

Abstract
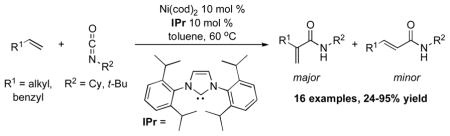
The nickel(0)-catalyzed coupling of α-olefins and isocyanates proceeds in the presence of the N-heterocyclic carbene ligand IPr to provide α,β-unsaturated amides. Carbon-carbon bond formation occurs preferentially at the 2-position of the olefin. The N-tert-butyl amide products can be converted to the corresponding primary amides under acidic conditions.
A major goal of organic chemistry is the elaboration of molecular complexity from simple, inexpensive precursors. Catalytic reactions that effect selective carbon-carbon bond formation facilitate this aim with economy of operations1 and materials.2
Methodology for the selective coupling of α-olefins and isocyanates to provide acrylamides has potential applications in the field of polymer science. Poly(N-alkylacrylamide)s and poly(N-alkylmethacrylamide)s have been extensively studied for their properties as temperature-sensitive aqueous microgels.3 The monomers are commonly prepared by reaction of (meth)acryloyl chloride with the corresponding amine. By comparison, direct synthesis of these unsaturated amides from alkenes and isocyanates would afford monomers with a greater variety of substitution patterns of the polymer backbone and avoid the formation of byproducts such as chloride salts.4
Several nickel(0)-mediated reactions of isocyanates have been described in the literature. Much of the seminal investigation in this area was done by Hoberg, who first reported the stoichiometric5a and catalytic5c coupling reactions of phenyl isocyanate and ethylene on nickel(0) with trialkyl phosphine ligands to give N-phenyl acrylamide. Hoberg proposed that the reaction proceeds via an azanickelacyclopentanone intermediate, which then undergoes β-hydrogen elimination (Scheme 1).
Scheme 1.

Hoberg’s Proposed Mechanism for the Nickel-Catalyzed Coupling of Ethylene and Phenyl Isocyanate.
Other α-olefins react with phenyl isocyanate with nickel(0) and phosphine ligands;5b,d–h the major product is the trans-disubstituted α,β-unsaturated amide. Formation of a 1,1-disubstituted acrylamide is observed only in two cases as a minor product, in 3% and 13% yield. In addition, isocyanates have been shown to react on nickel(0) with electronically activated alkenes,6 1,3-dienes,7 allenes,8 and aldehydes.9
Most recently, Louie has demonstrated that an NHC-nickel(0) system efficiently catalyzes the cycloaddition of diynes or alkynes and isocyanates to form pyridones.10a Cycloadditions run with an excess of isocyanate lead to the formation of pyrimidinediones.10b Free NHC ligands themselves are known to rapidly catalyze isocyanate trimerization to urethanes, as well as undergo reversible carboxylation in the presence of carbon dioxide.11
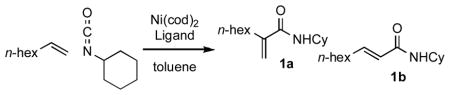
Recent work in our laboratory has shown that N-heterocyclic carbene (NHC) ligands can provide very high selectivity for reaction at the 2-position of an α-olefin in the nickel-catalyzed coupling of aldehydes, α-olefins, and silyl triflates.12 Herein we report the first example of a nickel(0)-catalyzed reaction of an alkene and an isocyanate in which carbon-carbon bond formation occurs selectively at the 2-position of the α-olefin, yielding N-alkylated acrylamides (Table 1).
Table 1.
Optimization of Nickel-Catalyzed Coupling of α-Olefin and Isocyanate.a
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | ligandb | Ni(cod)2 mol % | ligand mol % | time | temp | yieldc 1a (%) | yieldc 1b (%) |
| 1 | IPr | 10 | 10 | 4 d | rt | 61 | 24 |
| 2d | IPr | 10 | 10 | 20 h | 60 °C | 79 | 14 |
| 3d | IPr | 5 | 5 | 20 h | 60 °C | 54 | 15 |
| 4 | IAd | 10 | 10 | 20 h | 60 °C | 0 | 0 |
| 5 | ItBu | 10 | 10 | 20 h | 60 °C | 0 | 0 |
| 6 | PPh3 | 10 | 10 | 20 h | 60 °C | 0 | 0 |
| 7 | PCy2Ph | 10 | 10 | 20 h | 60 °C | 0 | 3 |
| 8 | P(n-Bu)3 | 10 | 10 | 20 h | 60 °C | 0 | 12 |
Reactions were run with 0.5 mmol 1-octene and 1.0 mmol cyclohexyl isocyanate in 2 mL toluene under Ar(g) in a sealed tube.
Abbreviations: IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-yildene; IAd = 1,3-bis(1-adamantyl)imidazol-2-ylidene; ItBu = 1,3-di-tert-butylimidazol-2-ylidene.
Isolated yields.
Reactions carried out with 0.5 mL toluene.
In the presence of the NHC ligand IPr, excellent conversions were obtained upon heating (Table 1, entry 2) or extended reaction time (entry 1) in the nickel-catalyzed coupling reaction. The NHC ligand gives products with the opposite sense of regioselectivity compared to those obtained when phosphine ligands are used (entries 7 and 8). The observed selectivity for carbon-carbon bond formation at the 2-position of the α-olefin may be attributed to a preference for the substituent on the olefin to be oriented away from the bulky NHC ligand (Figure 1).
Figure 1.

Model for Observed Regioselectivity in Nickel-Catalyzed Coupling of α-Olefin and Isocyanate.
A catalyst loading of 10 mol% with respect to the alkene is optimal for yield, although a slightly higher turnover number was noted with 5 mol% catalyst and ligand. Control experiments run in the absence of either Ni(cod)2 or IPr indicate that both species are necessary for catalysis.
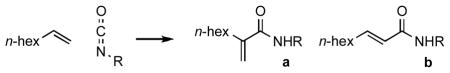
The reaction proceeds smoothly in a variety of solvents (Table 2). In the absence of solvent, the coupling of 1-octene and tert-butyl isocyanate occurs with good yield (entry 6); however, with cyclohexyl isocyanate a diminished yield is observed (entry 7), perhaps due to incomplete mixing associated with the precipitation of solid cyclohexyl amide products.
Table 2.
Solvent Screen for Nickel-Catalyzed Coupling of α-Olefin and Isocyanate.a
 | |||||
|---|---|---|---|---|---|
| entry | R | solvent | yield ab (%) | yield bb (%) | |
| 1 | t-Bu | toluene | 71 | 17 | |
| 2 | t-Bu | THF | 67 | 15 | |
| 3 | t-Bu | EtOAc | 69 | 15 | |
| 4 | t-Bu | benzene | 64 | 19 | |
| 5 | t-Bu | ether | 69 | 18 | |
| 6 | t-Bu | (neat) | 72 | 18 | |
| 7 | Cy | (neat) | 37 | 8 | |
Standard conditions (see Supporting Information): Reactions were run with 0.5 mmol 1-octene, 1.0 mmol tert-butyl isocyanate, 0.05 mmol Ni(cod)2, and 0.05 mmol IPr under Ar (g) in a sealed tube at 60 °C for 18–24 h.
Isolated yields.
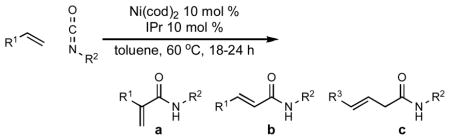
Table 3 illustrates the scope and selectivity of the reaction. Reactions of aliphatic olefins with branching at the allylic or homoallylic position proceed in high yields and with excellent selectivity for carboxamidation at the 2-position of the olefin. With allylbenzene, a third product is formed in which the double bond has moved into conjugation with the aromatic ring (entries 7 and 8). The catalytic reaction appears to be selective for monosubstituted olefins (entries 9 and 10). In other experiments, 1,1-disubstituted, cis-, and trans-olefins react poorly or not at all.13 Furthermore, esters and trialkylsilyl-protected alcohols are well tolerated under the reaction conditions. Hex-5-en-2-one reacts cleanly with tert-butyl isocyanate, but with cyclohexyl isocyanate the yield is substantially diminished (entries 13 and 14). Since the hexenone differs from other linear aliphatic olefins we have examined only at a position remote to the alkene, we hypothesize that the enhanced selectivity may be due to coordination of the carbonyl group to nickel at a selectivity-determining step in the catalytic cycle.14
Table 3.
Scope and Selectivity in Nickel-Catalyzed Coupling of α-Olefins and Isocyanates.a
 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | product(s) | yield a; b; c (%)b |
| 1 | n-hexyl | Cy | - | 1a, 1b | 79; 14; 0 |
| 2 | n-hexyl | t-Bu | - | 2a, 2b | 74; 17; 0 |
| 3c |
|
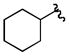
Cy | - | 3a | 72; 0; 0 |
| 4 |
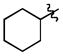
|
t-Bu | - | 4a | 91; 0; 0 |
| 5c |
|
Cy | - | 5a, 5b | 74; 5; 0 |
| 6 |
|
t-Bu | - | 6a, 6b | 71; 10; 0 |
| 7c | PhCH2 | Cy | Ph | 7a, 7b, 7c | 65; 8; 22 |
| 8c | PhCH2 | t-Bu | Ph | 8a, 8b, 8c | 83; 1d; 5d |
| 9c |
|
Cy | - | 9a | 86; 0; 0 |
| 10 |
|
t-Bu | - | 10a | 82; 0; 0 |
| 11 |
|
Cy | - | 11a, 11b | 74; 13; 0 |
| 12 |
|
t-Bu | - | 12a, 12b | 72; 12; 0 |
| 13 |
|
Cy | - | 13a | 24; 0; 0 |
| 14 |
|
t-Bu | - | 14a, 14b | 70; 2; 0 |
| 15 |
|
Cy | - | 15a, 15b | 68; 17; 0 |
| 16 |
|
t-Bu | - | 16a, 16b | 65; 9; 0 |
Standard conditions (see Supporting Information): Reactions were run with 0.5 mmol 1-octene, 1.0 mmol tert-butyl isocyanate, 0.05 mmol
Ni(cod)2, and 0.05 mmol IPr in 0.5 mL toluene under Ar(g) in a sealed
tube at 60 °C for 18–24 h.
Isolated yields.
Reaction was run using 2 mL toluene.
Isolated as a mixture of 8b and 8c, with relative ratios determined by 1H NMR.
The scope of the reaction with respect to the isocyanate appears to be limited to bulky, electron-rich alkyl isocyanates. In the reactions of ethyl and benzyl isocyanate with 1-octene, small amounts of coupling product can be isolated, but the major products obtained are isocyanate oligomers. Slow or portion-wise addition of isocyanate does little to circumvent this side reaction, and no improvement in yield is observed when Ni(cod)2 and IPr are allowed to equilibrate in solution for six hours prior to reaction, as done by Louie.10a Phenyl isocyanate, trichloromethyl isocyanate, and phenyl isocyanatoformate all gave no desired product under the reaction conditions.
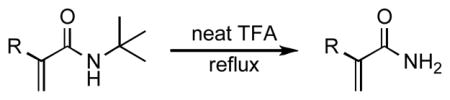
In recognition of the prevalence of primary amide motifs in the natural world, we investigated the possibility of forming such amides from our initial products. Deprotection of N-tert-butyl amides requires strongly acidic conditions, often coupled with heat;15 nevertheless, the reaction has been successfully utilized in the contexts of natural product synthesis16 and drug discovery.17 Still, it was unclear whether 1,1-disubstituted acrylamides would emerge unscathed from the forcing conditions necessary to effect deprotection. Gratifyingly, we were able to obtain the primary amides from several of the coupling products after heating in neat trifluoroacetic acid at reflux overnight (Table 4).
Table 4.
Deprotection of α,β-unsaturated amides.
 | |||
|---|---|---|---|
| entry | substrate | product | yield (%) |
| 1 | 2a |
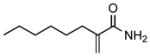 17 |
69 |
| 2 | 12a |
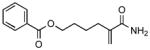 18 |
81 |
| 3 | 8a |
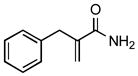 19 |
70 |
In summary, we have described a new preparation of acrylamides via the nickel(0)-IPr-catalyzed reaction of simple α-olefins and isocyanates. The NHC ligand gives products with the opposite sense of regioselectivity compared to those obtained using phosphine additives. Acid treatment converts the N-tert-butyl products to free amides. In addition to more conventional synthetic operations, compounds prepared by this method may find application as novel monomers for polymer synthesis.
Supplementary Material
Acknowledgments
This work was supported by the NIGMS (GM-063755). We are grateful to Ms. Li Li for obtaining mass spectrometric data for all compounds (MIT Department of Chemistry Instrumentation Facility, which is supported in part by the NSF (CHE-9809061 and DBI-9729592) and the NIH (1S10RR13886-01)).
Footnotes
Supporting Information Available. Experimental procedures and characterization data for all numbered compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Wender PA, Bi FC, Gamber GG, Gosselin F, Hubbard RD, Scanio MJC, Sun R, Williams TJ, Zhang L. Pure Appl Chem. 2002;74:25. [Google Scholar]; (b) Wender PA, Handy ST, Wright DL. Chem Ind (London) 1997:765. [Google Scholar]; (c) Wender PA, Miller BL. In: In Organic Synthesis: Theory and Applications. 2. Huldicky T, editor. JAI Press; Greenwich: 1993. p. 27. [Google Scholar]
- 2.Trost BM. Science. 1991;254:1471. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- 3.(a) Liang L, Feng X, Liu J, Rieke PC, Fryxell GE. Macromolecules. 1998;31:7845. [Google Scholar]; (b) Pelton R. Adv Colloid Interface Sci. 2000;85:1. doi: 10.1016/s0001-8686(99)00023-8. [DOI] [PubMed] [Google Scholar]; (c) Maeda Y, Nakamura T, Ikeda I. Macromolecules. 2001;34:1391. [Google Scholar]
- 4.Beak has also reported regioselective β′-lithiation and alkylation of α,β-unsaturated amides: Beak P, Kempf DJ, Wilson KD. J Am Chem Soc. 1985;107:4745.
- 5.(a) Hoberg H, Sümmermann K, Milchereit A. Angew Chem, Int Ed Engl. 1985;24:325. [Google Scholar]; (b) Hoberg H, Sümmermann K, Milchereit A. J Organomet Chem. 1985;288:237. [Google Scholar]; (c) Hoberg H, Hernandez E. J Chem Soc, Chem Commun. 1986:544. [Google Scholar]; (d) Hoberg H, Hernandez E. J Organomet Chem. 1986;311:307. [Google Scholar]; (e) Hernandez E, Hoberg H. J Organomet Chem. 1987;328:403. [Google Scholar]; (f) Hoberg H, Sümmermann K, Hernandez E, Ruppin C, Guhl D. [Google Scholar]; (g) Hoberg H. J Organomet Chem. 1988;358:507. [Google Scholar]; (h) Hoberg H, Guhl D. J Organomet Chem. 1990;384:C43. [Google Scholar]
- 6.(a) Hernandez E, Hoberg H. J Organomet Chem. 1986;315:245. [Google Scholar]; (b) Hoberg H, Hernandez E, Guhl D. J Organomet Chem. 1988;339:213. [Google Scholar]; (c) Hoberg H, Guhl D. J Organomet Chem. 1989;375:245. [Google Scholar]; (d) Hoberg H, Nohlen M. J Organomet Chem. 1990;382:C6. [Google Scholar]; (e) Hoberg H, Guhl D, Betz P. J Organomet Chem. 1990;387:233. [Google Scholar]; Hoberg H, Nohlen M. J Organomet Chem. 1991;412:225. [Google Scholar]
- 7.(a) Hoberg H, Hernanadez E. Angew Chem, Int Ed Engl. 1985;24:961. [Google Scholar]; (b) Hernandez E, Hoberg H. J Organomet Chem. 1987;327:429. [Google Scholar]; (c) Hoberg H, Bärhausen D. J Organomet Chem. 1990;397:C20. [Google Scholar]
- 8.Hoberg H, Hernandez E, Sümmermann K. J Organomet Chem. 1985;295:C21. [Google Scholar]
- 9.Hoberg H, Sümmermann K. J Organomet Chem. 1984;264:379. [Google Scholar]
- 10.(a) Duong HA, Cross MJ, Louie J. J Am Chem Soc. 2004;126:11438. doi: 10.1021/ja046477i. [DOI] [PubMed] [Google Scholar]; (b) Duong HA, Louie J. Tetrahedron. 2006;62:7552. [Google Scholar]
- 11.(a) Duong HA, Tekavec TN, Arif AM, Louie J. Chem Commun. 2004;1:112. doi: 10.1039/b311350g. [DOI] [PubMed] [Google Scholar]; (b) Duong HA, Cross MJ, Louie J. Org Lett. 2004;6:4679. doi: 10.1021/ol048211m. [DOI] [PubMed] [Google Scholar]
- 12.Ho CY, Jamison TF. Angew Chem Int Ed. 2007;46:782. doi: 10.1002/anie.200603907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.When reacted with cyclohexyl isocyanate, 10 mol% Ni(cod)2, and 10 mol% IPr in toluene in a sealed tube at 60 °C for 18–24 hours, methylenecyclohexane and cyclohexene afforded coupling products in less than 10% yield. No product was observed with trans-4-octene under identical conditions.
- 14.For related phenomena in coupling reactions of 1,6-enynes, see: Miller KM, Jamison TF. J Am Chem Soc. 2004;126:15342. doi: 10.1021/ja0446799.Moslin RM, Jamison TF. Org Lett. 2006;8:455. doi: 10.1021/ol052719n.Moslin RM, Miller KM, Jamison TF. Tetrahedron. 2006;62:7598.
- 15.Lacey RN. J Chem Soc. 1960:1633. [Google Scholar]
- 16.Conover LH, Butler K, Johnston JD, Korst JJ, Woodward RB. J Am Chem Soc. 1962;84:3222. [Google Scholar]
- 17.Huang W, Zhang P, Zuckett JF, Wang L, Woolfrey J, Song Y, Jia ZJ, Clizbe LA, Su T, Tran K, Huang B, Wong P, Sinha U, Park G, Reed A, Malinowski J, Hollenbach SJ, Scarborough RM, Zhu BY. Bioorg Med Chem Lett. 2003;13:561. doi: 10.1016/s0960-894x(02)00927-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.