

Abstract
The RAS/RAF/MEK/ERK signaling pathway has been largely unexplored as a potential therapeutic target in lymphoma. The novel 2nd generation anti-MEK small molecule, AZD6244, down-regulated its direct downstream target, phospho-ERK (pERK) in germinal center and nongerminal center diffuse large B-cell lymphoma (DLBCL) cell lines and primary cells. Similar decreased pERK levels were noted despite constitutive activation (CA) of MEK. Consequently, several lymphoma-related ERK substrates were down-regulated by AZD6244 including MCT-1, c-Myc, Bcl-2, Mcl-1, and CDK1/2. AZD6244 induced time- and dose-dependent antiproliferation and apoptosis in all DLBCL cell lines and fresh/primary cells (IC50 100nM-300nM). Furthermore, AZD6244 resulted in significantly less tumor compared with control in an in vivo DLBCL SCID xenograft model. Cell death was associated with cleaved PARP, caspases-8, -9, and -3, and apoptosis was caspase-dependent. In addition, there was stabilization of FoxO3a, activation of BIM and PUMA, and a significant decrease in c-Myc transcripts. Moreover, siRNA knockdown of BIM abrogated AZD6244-related apoptosis, while shRNA knockdown of ERK minimally sensitized cells. Finally, manipulation of AKT with transfection of OCI-LY3 cells with CA-AKT or through chemical inhibition (LY294002) had minimal effect on AZD6244-induced cell death. Altogether, these findings show that the novel anti-MEK agent, AZD6244, induced apoptosis in DLBCL and that cell death was BIM-dependent.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common lymphoid malignancy in adults accounting for nearly 35% of all non-Hodgkin lymphomas (NHL). Significant advances have been in the treatment of DLBCL, particularly with immunochemotherapy, however approximately 30%-40% of patients still die from this malignancy. In addition, short- and long-term toxicities of chemotherapy, including secondary malignancies and leukemias, continue to adversely impact the long-term prognosis of patients. Continued investigations of novel targeted therapeutic agents in DLBCL are warranted.
The RAS/RAF/mitogen-activated protein kinase 1/2 (MEK1/2)/extracellular signal-regulated kinase 1/2 (ERK1/2) plays a prominent role in cancer biology, including hematologic malignancies, in part through the regulation of cell growth and proliferation.1–5 Activating mutations in RAS and RAF lead to aberrant activation of their downstream target, MEK1/2. Directly downstream of MEK1/2, ERK1 and ERK2 are intimately involved in transducing signals from growth factor receptors and cytokine receptors after ligand binding.2 Furthermore, ERK is the only known catalytic substrate of MEK.6 We and others have shown that the MEK/ERK signaling pathway is constitutively active in a large number of cancers, including hematologic malignancies.3,4,7–9 Moreover, the MEK/ERK signal transduction cascade has been shown to be susceptible to pharmacologic intervention. Thus, MEK has emerged as an attractive therapeutic target in cancer.
The majority of preclinical, and especially clinical trial data studying MEK inhibitors to date have emerged largely from solid tumor studies.10–14 We recently showed in preclinical studies that inhibition of ERK1/2 phosphorylation by 1st generation MEK and ERK inhibitors correlated with significant cell death in a lymphoma tumor model,15 while others showed that sublethal concentrations of a 1st generation MEK antagonist potentiated the effect of sorafemib in lymphoma cells.16 Furthermore, we recently demonstrated that MCT-1, an oncogene directly downstream of MEK/ERK, is over-expressed in the majority of primary DLBCLs.15 MCT-1 is known to colocalize with ERK1/2, while phosphorylation of MCT-1 protein by ERK is critical for stabilization of MCT-1 protein and for its functional ability to promote cell proliferation.15,17
ARRY-142886 (AZD6244, selumetinib; Astra Zeneca) is a selective nonATP-competitive 2nd generation oral MEK inhibitor studied primarily in solid tumor studies with reported nanomolar activity against purified MEK1 enzyme.18–23 Furthermore, phase 1 and phase 2 solid tumor clinical trials have shown this agent to be well-tolerated and have encouraging clinical efficacy.24–27 To our knowledge, minimal data are available on newer generation MEK inhibitors in lymphoma and moreover, this anti-MEK agent has never been examined in lymphoma. We sought to examine the mechanisms of action and cytotoxic effect of the novel 2nd generation MEK small molecule antagonist, AZD6244, in lymphoma cell lines, primary cells, and an in vivo human DLBCL xenograft model.
Methods
Cell culture and treatment
DLBCL germinal center cell lines (SUDHL4, SUDHL6, SUDHL10, and OCI-LY19) and the nongerminal center cell line, OCI-LY3, were grown in RPMI 1640 (Invitrogen) containing 10% FBS. The 2nd generation MEK inhibitor AZD6244 was supplied from Astra Zeneca.
MTT proliferation analysis
In a 96-well flat bottom plate, approximately 104 cells/100uL were plated and treated for 24, 48, or 72 hours with vehicle or increasing concentrations of AZD6244 (50nM-400nM). After treatment 20 μL of MTS/PMS (Promega Cell Titer 96 Aqueous Non-Radioactive Cell Proliferation assay), solution was added to each well and incubated for 4 hours at 37°C. Plates were then analyzed at 490 nm wavelength. Data were plotted as growth percentage of control. This value was determined by comparing the absorbance reading of each set of control wells to which no drug was added.
Apoptosis assays
DLBCL cell lines were seeded at equal density and then treated with AZD6244 in complete RPMI 1640 medium. Forty-eight hours after treatment, cells were harvested and apoptosis was analyzed by flow cytometry using the annexin V/PI staining kit (BD Biosciences). The significance of differences between experimental conditions was determined using the Student t test.
Soft agar colony-forming assay
SUDHL4 and OCILY3 cells were incubated with nanomolar concentrations of AZD6244 in the soft agar to form colonies as previously described.15 The number of colonies in each well was counted. Colony formation (> 50 cells) was examined under phase-contrast microscopy. Images were taken at room temperature using a Nikon Eclipse TE-2000S microscope. Each experiment was conducted at least 3 times and the significance of differences between experimental conditions was determined using the Student t test.
Primary DLBCL cells
After approval by the Northwestern University Institutional Review Board (IRB) and written informed consent in accordance with the Declaration of Helsinki, peripheral blood was drawn from 3 patients with leukemic phase of DLBCL. Each of these 3 patients had relapsed disease after prior rituximab/cytotoxic chemotherapy. Two of the 3 patients had a CD10+ germinal center phenotype, while the third appeared to have a nongerminal center DLBCL subtype (ie, CD10-negative and Bcl-6-negative). All peripheral blood was diluted 1:1 with PBS (Ca2+ and Mg2+ free) and was layered on top of Ficoll-Paque Plus (Sigma-Aldrich). Samples were then centrifuged at 150g for 20 minutes at room temperature; the buffy coat layer was removed and washed with PBS twice and subsequently placed in culture with RPMI medium.
In vivo human lymphoma xenograft model
Female severe combined immunodeficient (SCID) beige mice were housed in a pathogen-free environment under controlled conditions of light and humidity and received food and water ad libitum. SUDHL6 cells (2 × 106) were resuspended in 100μL PBS and then mixed with an equal volume of Matrigel. The mixture was injected subcutaneously into the left and right dorsal flanks of 5- to 7-week-old female SCID mice. When the tumor reached the size of 60-160 mm,3 the drug (AZD6244) was administered by intraperitoneal injection every other day at a dose of 10 mg/kg of body weight for a total of 3 weeks. Injection of the vehicle alone (5% DMSO in 0.05M PBS) was used as a control. The significance of differences between treatment arms was determined using the Student t test.
Cell-cycle analysis
Distinct phases of the cell cycle were distinguished by DNA staining with the fluorescent dye propidium iodide and measured by flow cytometry. Cells were washed in ice cold PBS, fixed in 70% ethanol, and stained for 30 minutes at 37°C with propidium iodide (50μg/mL propidium iodide in hypotonic sodium citrate solution containing 50 μg/mL RNase) followed by flow cytometric analysis. The percentages of cells in G1, S, and G2/M phases were determined using the cell-cycle analysis program Modfit LT 3.2 (Verity Software House).
Western blot analysis
Cells were centrifuged, washed with cold PBS, and lysed on ice for 30 minutes in lysis buffer containing protease and phosphatase inhibitors. Protein concentrations were determined with the Bio-Rad protein assay kit (Bio-Rad). Total protein (50 μg) was electrophoresed on 12% SDS polyacrylamide gels and transferred to nitrocellulose membranes, blocked for 1 hour with 50mM Tris buffer, pH 7.5 containing 0.15M NaCl, 0.05% Tween 20 (TBST) and 5% (wt/vol) nonfat dry milk and probed overnight at 4°C with TBST containing primary antibodies. After three 10-minute washes in TBST, the filters were incubated HRP-conjugated secondary antibody in the blocking buffer for 1 hour. After three 10-minute washes in TBST, proteins were detected by enhanced chemiluminescence detection reagents (Amersham Biosciences). The following antibodies used for immunoblotting were purchased from Cell Signaling Technology: pERK1/2, ERK1/2, MEK2, Bcl2, Mcl1, BIM, PUMA, pFoxO3a, FoxO3a, caspases 3, 8, 9, and PARP. c-Myc antibody was purchased from BD Biosciences, and MCT-1 antibody was purchased from Research Genetics Inc. Blots were stripped and reprobed with β-actin (Santacruz Biotechnology) used as the loading control.
Plasmids and transfections
ERK1, ERK2, and BIM were knocked down using GIPZ lentiviral shRNA from Open Biosystem. After transduction, cells were selected in puromycin-containing media for 14 days. Stably transduced cells that were viable after knockdown were selected for further analysis. This included transfection with Bcl-2 (Addgene plasmid 8768) or Mcl-1 (Addgene plasmid 25375) expressing plasmids. Wild-type MEK2 (WT MEK2) and constitutively active MEK2 constructs have been previously described.28 Constitutively active MEK1 (plasmid L1E-1) was obtained from Addgene. Cells were transfected with empty vector, wild-type MEK2, and constitutively active MEK1 or MEK2 constructs using Amaxa Nucleofector kit V (Amaxa) and program M013. After transfection cells were selected in puromycin or hygromycin, respectively. Stably transfected cells were used for further studies. Constitutively active AKT construct or Myr-AKT (plasmid 1036) was purchased from Addgene. OCI-LY3 cells were transfected with Myr- AKT or empty vector using Amaxa nucleofection kit L (Amaxa) and program A20. After transfection cells were selected in neomycin. Viable stably transfected cells that expressed constitutively active AKT were selected for further analysis. BIM and AKT siRNA were purchased from QIAGEN. Cells were transiently transfected with control, BIM or AKT siRNA using Amaxa nucleofection kit L. After 24 hours of transfection, cells were treated with AZD6244 for 48 hours. The c-Myc promoter-luciferase reporter plasmid containing 2100 bp of human c-Myc promoter sequences was a kind gift from Dr Bayar Thimmapaya (Northwestern University). DLBCL cells were incubated with AdM4 Myc reporter or mutant Myc reporter adenovirus for 4 hours. After 24-hour transduction, cells were incubated with AZD6244 for 24 hours followed by luciferase assay.
Results
AZD6244 inhibits ERK phosphorylation levels in DLBCL cells
Because ERK is the only known direct substrate for MEK, we examined whether inhibition of MEK with AZD6244 affected ERK phosphorylation. Figure 1A shows prompt (as early as 3 hours) and marked reduction of ERK phosphorylation in SUDHL6, SUDHL10, OCI-LY19, and OCI-LY3 DLBCL cells after exposure to 200nM AZD6244. Figure 1B shows dose-dependent reduction in pERK and MCT-1 after incubation with nanomolar concentrations of AZD6244. We further examined the effect of AZD6244 on fresh/primary DLBCL cells. A notable decrease in pERK level was observed with as little as 50nM AZD6244, while there was only a slight decrease in MCT-1 expression with 200nM AZD6244 in DLBCL primary cells (Figure 1C). Interestingly, minimal decrease of MCT-1 was also noted in OCI-LY19 (Figure 1B) as well as OCI-LY3 cells (data not shown).
Figure 1.

ERK and MCT-1 in DLBCL cell lines and primary cells. (A) SUDHL6, SUDHL10, OCI-LY19, and OCI-LY3 cells were treated with 200nM AZD6244 for the indicated periods of time. Cell lysates were subjected to Western blotting using pERK and ERK antibodies. Actin was used as an internal control. (B) SUDHL4, SUDHL6, and OCI-LY19 cells were treated with indicated concentration of AZD6244 for 18 hours. pERK, ERK, and MCT-1 protein levels were measured by Western blotting using the respective specific antibodies. (C) Western blot showing down-regulation of pERK and MCT-1 in primary DLBCL cells afterAZD6244 exposure for 6 and 16 hours. Peripheral blood monocytes (PBMC) were obtained from 3 DLBCL patients (each with relapsed/refractory transformed DLBCL with leukemia involvement). PBMCs were incubated with indicated concentrations of AZD6244 for 6 or 16 hours. This patient/figure is representative of 3 primary DLBCL subjects. Cell lysates were subjected to Western blotting using specific antibodies for pERK, ERK or MCT-1. (D) Over-expression of constitutively active (CA) MEK2 increased ERK and MCT-1 protein levels. Raji cells were transfected with wild-type (WT) MEK2, CA MEK2 construct, or the vector control (V). After 24 hour transfection, cells were incubated with 200nM of AZD6244 for 24 hours. The cell lysates were subjected to Western blot analysis. (E) OCI-LY3 cells were transduced with scrambled (sc) shRNA or ERK2 shRNA by spin infection using GIPZ lentivirus system. After puromycin selection cells were subjected to Western blotting to check the protein level. (F) OCI-LY3 cells after positive selection were treated with 200nM or 300nM AZD6244 for 48 hours, which was followed by annexin V/PI staining and analyzed by flow cytometry.
AZD6244-induced apoptosis is mediated through MEK/ERK inhibition
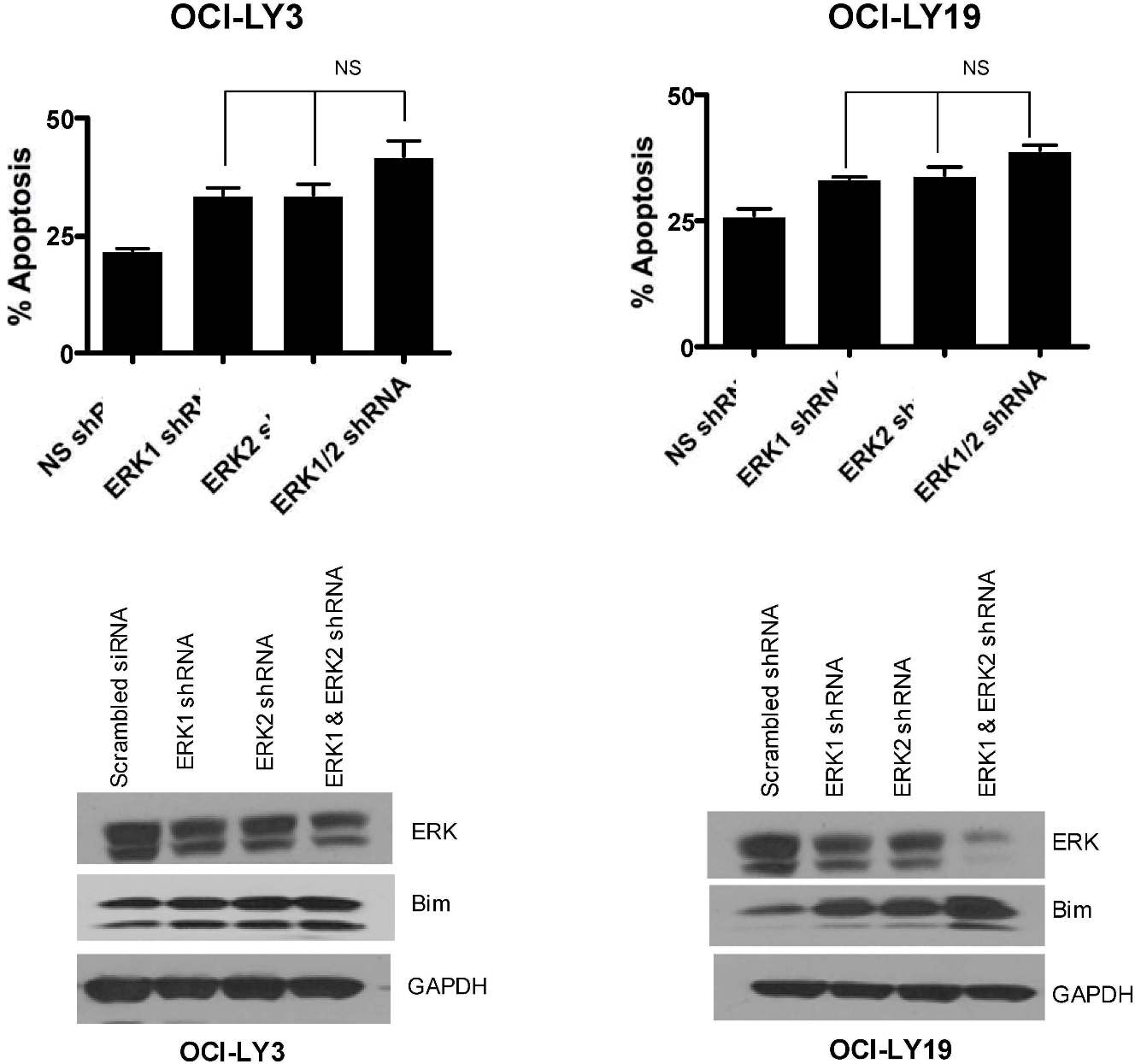
To further examine the specificity of AZD6244 and the MEK/ERK signaling pathway, we determined the effect of AZD6244 on ERK phosphorylation in Raji cells transfected with wild-type or constitutively active MEK (CA MEK). As expected, CA MEK up-regulated pERK expression. Of note, AZD6244 overcame the effect of constitutively active MEK; in Raji cells transfected with CA MEK, ERK phosphorylation remained markedly diminished (Figure 1D). Furthermore, we corroborated our small inhibitory molecule inhibition findings employing a complementary genetic approach. ERK2 was knocked down in OCI-LY3 cells using lentivirus-based ERK2 shRNA, which was confirmed by Western blotting (Figure 1E). OCI-LY3 cells were treated with 200nM or 300nM of AZD6244 for 48 hours after transduction of ERK2 shRNA. AZD6244-induced apoptosis was increased in the presence of ERK2 shRNA at both concentrations compared with control, however this difference was not significant (P > .05; Figure 1F). We further investigated cell death after knockdown of both ERK1 and ERK2 concurrently in OCI-LY3 and OCI-LY19 cell lines. Knockdown with ERK1 and ERK2 together appeared to cause slightly increased apoptosis in these cells compared with either construct alone (OCI-LY3 more so than OCI-LY19), however these results were not significant (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Disruption of cell-cycle progression, growth inhibition, reduction in clonogenic capacity, and induction of apoptosis
Flow cytometric analysis revealed significant changes in the cell-cycle distribution profile of DLBCL cells after exposure to AZD6244, which occurred in a concentration-dependent manner. The treatment of cells with AZD6244 resulted in G0/G1 arrest with an associated decrease in G2/M and S phase cell population (Figure 2A). We hypothesized that disruption of MEK/ERK pathway would result in growth inhibition of DLBCL cells. To test this hypothesis, we treated cell lines with increasing concentrations of AZD6244 for 24 to 72 hours and measured cell growth inhibition. As shown in Figure 2B, there is significant growth inhibition in all cell lines after 48 hours. In addition, we noted a statistically significant decrease in the colony formation of SUDHL4 and OCILY3 cells after 200 nm (P < .01) or 300 nm (P = .006) AZD6244 exposure (data not shown).
Figure 2.

AZD6244 induces cell cycle and growth arrest in DLBCL cell lines, primary cells, and in a SCID xenograft model. (A) All cell lines were treated with the indicated concentrations of AZD6244 for 24 hours. After fixation in 70% ethanol, cells were stained with propidium iodide (final concentration 50 μg/mL) in hypotonic solution containing RNase (180 units/mL) for 30 minutes. Cells were analyzed by flow cytometry. The bar graph shows significant G1 arrest after AZD6244 treatment. (B) AZD6244 induces growth inhibition. All cell lines were treated with variousconcentration (50nM to 400nM) for 24, 48, and 72 hours. Cell growth was measured by MTT assay. P values are inserted for each cell line regarding concentration- and time-dependent comparisons. (C) AZD6244 induces concentration-dependent apoptosis. All cell lines were treated with indicated concentrations of AZD6244 for 48 hours. Apoptosis was measured by annexin V/PI staining by flow cytometry. (D) Apoptotic induction in primary/fresh DLBCL cells afterAZD6244. PBMCs were incubated with the indicated concentrations for 24 (left) or 72 hours (right). Apoptosis was measured by annexin V/PI staining followed by flow cytometric analysis. Data were analyzed by FACS express software. (E) SUDHL-6 cells were subcutaneously injected into left and right dorsal flanks of 7-week-old female SCID mice. When the tumor reached the size of approximately 60-160 mm,3 AZD6244 was administered intraperitoneally every other day at a dose of 10 mg/kg body weight for a total of 3 weeks. Tumors were measured 3 times weekly. Ctrl indicates control; and Hr, hours. *P < .05; **P < .01, and ***P < .001.
We next examined induction of apoptosis in DLBCL cell lines after AZD6244 treatment. Figure 2C shows > 70% apoptosis in SUDHL4 and > 40% apoptosis in SUDHL6 with 200nM, while OCI-LY19 and OCI-LY3 show approximately 60% apoptosis with 300nM AZD6244. DLBCL patient peripheral blood monocytes were also exposed to increasing concentrations of AZD6244 (ie, 25nM to 400nM) for 24 and 72 hours (Figure 2D). AZD6244 induced apoptosis in primary cells at concentrations as low as 100nM. Altogether, the half maximal inhibitory concentration (IC50) for in vitro and primary cells was 100nM-300nM. Notably, there were no differences in apoptosis noted based on germinal center versus nongerminal center DLBCL subtype (data not shown).
Antitumor efficacy of AZD6244 in a DLBCL SCID xenograft model
We examined whether AZD6244 inhibits lymphoma growth in a SCID xenograft DLBCL mouse model. As shown in Figure 2E, 10mg/kg of AZD6244 effectively inhibited tumor growth in a SUDHL6 xenograft. At this drug dose, no lethal toxicity or significant weight loss were observed among treated animals compared with control mice (data not shown). From 28 weeks on, mice treated with AZD6244 had significantly decreased average tumor volume compared with control (P < .05).
Drug inhibition and genetic manipulation of MEK
We assessed pERK levels in OCI-LY3 cells transfected with constitutively active MEK1 followed by treatment with the first generation MEK inhibitor, PD098059, or AZD6244. Unlike AZD6244-treated cells, appreciable pERK levels were detectable with PD98059 exposure in the presence of constitutively active MEK1 in OCI-LY3 cells, albeit less than untreated cells (Figure 3A). Furthermore, constitutively active MEK1 transfection resulted in partial inhibition of AZD6244-induced apoptosis. Interestingly, PD98059 had minimal apoptotic effect in Raji or OCI-LY3 cells (Figure 3B) as well as other cell lines (data not shown). We also transfected OCI-LY19 and OCI-LY3 cells with dominant negative MEK1. Dominant negative MEK1 resulted in increased levels of apoptosis compared with vector control (Figure 3C).
Figure 3.

MEK overexpression and deletion. (A) DLBCL cells were transduced with wild-type or constitutively active MEK1 using retroviral delivery. After transduction of constitutively active MEK1, cells were treated with PD98059 (100μM) or AZD6244 (200nM for Raji and 300nM for OCI-LY3). Expression of pERK and ERK was determined by Western blot analysis using specific antibodies in OCI-LY3 cells with constitutively active MEK1 in the presence of PD98059 or AZD6244. (B) Raji and OCI-LY3 cells were treated with PD98059 or AZD for 48 hours after transduction with MEK1-CA retrovirus. Apoptosis was measured by annexin V/PI staining followed by flow cytometry. (C) OCI-LY3 and OCI-lY19 cells were transfected with vector alone or dominant negative (DN)–MEK1. After 72-hour transfection, apoptosis was measured by annexin V/PI staining and assessed by flow cytometry. Expression of DN-MEK1 was confirmed by Western blotting using HA antibody. CA indicates constitutively active; wt, wild-type; AZDAZD6244; and PD, PD98059.
Caspase-dependent cell death
To assess the role of caspase activation in AZD6244-induced apoptosis, caspases and PARP were measured. As shown in Figure 3A, increasing concentrations of AZD6244 induced cleavage of caspases 9 and 8 with associated decreases in full-length caspases. In addition, cleavage of caspase 3 and PARP were observed in cell lines (Figure 4A). In primary/fresh DLBCL cells, caspases 3 and 8 and PARP were cleaved (Figure 4B). To further examine the importance of caspase activation in AZD6244 induced cell death, cells were preincubated with the caspase inhibitors. In OCI-LY3 cells (Figure 4C), there was partial inhibition of apoptosis with caspase 9 and pan-caspase inhibition, while in SUDHL6 cells (Figure 4D), apoptosis was blocked most prominently with the pan-caspase inhibitor Z-VAD. Collectively, these data suggest that AZD6244-induced apoptosis is in part caspase-dependent regulated through the esctrinsic and moreso intrinsic pathway.
Figure 4.

AZD6244 induces caspase-dependent apoptosis. (A) All cell lines were treated with the indicated concentrations of AZD6244 for 18 hours. Protein levels were measured by immunoblotting using specific antibodies. (B) Caspase and PARP cleavage in PBMCs from a DLBCL patient. These results are representative of 3 total DLBCL patients. Cells were incubated with the indicated concentrations of AZD6244 for 6 and 16 hours. Cell lysates was subjected to Western blotting using caspase and PARP antibodies. (C) OCI-LY3 and (D) SUDHL6 cells were preincubated with 50μM caspase inhibitor for 2 hours followed by incubation with 200nM or 300nM for 48 hours. Apoptosis was determined by annexin V/PI staining followed by flow cytometric analysis.
AZD6244 modulates the expression of cell-cycle and apoptosis regulating proteins
We next analyzed the effect of AZD6244 on the expression of key regulators of cell-cycle progression and apoptosis by immunoblot analysis. Figure 5A shows a significant decrease in c-Myc, Bcl-2 and Mcl-1 after AZD6244 treatment in all DLBCL cell lines. Consistent with the observed G1 accumulation, protein expression of cyclin D1 and CDK1/2 was strikingly decreased in AZD6244-treated cells. Similar to all DLBCL cell lines, we noted down regulation of antiapoptotic proteins (Mcl-1 and Bcl-2) and c-MYC in primary DLBCL cells after AZD6244 exposure (Figure 5B). Conversely, the cyclin-dependent kinase inhibitor p27KIP1 accumulated in AZD6244 treated cells in a time-dependent fashion (Figure 5C). To determine whether AZD6244 could reduce c-Myc activity in DLBCL cells, we quantified the c-Myc transcriptional activation using AdM4, a Myc-reporter virus, after AZD6244 treatment. As shown in Figure 5D, Myc reporter activity was reduced 3- to 10-fold compared with control. Further, AZD6244 induced more prominent down-regulation of Myc in the germinal center cell line, SUDHL6, compared with the nongerminal center line, OCI-LY3.
Figure 5.

AZD6244 induces decrease in cell-cycle regulatory and antiapoptotic proteins. (A) Cells were incubated with 200nM and 300nM AZD6244 for 18 hours. Protein levels were measured in whole cell lysates by immunoblotting using indicated antibodies. (B) Down-regulation of MCL1, BCL2 and c-MYC in the whole cell lysate of PBMC afterAZD6244 exposure for 18 hours. (C) Induction of p27 by AZD6244 in all the cell lines afterincubation with 200nM AZD6244 for 18 hours. (D) SUDHL6 and OCI-LY3 cells were incubated with AdM4 Myc reporter or mutant Myc reporter adenovirus for 4 hours. After 24-hour transduction, cells were incubated with 200nM AZD6244 in SUDHL6 and 300nM in OCI-LY3 cells for 24 hours followed by luciferase assay.
MEK blockade modulates FoxO3a, PUMA, and BIM
To elucidate further the mechanisms of cell death, AZD6244-related apoptotic proteins were investigated by Western blot. Based in part on prior observations that FoxO3a may be down-regulated by ERK,29–31 we were interested in examining whether FoxO3a is a target for AZD6244-mediated cell-cycle arrest and apoptosis. Indeed, we found that AZD6244 enhanced down-regulation of FoxO3a phosphorylation in AZD6244-treated cancer cell lines (Figure 6A) resulting in stabilization of FoxO3a. Furthermore, time-dependent up-regulation of FoxO3a target proteins, p53-up-regulated modulator of apoptosis (PUMA), and Bcl-2–interacting mediator of cell death (BIM) were observed after AZD6244 treatment (Figure 6B). An increase in PUMA and BIM expression was also observed in primary DLBCL cells after AZD6244 exposure (Figure 6C).
Figure 6.

FOXO3a and BIM expression and function. (A) Reduction of FoxO3a activity. SUDHL4, SUDHL10, and OCI-LY3 cells were treated with indicated concentration of AZD6244 for 24 hours. Cell lysates were subjected to Western blotting using p-FoxO3a and FoxO3a antibodies. (B) Induction of proapoptotic proteins by AZD6244. SUDHL4 and OCI-LY-19 cells were treated with 200nM AZD6244 for indicated periods of time. PUMA and BIM proteins were detected by immunoblotting using specific antibodies. (C) Increase in proapoptotic proteins PUMA and BIM in PBMCs from DLBCL patients by Western blotting after AZD6244 treatment for 18 hours. Actin was used as a loading control for all blots. (D) OCI-LY3 and (E) SUDHL4 cells were transfected with BIM siRNA or scrambled siRNA, using Amaxa nucleofection kit, followed by incubation with 100nM or 200nM AZD6244 for 48 hours. Knockdown of BIM is shown by Western blot. Apoptosis was measured by annexin V/PI staining followed by flow cytometry. (F) OCI-LY3 cells were transduced with BIM shRNA using lentivirus system. Stably transduced cells that were subsequently transfected with Bcl-2 or Mcl-1 expressing plasmids. After 24-hour transfection, cells were treated with 200nM of AZD6244 for 48 hours and annexin V/PI staining and analyzed by flow cytometry.
Furthermore, to confirm the role of BIM in AZD6244-induced apoptosis, we knocked down BIM in SUDHL4 and OCI-LY3 cells using BIM siRNA. As shown in Figure 6D (OCI-LY3) and E (SUDHL4), BIM knockdown diminished the apoptotic effect of AZD6244. In addition, to assess the contribution of Mcl-1 and Bcl-2 in the cell death process, BIM shRNA DLBCL cells were transfected with Mcl-1 or Bcl-2 plasmids. Figure 6F shows that overexpression of Mcl-1 in BIM knockout cells enhanced cell survival, while Bcl-2 overexpression had no significant effect on survival compared with BIM knockdown alone.
Inhibition of AKT minimally sensitizes DLBCL cells to AZD6244
To determine the extent, if any, that AKT is involved in the regulation of MEK-induced cell death, we analyzed the effect of AZD6244 on AKT activation. Treatment of cells with AZD6244 showed reduction in AKT activation at 300nM (Figure 7A). To investigate further the potential of AKT-mediated resistance to AZD6244-induced cell death, OCI-LY3 were transfected with siRNA against AKT. Knockdown of AKT minimally sensitized cells to AZD6244-induced apoptosis (Figure 7B). Similar results were obtained by inhibiting the PI3K-AKT pathway using the chemical inhibitor, LY294002 (Figure 7C). In addition, we further confirmed AKT-mediated resistance by transfecting OCI-LY3 cells with constitutively active AKT construct followed by incubation with AZD6244. Cells transfected with constitutively active AKT showed a slight reduction in AZD6244-induced apoptosis (Figure 7D). These findings showed that AKT activation has minimal effect on anti-MEK AZD6244-induced apoptosis.
Figure 7.

Effect of AKT signaling. (A) Western blotting showing p-AKT and AKT expression. SUDHL4 and OCI-LY3 cells were treated with indicated concentrations of AZD6244 for 24 hours. Whole cell lysates were used to determine protein expression by Western blotting using specific antibodies against p-AKT and AKT. (B) AKT knockdown using AKT siRNA. OCI-LY3 cell were transfected with AKT siRNA or scrambled siRNA using Amaxa nucleofection kit. Knockdown of AKT is shown by Western blotting. After 24 hour transfection, cells were treated with AZD6244 for 48 hours. Apoptosis was measured by annexin V/PI by flow cytometry. (C) Chemical blockade of the PI3/AKT pathway. OCI-LY3 cells were pretreated with 20μM of LY294002 (LY) for 1 hour followed by incubation with 200nM AZD6244 for 48 hours. Apoptosis was measured by annexin V/PI staining followed by flow cytometry. (D) Constitutive activation of AKT. OCI-LY3 cells were transfected with either constitutively active AKT (Myr-Akt) or vector (pcDNA) alone. After selection in neomycin for 14 days, positively selected cells were treated with indicated concentration of AZD6244 for 48 hours. Apoptosis was measured by annexin V/PI staining followed by flow cytometry.
Discussion
Altogether, these data show that the 2nd generation MEK small molecule inhibitor, AZD6244, induced significant cell death at nanomolar (and clinically achievable) concentrations in multiple DLBCL cells lines, primary DLBCL cells, and in a preclinical human lymphoma xenograft model. This was associated with markedly decreased phosphorylation of ERK, the only known substrate downstream of MEK. Apoptosis was caspase-dependent and cell death was associated with cell-cycle dysregulation. Blockade of the MAP kinase MEK/ERK pathway resulted in stabilization of FoxO3a and hence down-regulation of p27, BIM, and PUMA. This was associated with cell-cycle arrest and induction of apoptosis. Moreover, BIM siRNA knockout blocked AZD6244-related apoptosis, while manipulation of ERK or AKT minimally affected cell death. In interpreting these findings, several factors should be considered.
The MAP kinase RAS/RAF/MEK/ERK signal transduction cascade plays a prominent role in the regulation of cell growth and proliferation.1,4,5,32,33 The MEK/ERK signaling pathway is stabilized or up-regulated in a large number of cancers, including lymphoma.3,34,35 ERK1 and ERK2, also known as p44/42 MAPK, are intimately involved in transducing signals from growth factor receptors and cytokine receptors after ligand binding.2 We and others have shown that ERK is constitutively activated in the majority of B-cell lymphomas.15,36,37 Furthermore, several studies from our group have shown that an oncogene directly downstream of MEK/ERK, MCT-1, is involved in cell proliferation, suppression of apoptosis, enhancement of cell survival signaling, and enhanced G1 cyclin/CDK kinase activity.8,9,38–40 Previous work from our group showed in a large-scale immunohistochemical (IHC) screen that MCT-1 protein was strongly expressed in 85% of DLBCL samples (weak and strong expression > 95%) compared with only 6% of follicular lymphoma cases.15 Moreover, we showed that genetic knockdown of MCT-1 resulted in apoptosis and tumor regression.15 There are currently no clinically available specific small inhibitor molecules that can directly modulate MCT-1 or ERK1/2 function, however several drug candidates, to target MEK to cripple the MEK/ERK pathway, have been developed. Small-molecule MEK inhibitors represent the most specific and effective strategy tested to date to suppress MAPK activity.
PD184352 (CI-1040) was the first MEK inhibitor studied in oncology trials, however drug-related toxicities have precluded further clinical development of this agent.11,12 Two 2nd generation oral MEK inhibitors, which are more potent and better tolerated, have been clinically developed (ie, PD0325901 and AZD6244).10,14,18–23 AZD6244 is a benzimidazole and selective 2nd generation MEK inhibitor with reported activity at nanomolar concentrations against purified MEK1 enzyme in preclinical solid tumor studies.18–23 Further, AZD6244 is a noncompetitive MEK inhibitor with preclinical antitumor activity in solid tumor models including hepatocellular, colon, myeloma, thyroid, pancreatic, melanoma, and breast cancers19,20,41 and tested clinically in phase 124 and phase 2 trials of advanced, refractory colorectal, melanoma, and lung cancer.25–27 AZD6244 has been examined in leukemia and myeloma models,42–44 however to our knowledge, has never been tested in lymphoma.
We showed that nanomolar concentrations of AZD6244 resulted in marked down-regulation of pERK in DLBCL cell lines (ie, germinal center and nongerminal center). MCT-1, a substrate downstream of ERK was variably affected by AZD6244. However, several other target substrates of MEK/ERK were effectively down-regulated including c-Myc, Mcl-1, and Bcl-2, including in primary DLBCL cells. The precise contribution of MCT-1 in AZD6244-induced cell death is an area of active investigation in our laboratory. It is noteworthy that AZD6244 had a profound effect on c-Myc transcriptional activity; further, c-Myc activity was preferentially decreased in germinal center DLBCL cells, however the effect was significant in nongerminal centers as well.
At the cellular and molecular level, the Bcl-2 family represents a critical checkpoint for the hierarchical regulation of apoptosis.45–47 Others have shown that cell death of acute myelogenous leukemia cells with the 1st generation MEK inhibitor PD184352 (CI-1040) in combination with other agents (eg, perifosine48) was dependent in part on the Bcl-2 family. Meng et al showed that BIM in particular was key to AZD6244-induced apoptosis in a lung cancer model.48 We found here that AZD6244 resulted in up-regulation of several proapoptotic mediators (ie, PUMA and BIM) and down-regulation of antiapoptotic proteins (ie, Mcl-1 and Bcl-2). Knockdown of BIM in germinal center (SUDHL4) and nongerminal center (OCI-LY3) DLBCL cells diminished the effect of AZD6244-induced apoptosis. Furthermore, Mcl-1 appears to be an important component for DLBCL cell survival as overexpression of Mcl-1 with BIM shRNA knockout decreased apoptosis, while Bcl-2 over expression did not.
An exciting finding in this study was that forced AKT activation through a constitutively active construct or inhibition of AKT (through genetic silencing or chemical inhibition) had no significant effect on AZD6244-induced apoptosis. However, other pathways we found to be affected by AZD6244 included FOXO3a, p27KIP1, and c-Myc. Continued examination of these and other signaling pathways will be important in further delineating the cell death mechanisms for this and other novel anti-MEK small molecule agents as well as to aid in the identification of potential rational combinations. In addition, clinical study of AZD6244 in DLBCL is warranted, while the overall role of the MEK/ERK signaling cascade in lymphomagenesis should continue to be investigated.
Supplementary Material
Acknowledgments
The authors thank the flow cytometry core facility at Northwestern University, as well as the RNAi core facility for providing lentivirus/constructs and DNA/RNA delivery cores for providing lentivirus for ERK2 shRNA.
This work was supported in part by a K23 CA109613 award from the National Cancer Institute (A.M.E.), a Merit Review award from the Department of Veterans Affairs (R.B.G.), and R01AA017972 (R.B.G.) from the National Institutes of Health.
Footnotes
Presented at the 51st Annual Meeting of the American Society of Hematology, New Orleans, LA, December 2009.49
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.B. designed and performed research, analyzed data, and wrote the paper; A.M.E. designed and performed research, analyzed data, and wrote the paper; B.D. performed research and analyzed data; S.P. performed research and analyzed data; L.I.G. designed research and analyzed data; and R.B.G. designed and performed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Andrew M. Evens, DO, MSc, Division of Hematology/Oncology, 55 Lake Ave, North Worcester, MA 01655; e-mail: andrew.evens@umassmed.edu; or Ronald B. Gartenhaus, MD, Division of Hematology/Oncology, 22 S Greene St, Baltimore, MD 21201; e-mail: rgartenhaus@som.umaryland.edu.
References
- 1.Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 2.Chang F, Steelman LS, Lee JT, et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17(7):1263–1293. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- 3.Dent P, Grant S. Pharmacologic interruption of the mitogen-activated extracellular-regulated kinase/mitogen-activated protein kinase signal transduction pathway: potential role in promoting cytotoxic drug action. Clin Cancer Res. 2001;7(4):775–783. [PubMed] [Google Scholar]
- 4.Lee JT, Jr., McCubrey JA. The Raf/MEK/ERK signal transduction cascade as a target for chemotherapeutic intervention in leukemia. Leukemia. 2002;16(4):486–507. doi: 10.1038/sj.leu.2402460. [DOI] [PubMed] [Google Scholar]
- 5.Shapiro P. Ras-MAP kinase signaling pathways and control of cell proliferation: relevance to cancer therapy. Crit Rev Clin Lab Sci. 2002;39(4–5):285–330. doi: 10.1080/10408360290795538. [DOI] [PubMed] [Google Scholar]
- 6.Seger R, Ahn NG, Posada J, et al. Purification and characterization of mitogen-activated protein kinase activator(s) from epidermal growth factor-stimulated A431 cells. J Biol Chem. 1992;267(20):14373–14381. [PubMed] [Google Scholar]
- 7.Hahn M, Li W, Yu C, Rahmani M, Dent P, Grant S. Rapamycin and UCN-01 synergistically induce apoptosis in human leukemia cells through a process that is regulated by the Raf-1/MEK/ERK, Akt, and JNK signal transduction pathways. Mol Cancer Ther. 2005;4(3):457–470. doi: 10.1158/1535-7163.MCT-04-0137. [DOI] [PubMed] [Google Scholar]
- 8.Shi B, Hsu HL, Evens AM, Gordon LI, Gartenhaus RB. Expression of the candidate MCT-1 oncogene in B- and T-cell lymphoid malignancies. Blood. 2003;102(1):297–302. doi: 10.1182/blood-2002-11-3486. [DOI] [PubMed] [Google Scholar]
- 9.Hsu HL, Shi B, Gartenhaus RB. The MCT-1 oncogene product impairs cell cycle checkpoint control and transforms human mammary epithelial cells. Oncogene. 2005;24(31):4956–4964. doi: 10.1038/sj.onc.1208680. [DOI] [PubMed] [Google Scholar]
- 10.Haura EB, Ricart AD, Larson TG, et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced nonsmall cell lung cancer. Clin Cancer Res. 2010;16(8):2450–2457. doi: 10.1158/1078-0432.CCR-09-1920. [DOI] [PubMed] [Google Scholar]
- 11.Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol. 2005;23(23):5281–5293. doi: 10.1200/JCO.2005.14.415. [DOI] [PubMed] [Google Scholar]
- 12.Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced nonsmall-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22(22):4456–4462. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 13.Wang D, Boerner SA, Winkler JD, LoRusso PM. Clinical experience of MEK inhibitors in cancer therapy. Biochim Biophys Acta. 2007;1773(8):1248–1255. doi: 10.1016/j.bbamcr.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 14.LoRusso PM, Krishnamurthi SS, Rinehart JJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16(6):1924–1937. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- 15.Dai B, Zhao XF, Hagner P, et al. Extracellular signal-regulated kinase positively regulates the oncogenic activity of MCT-1 in diffuse large B-cell lymphoma. Cancer Res. 2009;69(19):7835–7843. doi: 10.1158/0008-5472.CAN-09-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen TK, Jordan N, Friedberg J, Fisher RI, Dent P, Grant S. Inhibition of MEK/ERK1/2 sensitizes lymphoma cells to sorafenib-induced apoptosis. Leuk Res. 2010;34(3):379–386. doi: 10.1016/j.leukres.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nandi S, Reinert LS, Hachem A, et al. Phosphorylation of MCT-1 by p44/42 MAPK is required for its stabilization in response to DNA damage. Oncogene. 2007;26(16):2283–2289. doi: 10.1038/sj.onc.1210030. [DOI] [PubMed] [Google Scholar]
- 18.Davies BR, Logie A, McKay JS, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6(8):2209–2219. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 19.Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 20.Meng J, Peng H, Dai B, et al. High level of AKT activity is associated with resistance to MEK inhibitor AZD6244 (ARRY-142886). Cancer Biol Ther. 2009;8(21):2073–2080. doi: 10.4161/cbt.8.21.9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoon YK, Kim HP, Han SW, et al. KRAS mutant lung cancer cells are differentially responsive to MEK inhibitor due to AKT or STAT3 activation: implication for combinatorial approach. Mol Carcinog. 2010;49(4):353–362. doi: 10.1002/mc.20607. [DOI] [PubMed] [Google Scholar]
- 22.Gopal YN, Deng W, Woodman SE, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70(21):8736–8747. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tentler JJ, Nallapareddy S, Tan AC, et al. Identification of predictive markers of response to the MEK1/2 inhibitor selumetinib (AZD6244) in K-ras-mutated colorectal cancer. Mol Cancer Ther. 2010;9(12):3351–3362. doi: 10.1158/1535-7163.MCT-10-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerji U, Camidge DR, Verheul HM, et al. The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res. 2010;16(5):1613–1623. doi: 10.1158/1078-0432.CCR-09-2483. [DOI] [PubMed] [Google Scholar]
- 25.Bennouna J, Lang I, Valladares-Ayerbes M, et al. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens [published online ahead of print February 2, 2010]. Invest New Drugs. doi: 10.1007/s10637-010-9392-8. doi: 10.1007/s10637-010-9392-8. [DOI] [PubMed] [Google Scholar]
- 26.Hainsworth JD, Cebotaru CL, Kanarev V, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with nonsmall cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol. 2010;5(10):1630–1636. doi: 10.1097/JTO.0b013e3181e8b3a3. [DOI] [PubMed] [Google Scholar]
- 27.Board RE, Ellison G, Orr MC, et al. Detection of BRAF mutations in the tumour and serum of patients enrolled in the AZD6244 (ARRY-142886) advanced melanoma phase II study. Br J Cancer. 2009;101(10):1724–1730. doi: 10.1038/sj.bjc.6605371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mansour SJ, Candia JM, Gloor KK, Ahn NG. Constitutively active mitogen-activated protein kinase kinase 1 (MAPKK1) and MAPKK2 mediate similar transcriptional and morphological responses. Cell Growth Differ. 1996;7(2):243–250. [PubMed] [Google Scholar]
- 29.Yang JY, Zong CS, Xia W, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10(2):138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang W, Konopleva M, Burks JK, et al. Blockade of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase and murine double minute synergistically induces Apoptosis in acute myeloid leukemia via BH3-only proteins Puma and Bim. Cancer Res. 2010;70(6):2424–2434. doi: 10.1158/0008-5472.CAN-09-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy SK, Srivastava RK, Shankar S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J Mol Signal. 2010;5:10. doi: 10.1186/1750-2187-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71(3–4):479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 33.Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 34.Jazirehi AR, Vega MI, Chatterjee D, Goodglick L, Bonavida B. Inhibition of the Raf-MEK1/2-ERK1/2 signaling pathway, Bcl-xL down-regulation, and chemosensitization of nonHodgkin's lymphoma B cells by Rituximab. Cancer Res. 2004;64(19):7117–7126. doi: 10.1158/0008-5472.CAN-03-3500. [DOI] [PubMed] [Google Scholar]
- 35.Scholl FA, Dumesic PA, Khavari PA. Effects of active MEK1 expression in vivo. Cancer Lett. 2005;230(1):1–5. doi: 10.1016/j.canlet.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 36.Hollmann CA, Owens T, Nalbantoglu J, Hudson TJ, Sladek R. Constitutive activation of extracellular signal-regulated kinase predisposes diffuse large B-cell lymphoma cell lines to CD40-mediated cell death. Cancer Res. 2006;66(7):3550–3557. doi: 10.1158/0008-5472.CAN-05-2498. [DOI] [PubMed] [Google Scholar]
- 37.Ogasawara T, Yasuyama M, Kawauchi K. Constitutive activation of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase in B-cell lymphoproliferative disorders. Int J Hematol. 2003;77(4):364–370. doi: 10.1007/BF02982645. [DOI] [PubMed] [Google Scholar]
- 38.Dierov J, Prosniak M, Gallia G, Gartenhaus RB. Increased G1 cyclin/cdk activity in cells overexpressing the candidate oncogene, MCT-1. J Cell Biochem. 1999;74(4):544–550. [PubMed] [Google Scholar]
- 39.Prosniak M, Dierov J, Okami K, et al. A novel candidate oncogene, MCT-1, is involved in cell cycle progression. Cancer Res. 1998;58(19):4233–4237. [PubMed] [Google Scholar]
- 40.Reinert LS, Shi B, Nandi S, et al. MCT-1 protein interacts with the cap complex and modulates messenger RNA translational profiles. Cancer Res. 2006;66(18):8994–9001. doi: 10.1158/0008-5472.CAN-06-1999. [DOI] [PubMed] [Google Scholar]
- 41.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 42.Nishioka C, Ikezoe T, Yang J, Yokoyama A. Inhibition of MEK/ERK signaling induces apoptosis of acute myelogenous leukemia cells via inhibition of eukaryotic initiation factor 4E-binding protein 1 and down-regulation of Mcl-1. Apoptosis. 2010;15(7):795–804. doi: 10.1007/s10495-010-0483-y. [DOI] [PubMed] [Google Scholar]
- 43.Park MA, Zhang G, Mitchell C, et al. Mitogen-activated protein kinase kinase 1/2 inhibitors and 17-allylamino-17-demethoxygeldanamycin synergize to kill human gastrointestinal tumor cells in vitro via suppression of c-FLIP-s levels and activation of CD95. Mol Cancer Ther. 2008;7(9):2633–2648. doi: 10.1158/1535-7163.MCT-08-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tai YT, Fulciniti M, Hideshima T, et al. Targeting MEK induces myeloma-cell cytotoxicity and inhibits osteoclastogenesis. Blood. 2007;110(5):1656–1663. doi: 10.1182/blood-2007-03-081240. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Willis S, Day CL, Hinds MG, Huang DC. The Bcl-2-regulated apoptotic pathway. J Cell Sci. 2003;116(pt 20):4053–4056. doi: 10.1242/jcs.00754. [DOI] [PubMed] [Google Scholar]
- 46.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9(5):351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 47.Kim H, Rafiuddin-Shah M, Tu HC, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8(12):1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 48.Meng J, Fang B, Liao Y, Chresta CM, Smith PD, Roth JA. Apoptosis induction by MEK inhibition in human lung cancer cells is mediated by Bim. PLoS ONE. 2010;5(9):e13026. doi: 10.1371/journal.pone.0013026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhalla S, Gartenhaus R, Dai B, et al. The novel 2nd generation small molecule MEK inhibitor, AZD-6244, induces cell death in lymphoma cell lines, primary cells, and in a human lymphoma xenograft model [abstract]. Blood (ASH Annual Meeting Abstracts) 2009;114:285a. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}