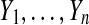 be distinct numbers and let
be distinct numbers and let 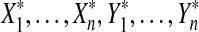 be those same values after a possible relabeling. Let
be those same values after a possible relabeling. Let 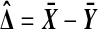 , let t be the t statistic in (2) and take
, let t be the t statistic in (2) and take 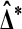 and t* to be the analogous quantities on the relabeled values. Then
and t* to be the analogous quantities on the relabeled values. Then 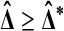 holds if and only if t ≥ t*
holds if and only if t ≥ t*
Proof
The value 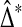 is a strictly increasing function of
is a strictly increasing function of 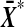 , as we vary the labelling. The proof follows by showing that t* is also strictly increasing in
, as we vary the labelling. The proof follows by showing that t* is also strictly increasing in 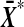 . For notational simplicity we show that t is strictly increasing in
. For notational simplicity we show that t is strictly increasing in 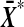 .
.
Let 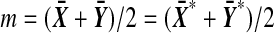 . Then
. Then 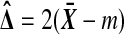 , where m does not depend on the labeling. Let D be the positive square root of
, where m does not depend on the labeling. Let D be the positive square root of 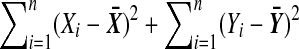 , so that
, so that 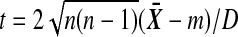 . We need to show that
. We need to show that 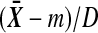 is strictly increasing in
is strictly increasing in 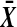 , where D depends on the labeling.
, where D depends on the labeling.
Introduce 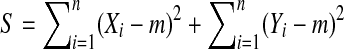 which does not depend on labeling. Replacing
which does not depend on labeling. Replacing 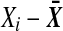 by
by 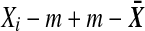 in D2, and similarly for the Y values, we get
in D2, and similarly for the Y values, we get 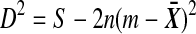 after simplification. Now
after simplification. Now
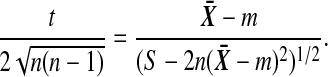 |
(5) |
The derivative of (5) with respect to 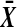 is
is 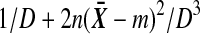 , which is strictly positive, and so t is increasing in
, which is strictly positive, and so t is increasing in 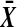 . ▪
. ▪
Theorem 3 shows that we get the same calibration problem whether we use t or 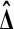 when using a one tailed criterion. The same equivalence holds for the two tailed criteria
when using a one tailed criterion. The same equivalence holds for the two tailed criteria 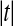 and
and 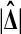 . Both
. Both 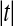 and
and 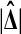 are increasing in
are increasing in 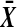 over the range
over the range 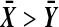 both are decreasing in
both are decreasing in 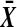 when
when 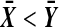 , and both are 0 when
, and both are 0 when 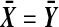 .
.
The assumption that all 2n values are distinct can be weakened. It ensures that D is never 0, but that can be enforced with a weaker condition.
3.5. Practical consequences
What we see in the figures is that the reference histogram is roughly uniform but with a spike in the middle and two more at the ends that grow quickly with n.
The spikes at the ends are the most serious because they strongly affect p-values. Suppose for example that n = 10. Then when the treatment effect beats all balanced permutations we might naively quote a one-tailed p value of 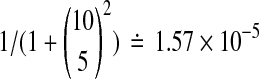 . However, the proper p-value in that setting is about 1.98 × 10−3, roughly 125 times larger.
. However, the proper p-value in that setting is about 1.98 × 10−3, roughly 125 times larger.
In studies that screen for interesting genes, using a small number n of arrays per group, the case where the observed effect beats all permutations is the most important one. When many genes are investigated, one has to make adjustments for multiple comparisons. Step down methods start by looking at the most significant genes first before choosing a cutoff and quoting a false discovery rate. For an account of multiple testing procedures, see Dudoit and van der Laan (2008). When the very smallest p-values are permissive, the whole process will be adversely affected.
The problem is not confined to the very smallest p-value. If the original treatment effect beats all but one of the balanced permutations, then the claimed p-value is about (125 + 1) / 2 = 63 times as small as it should be, assuming that the second bar from the right in the reference histogram has nearly its proper height. Similarly, outcomes that are near the ends of the histogram will get permissive p-values.
4. Pooled Balanced Permutations
As mentioned previously, microarray technologies can simultaneously measure the expression levels of thousands of genes at once. To this end, most microarrays use multiple measuring points (probes) for every gene. The main use of balanced permutations in high throughput problems is to calibrate a p value for one probe, by pooling data from a large number N of similar probes. It is also used to compute summaries of N linked tests, such as estimated FDRs. Sometimes there is a near one to one match relating probes to genes, but in other settings there can be numerous probes per gene. Typically, N is in the thousands, while n is much smaller.
Fan et al. (2004) use balanced permutations to get a pooled reference distribution to set N p-values. Jones-Rhoades et al. (2007) use them to estimate a rate for false discovery of genes, in a procedure that merges nearby differentially expressed probes into up and downregulated genomic intervals and then searches for new genes in those intervals.
Suppose that we are making N tests of type H0, one for each probe. For the i'th probe we get a t statistic ti. The null distribution of ti will not be exactly the t(2n−2) distribution, if the underlying data are not normally distributed. Furthermore, one might replace the t test by some quantity that is more robust to very noisy estimates of probe specific standard deviations.
Either a permutation or a balanced permutation analysis generates B permutations of the data and forms a reference distribution of BN test statistics tij for probes 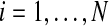 and permutations πj for
and permutations πj for 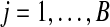 . With such a reference distribution, the p value for ti′ is
. With such a reference distribution, the p value for ti′ is
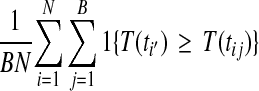 |
(6) |
where the usual choice for T is 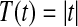 . In using equation (6) the original permutation of the data should be one of the B included.
. In using equation (6) the original permutation of the data should be one of the B included.
4.1. Non-null genes
There are known problems with pooling permutation-based statistics from N problems, whether the permutations are balanced or not. The main one is that the presence of non-null genes among the N cases tested distorts the reference distribution. In a full permutation analysis, many of the sampled permutations are very unbalanced for the treatment effect and they generate a heavy tailed reference distribution. The result is a conservative method that over-estimates FDR. Fan et al. (2005) and Xie et al. (2005) both raise this issue and seek to apply the permutations only to those genes that are not differentially-expressed. Identifying null genes is at least as hard as identifying non-null genes. Yang and Churchill (2007) compare methods that attempt to remove non-null genes.
The opposite occurs in balanced permutations. The balance has the effect of inflating the denominators of t statistics while the treatment effect cancels out of the numerators. The result is a resampled test statistic distribution with tails that are too short. Then, to the extent that these genes are included in the permutation, the FDR will be underestimated.
Table 3 shows some results of this type. Let the non-null data have Xi∼ N(μ, 1) and Yi ∼ N(0, 1) all independent, and suppose that the null data have a common N(τ, σ2) for all of Xi and Yi. The table presents the probability that t computed from the null data beats all the balanced permutation t statistics from the non-null data. The effect grows with μ and with n. The case with μ = 0 and n = 4 is not permissive, but μ = 0 is a null case, described in more detail below.
Table 3.
This Table Presents the Probability that a Null t Statistic Beats the Largest t Statistic Among Balanced Permutations of Independent Non-null Data
| |
n = 4 |
|
|
n = 6 |
|
|---|---|---|---|---|---|
| μ = 0 | μ = 1 | μ = 2 | μ = 0 | μ = 1 | μ = 2 |
| 0.971 | 1.936 | 4.234 | 1.813 | 6.144 | 20.64 |
| 0.030 | 0.025 | 0.017 | 0.043 | 0.037 | 0.021 |
The values shown in the top row are  , which should be 1 for a properly calibrated reference set. Here t is a t statistic computed with independent Xi,
, which should be 1 for a properly calibrated reference set. Here t is a t statistic computed with independent Xi,  for
for  and
and  are all of the balanced permutations of a t statistic, on data where
are all of the balanced permutations of a t statistic, on data where 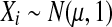 and
and 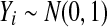 independently of each other and of t. The second row gives 99% relative error values. There were 10,000 simulations of each case.
independently of each other and of t. The second row gives 99% relative error values. There were 10,000 simulations of each case.
There is partial remedy for the non-null case, described by Pan (2003), which should be helpful when n is large. Notice that each observation belongs to either the test or control group in the original data and to either the test or control group in the permuted data. This yields four different sets of observations in any balanced permutation. The idea of Pan (2003) is to form the variance estimate in the denominator of the t statistic by pooling sample variances from these four subsamples instead of just the permuted treatment and control groups. This will remove mean differences from variance estimate in the denominator of t. Unfortunately, the degrees of freedom for the resulting reference distribution are 2n − 4 instead of 2n − 2, which will be an important difference when n is small.
Scheid and Spang (2006, 2007) also note the problem from including non-null genes in the reference distribution. They have proposed a method of filtered permutations. Instead of filtering the list of genes to be mostly null, they filter the set of permutations to be the ones π for which a subsequent permutation 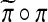 gives nearly U(0, 1) distributed p values for a comparison of the two permuted groups. That is, they employ an iterated permutation analysis. By considering permutations of permutations their method might be accomplishing numerically what the group structure provides mathematically. But there is as yet no theoretical analysis of filtered permutations.
gives nearly U(0, 1) distributed p values for a comparison of the two permuted groups. That is, they employ an iterated permutation analysis. By considering permutations of permutations their method might be accomplishing numerically what the group structure provides mathematically. But there is as yet no theoretical analysis of filtered permutations.
4.2. Null genes
In the case of balanced permutations, there are also difficulties in the distribution of p values from the null genes. For any probe i, we know that the uniform distribution on 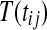 for
for 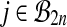 or
or 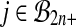 fails to be a good reference distribution. However, when we combine all N of these test distributions into a single reference distribution, then for whichever probe i′ we are testing, our reference distribution is dominated by data from probes i ≠ i′. Hence we need to look at how a reference distribution from one probe works for data from another probe.
fails to be a good reference distribution. However, when we combine all N of these test distributions into a single reference distribution, then for whichever probe i′ we are testing, our reference distribution is dominated by data from probes i ≠ i′. Hence we need to look at how a reference distribution from one probe works for data from another probe.
A precise comparison of T(ti′) with π(T (ti)) depends on the dependence between data from probes i and i′. There is usually some dependence, because different genes on the same array tend to be correlated. The case where probes i and i′ are highly correlated should be similar to that in which a probe is compared to its own balanced permutations. The case of independent probes is easier to study than probes with arbitrary dependence, and this case should be favorable to the use of balanced permutations. So we consider 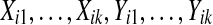 that are independent of
that are independent of 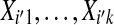 ,
, 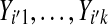 .
.
For the test statistic 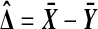 , on normal data, we find from Lemma 1 that it makes no difference whether probe i is compared to balanced permutations of itself or of another null probe i′.
, on normal data, we find from Lemma 1 that it makes no difference whether probe i is compared to balanced permutations of itself or of another null probe i′.
As mentioned above, when multiple genes are used to calibrate each other, it is more reasonable to permute t statistics. In Table 3, the values for μ = 0 correspond to independent null probes. Figure 3 shows histograms of the number of balanced permuted t statistics from one sample that are beaten by a t statistic from another independently generated sample. Because the second sample is independent of the first we can use the known t distribution to advantage. In this simulation, we generated the list of balanced t values 1000 times. For each of these times, we sorted the 36 possible values, splitting the real line into 37 bins, and evaluated the probability that an independent t random variable would land in each of those bins. The bars shown are the averages of these probabilities over the 1000 simulations.
FIG. 3.

These plots show histograms of the number of balanced permutations of the t statistic from one data set that are beaten by a t test from an independently sampled data set. All data values were independent N(0,1) in both data sets. The top figure is for n = 4, and the bottom one is for n = 6.
When n = 4 the spikes at the ends have vanished, but the one in the middle remains at about 2.0. So for this case we would get very nearly the desired flat histogram from the device of inserting 0 as a pseudo value. When we repeat the simulation for n = 6, there are spikes at the end. The t statistic computed on one data set with n = 6 has about twice the chance it should have of beating all the balanced permutations from another data set.
4.3. Multiple independent null genes
Now suppose that probe i′ under test is compared to a reference distribution made up of numerous probes indexed by 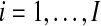 and that all I + 1 of the probes are statistically independent. Probe i′ may tend to beat all balanced permutations of any probe i more often than it should. But when the I probes are independent of each other it becomes less likely that i′ beats all balanced permutations of all I other probes.
and that all I + 1 of the probes are statistically independent. Probe i′ may tend to beat all balanced permutations of any probe i more often than it should. But when the I probes are independent of each other it becomes less likely that i′ beats all balanced permutations of all I other probes.
We generated 1,000,000 normally distributed data sets of two groups with n = 6 observations each. From each of these, we found 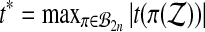 , the maximum of 200 two-tailed t statistics on 10 degrees of freedom each. These million values were then arranged into roughly 106/I lists of I values each. The probability that an independently generated value of |t(10)| exceeds the largest of these I values should average to 1/(200I) for proper calibration. Figure 4 shows corresponding sample averages presented as a multiple of 1/(200I), as I varies from 1 to 25. We see that pooling independent null data from balanced permutations is somewhat permissive but that the effect disappears by around I = 15 to 25.
, the maximum of 200 two-tailed t statistics on 10 degrees of freedom each. These million values were then arranged into roughly 106/I lists of I values each. The probability that an independently generated value of |t(10)| exceeds the largest of these I values should average to 1/(200I) for proper calibration. Figure 4 shows corresponding sample averages presented as a multiple of 1/(200I), as I varies from 1 to 25. We see that pooling independent null data from balanced permutations is somewhat permissive but that the effect disappears by around I = 15 to 25.
FIG. 4.

This figure shows that a reference distribution based on balanced permutation t tests of I > 1 independent null probes is not as permissive as one using a single independent null probe. The horizontal axis shows the number I of independent probes used in a simulation of balanced permutation t tests for two groups of n = 6 observations each. The vertical axis shows 200 I times the probability that the original data has a value of |t| larger than that of all 200 I balanced permutation |t| values in the reference set. The dots show estimated probability ratios, the curves show upper and lower 99% pointwise confidence limits, and there is a horizontal reference line at 1.
The same pattern holds for n = 8 observations per group. There the ratio starts nearer to 4.0 but is again close to 1 for I = 20.
If we can successfully eliminate the non-null cases and then pool a large number of independent probes, we will get a suitable reference distribution. Then again we might also do that with ordinary permutations, and should we miss some non-null cases, the consequence would be a conservative test instead of a permissive one.
5. Discussion
We find no reason to prefer balanced permutations to ordinary permutations, for genomic applications. The intuitive argument that leads one to expect greater power for them, is countered by them producing permissive p-values when there is no effect. At a minimum, more work is required to justify their use or properly calibrate them.
When used to judge the sampling distribution of a test statistic from N different genes, balanced permutation reference sets are adversely affected by non-null cases among those permuted. So are ordinary permutations. But where ordinary permutations yield a conservative reference distribution, balanced permutations provide a permissive one, when based on the t statistic or on a difference in means. Such permissive methods are more likely to lead to false discoveries.
In practice, it is important to attempt to remove the non-null cases before constructing a permutation-based reference distribution. When the balanced permutations are applied to null cases, we find that balanced permutations can still be mildly permissive. Fortunately, the effect seems to disappear when a large number of null cases have been pooled. However, that disappearance was for null cases that were independent of the case being tested, a condition not required for ordinary permutations. Correlations among the null cases and between the null cases and the gene being tested could result in a distortion that does not decrease for large numbers of pooled reference genes.
For the problems that we have analyzed, it is clear that balanced permutations cannot simply be used as a substitute for full permutation methods. Our analysis does not cover very complicated methods such as the one Jones-Rhoades et al. (2007) used or the empirical Bayes analysis proposed by Efron et al. (2001). We would hesitate to use balanced permutations as an ingredient in more complicated analyses, because they work poorly in simple situations.
We started looking at balanced permutations as an ingredient in a similarly complicated analysis, involving data from the AGEMAP study of Zahn et al. (2007). We considered near balanced permutations with respect to age (old versus very old mice) within strata defined by male and female mice. It could only be near balance because there were 5 mice of each sex at each age. For each mouse, there were 14 tissues. The effects of interest were related to differences in the gene-gene correlation matrices at the two ages. Simulations of even a simple before versus after correlation for just one pair of genes whose correlation does not change revealed permissive p-values like the ones we report here. As a consequence that work continued with ordinary permutations instead of balanced ones.
A final disadvantage of balanced permutations is the one mentioned at the NCI website. There are fewer balanced permutations, so that granularity properties are exacerbated.
Acknowledgments
We thank Sarah Emerson for helpful discussions. This work was supported by the U.S. National Science Foundation (grant DMS-0604939), the U.S. National Institutes of Health, and a training fellowship from the National Library of Medicine to the Biomedical Informatics Training Program at Stanford (NLM grant no. 07033).
Disclosure Statement
No competing financial interests exist.
References
- Agresti A. Categorical Data Analysis. 2nd. Wiley; New York: 2002. [Google Scholar]
- Bahadur R.R. Savage L.J. The nonexistence of certain statistical procedures in nonparametric problems. Ann. Math. Stat. 1956;27:1115–1122. [Google Scholar]
- Dudoit S. van der Laan M.J. Multiple Testing Procedures with Applications to Genetics. Springer-Verlag; New York: 2008. [Google Scholar]
- Edgington E.S. Onghena P. Randomization Tests. 4th. Chapman and Hall/CRC; Boca Raton, FL: 2007. [Google Scholar]
- Efron B. Tibshirani R. Storey J.D., et al. Empirical Bayes analysis of a microarray experiment. J. Am. Stat. Assoc. 2001;96:1151–1160. [Google Scholar]
- Fan J. Tam P. Woude G.V., et al. Normalization and analysis of cDNA microarrays using within-array replications applied to neuroblastoma cell response to a cytokine. Proc. Natl. Acad. Sci. USA. 2004;101:1135–1140. doi: 10.1073/pnas.0307557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J. Chen Y. Chan H.M., et al. Removing intensity effects and indentifying significant genes for Affymetrix arrays in macrophage migration inhibitory factor-suppresed neuroblastoma cells. Proc. Natl. Acad. Sci. USA. 2005;102:17751–17756. doi: 10.1073/pnas.0509175102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good P. Permutation Tests: A Practical Guide to Resampling Methods for Testing Hypotheses. 2nd. Springer-Verlag; New York: 2000. [Google Scholar]
- Jones-Rhoades M.W. Borevitz J.O. Presus D. Genome-wide expression profiling of the Arabidopsis female gametophyte identifies families of small, secreted proteins. PLOS Genet. 2007;3:1848–1861. doi: 10.1371/journal.pgen.0030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann E.L. Romano J.P. Testing Statistical Hypotheses. 3rd. Springer; New York: 2005. [Google Scholar]
- Miller R.G. Beyond ANOVA, Basics of Applied Statistics. Wiley; New York: 1986. [Google Scholar]
- Pan W. On the use of permutation in and the performance of a class of nonparametric methods to detect differential gene expression. Bioinformatics. 2003;19:1333–1340. doi: 10.1093/bioinformatics/btg167. [DOI] [PubMed] [Google Scholar]
- Scheid S. Spang R. Permutation filtering: a novel concept for significance analysis of large-scale genomic data. Lect. Notes Comput. Sci. 2006;3909:338–347. [Google Scholar]
- Scheid S. Spang R. Compensating for unknown confounders in microarray data analysis using filtered permutations. J. Comput. Biol. 2007;14:669–681. doi: 10.1089/cmb.2007.R009. [DOI] [PubMed] [Google Scholar]
- Tusher V.G. Tibshirani R. Chu G. Significance analysis of microarrays applied to the ioninzing radiation response. Proc. Natl. Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y. Pan W. Khodursky A.B. A note on using permutation-based false discovery estimates to compare different analysis methods for microarray data. Bioinformatics. 2005;21:4280–4288. doi: 10.1093/bioinformatics/bti685. [DOI] [PubMed] [Google Scholar]
- Yang H. Churchill G. Estimating p-values in small microarray experiments. Bioinformatics. 2007;23:38–43. doi: 10.1093/bioinformatics/btl548. [DOI] [PubMed] [Google Scholar]
- Zahn J. Poosala S. Owen A.B., et al. AGEMAP: a gene expression database for aging in mice. PLOS Genet. 2007;3:2326–2337. doi: 10.1371/journal.pgen.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]