

Abstract
Described are several classes of unusual or unprecedented carbonyl-ene-type reactions, including those between alpha olefins and aromatic aldehydes. Catalyzed by nickel, these processes complement existing Lewis acid-catalyzed methods in several respects. Not only are monosubstituted alkenes, aromatic aldehydes, and tert-alkyl aldehydes effective substrates, but monosubstituted olefins also react faster than those that are more substituted, and large or electron-rich aldehydes are more effective than small or electron-poor ones. Conceptually, in the presence of a nickel-phosphine catalyst, the combination of off-the-shelf alkenes, silyl triflates, and triethylamine functions as a replacement for an allylmetal reagent.
Carbonyl addition reactions are among the most utilized carbon-carbon bond-forming transformations. In many of these the nucleophile is an organometallic reagent, whereas an alkene serves in this capacity in the carbonyl-ene reaction.i Although alkenes are among the most readily available classes of organic molecules, the full potential of this advantage has yet to be realized in the context of this transformation. Despite decades of research, the chief limitation of this otherwise versatile process is still one of scope. The most efficient reactants are electron-rich olefins (e.g., 1,1-disubstituted alkenes or 2-methoxypropene) and small and/or highly electron-deficient aldehydes (e.g., chloral, formaldehyde, or glyoxylate esters). Few carbonyl-ene reactions of aromaticii, iii or sterically demanding aldehydesiv have been reported. Equally rare are those of monosubstituted alkenes, and the vast majority of these are with electron-deficient aldehydes.v In short, current carbonyl-ene technology is effective for only a small subset of the plethora of possible coupling partners.
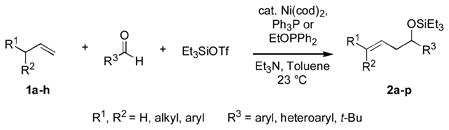 |
(1) |
Herein we describe a general means for catalyzing carbonyl-ene-type reactions (eq 1) of several types of compounds that heretofore were of very limited or nonexistent utility, including the most readily available alkenes (alpha olefinsvi) and several important families of aldehydes (aromatic, heteroaromatic, and tertiary aliphatic aldehydes). Catalyzed by a nickel-phosphine complex,vii these not only are the first intermolecular carbonyl-ene reactions between alpha olefins and aromatic aldehydes,iii but also the first between these alkenes and tert-alkyl aldehydesiv (t-BuCHO). These are also the first catalytic carbonyl-ene reactions in which a monosubstituted alkene reacts preferentially over a more substituted double bond,viii and the first in which electron-rich aldehydes are more efficient than those bearing electron-withdrawing substituents.
We recently reported that allylic alcohol derivatives can be prepared directly from alpha olefins, aldehydes, silyl triflates, and an amine base under nickel catalysis, and that homoallylic byproducts are formed in some cases.ix, x We have since found that certain organophosphorus additives (Ph3P or EtOPPh2) invert the selectivity, providing an efficient entry into synthetically valuable homoallylic alcohols that previously were unavailable by way of carbonyl-ene processes.
Under these conditions propene (1a) itself undergoes a nickel-catalyzed, carbonyl-ene reaction (Table 1, entry 1), yielding the triethylsilyl ether of allyl phenyl carbinol (2a). The minor product in this case is an allylic alcohol derivative (3a, not shownxi), but when the alpha olefin 1-octene (1b) is used, the analogous allylic byproducts are formed in only trace amounts (entries 2–8). The E configuration of the double bond is favored over the Z by a factor of 3 to 5 in all cases examined in this series.
Table 1.
Nickel-Catalyzed, Carbonyl-Ene-Type Reactions of Monosubstituted Alkenes.a
| entry | alkene (1) | aldehyde | major product (2) | yield (%)(2:3) b, c | E:Z (2) b |
|---|---|---|---|---|---|
| 1 d |
1a |
PhCHO |
2a
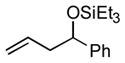 |
73 (89:11) | n.a. |
| 2 |
1b |
PhCHO |
2b
|
85 (95:5) | 75:25 |
| 3 e | 72 (>95:5) | 75:25 | |||
| 4 e | p-anisaldehyde |
2c
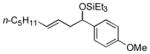 |
85 (>95:5) | 75:25 | |
| 5 e | p-Cl(C6H4)CHO |
2d
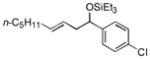 |
37 (>95:5) | 74:26 | |
| 6 f | 2-naphthaldehyde |
2e
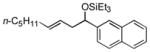 |
88 (>95:5) | 70:30 | |
| 7 | 1-methyl-2-indole-carboxaldehyde |
2f
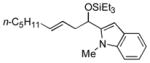 |
56 (>95:5) |
83:17 | |
| 8 f | t-BuCHO |
2g
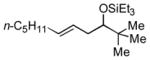 |
64 (>95:5) | 78:22 | |
| 9 |
1c |
PhCHO |
2h
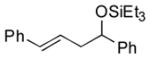 |
86 (92:8) | >95:5 |
| 10 | p-anisaldehyde |
2i
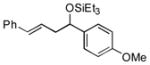 |
99 (92:8) | >95:5 | |
| 11 f, g | 98 (92:8) | >95:5 | |||
| 12 f | 2-naphthaldehyde |
2j
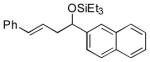 |
88 (95:5) |
>95:5 | |
| 13 | 1-methyl-2-indole-carboxaldehyde |
2k
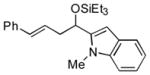 |
57 (>95:5) | >95:5 | |
| 14 f |
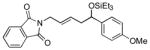
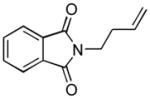 1dh |
p-anisaldehyde |
2l
 |
76 (>95:5) | 83:17 |
| 15 |
 1e |
PhCHO |
2m
 |
82 (>95:5) | 81:19 |
| 16 |
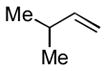 1f |
2n
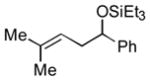 |
95 (86:14) | n.a. | |
| 17 |
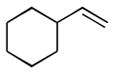 1g |
2o
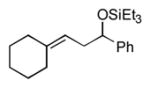 |
99 (75:25) | n.a. |
See Supporting Information and eq 1. Standard conditions (entries 1–7, 15–17): To a solution of Ni(cod)2 (0.1 mmol) and EtOPPh2 (0.2 mmol) in toluene (2.5 mL) at 23 °C under Ar were added the alkene (0.5 mL), triethylamine (3.0 mmol), the aldehyde (0.5 mmol), and triethylsilyltriflate (0.875 mmol). The mixture was stirred 48 h at room temperature and purified by chromatography (SiO2). Entries 8–14: Ph3P was used in place of EtOPPh2.
Determined by 1H NMR.
See Supporting Information for structures of allylic products (3a-3o).
Propene (1a, 1 atm) was used in place of Ar.
Reaction time 18 h.
Reaction temperature 35 °C.
Fivefold larger reaction scale (see text).
3 equiv of 1d was employed.
The analogous reactions of allylbenzene (1c) are highly selective with respect to both product distribution and olefin geometry (entries 9–13). Identical results (nearly quantitative yield) are obtained when the reaction is performed on a fivefold larger scale and with only 1.5 equiv of allylbenzene relative to p-anisaldehyde (entries 10 and 11). Imide carbonyl groups are tolerated in the reaction (entry 14), as are those bearing β- or α-branching (entries 15 and 16–17, respectively).
Several observations concerning several of the aldehydes deserve further comment. Heteroaromatic aldehydes such as 1-methyl-2-indolecarboxaldehyde are tolerated (entries 7 and 13), despite the fact that the silyl triflate used in the reaction is highly electrophilic. Noteworthy also is the fact that pivaldehyde (t-BuCHO) may be employed in this transformation (entry 8).xii Silyl ethers of homoallylic alcohols derived from these very sterically demanding aldehydes may thus be accessed directly from the alkene, without preparation of an allylsilane reagent.xiii Moreover, we are aware of no other examples of intermolecular carbonyl-ene reactions involving a tertiary aliphatic aldehyde.iv
While reactions of benzaldehyde require 48 h at room temperature to reach completion (compare entries 2 (48 h) and 3 (18 h)), those involving p-anisaldehyde can be complete within 18 h (entry 4) and are generally higher yielding (compare entries 3 and 4 and entries 9 and 10). Furthermore, aromatic aldehydes bearing electron-withdrawing substituents are much less efficient (entry 5).xiv While we have yet to conduct an exhaustive Hammett analysis, all evidence thus far points to the likelihood that there is a strong dependence of reaction rate upon the electronic nature of the aldehyde. Whatever the cause, we are unaware of other cases of carbonyl-ene reactions in which electron-rich aldehydes are more efficient than electron-poor.
In a similar vein, we have observed that substitution on the alkene has a profound impact on the efficiency of the transformation. Whereas 1,1-disubstituted alkenes are among the most effective olefins in Lewis acid-catalyzed carbonyl-ene reactions, they do not undergo coupling to any noticeable degree with the nickel-catalyzed system. Trans- and cis-disubstituted alkenes are similarly unreactive.xv
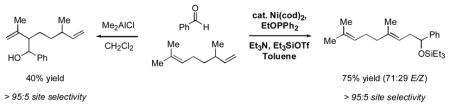
A profound demonstration of this complementary selectivity is illustrated in Scheme 1. When citronellene (1h) and benzaldehyde are treated with Me2AlCl, only the trisubstituted alkene reacts, and no detectable amount of reaction of the terminal olefin is observed. On the other hand, under nickel-catalyzed conditions, this selectivity is completely reversed. Products corresponding to reaction of the terminal alkene (2p) are the only ones detectable. To the best of our knowledge, this is the first example of a catalytic carbonyl-ene-like reaction that is faster for a monosubstituted alkene than for one more highly substituted.viii
Scheme 1.

In summary, the nickel-catalyzed carbonyl-ene reactions described here complement Lewis acid-catalyzed methods in several respects (Figure 1). In particular, alpha olefins, aromatic aldehydes, and tert-alkyl aldehydes are excellent starting materials, whereas previously they had not been utilized at all or only to a limited extent. That is, using only off-the-shelf reagents and catalysts, this process effects several classes of unprecedented carbonyl-ene reactions and expands the scope of this venerable transformation significantly. Currently we are investigating the mechanistic basis of the unusual selectivity and reactivity patterns, as well as further demonstration of the general concept of simple, unactivated alkenes functioning as nucleophiles in carbon-carbon bond-forming reactions.ix
Figure 1.

Complementarity of Catalytic Carbonyl-Ene Reactions
Supplementary Material
Acknowledgments
Support for this work was provided by the National Institute of General Medical Sciences (GM-063755). C. Y. H. thanks The Croucher Foundation for a postdoctoral fellowship. We are grateful to Dr. Li Li for obtaining mass spectrometric data for all compounds (MIT Department of Chemistry Instrumentation Facility, which is supported in part by the NSF (CHE-9809061 and DBI-9729592) and the NIH (1S10RR13886-01)).
Footnotes
Supporting Information Available. Experimental procedures and data for all new compounds (PDF). This information is available free of charge via the Internet at http://pubs.acs.org
References
- i.Reviews: Hoffmann HMR. Angew Chem, Int Ed. 1969;8:556–577.Snider BB. Acc Chem Res. 1980;13:426–432.Snider B. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 2. Pergamon Press; Oxford: 1991. pp. 527–561.Mikami K, Shimizu M. Chem Rev. 1992;92:1021–1050.Dias LC. Curr Org Chem. 2000;4:305–342.
- ii.(a) Snider BB, Rodini DJ. Tetrahedron Lett. 1980;21:1815–1818. [Google Scholar]; (b) Snider BB, Rodini DJ, Kirk TC, Cordova R. J Am Chem Soc. 1982;104:555–563. [Google Scholar]; (c) Majewski M, Bantle GW. Synth Comm. 1990;20:2549–2558. [Google Scholar]; (d) Houston TA, Tanaka Y, Koreeda M. J Org Chem. 1993;58:4287–4292. [Google Scholar]; (e) Aggarwal VK, Vennall GP, Davey PN, Newman C. Tetrahedron Lett. 1998;39:1997–2000. [Google Scholar]; (f) Ellis WW, Odenkirk W, Bosnich B. Chem Commun. 1998:1311–1312. [Google Scholar]; (g) Loh TP, Feng LC, Yang JY. Synthesis. 2002;7:937–940. [Google Scholar]
- iii.One isolated example of a carbonyl-ene reaction of an aromatic aldehyde and a monosubstituted alkene has been described (yield not reported): Epifani E, Florio S, Ingrosso G. Tetrahedron. 1988;44:5869–5877.
- iv.For intramolecular examples, see: Andersen NH, Hadley SW, Kelly JD, Bacon ER. J Org Chem. 1985;50:4144–4151.Fujita M, Shindo M, Shishido K. Tetrahedron Lett. 2005;46:1269–1271.
- v.Pioneering examples with aliphatic aldehydes: Snider BB, Phillips GB. J Org Chem. 1983;48:464–469.
- vi.Lappin GR, Sauer JD, editors. Alpha Olefins Applications Handbook. M. Dekker; New York: 1989. [Google Scholar]
- vii.Ni(II)-catalyzed carbonyl-ene reactions of 1,1-disubstituted alkenes and glyoxylate esters: Mikami K, Aikawa K. Org Lett. 2002;4:99–101. doi: 10.1021/ol016968x.
- viii.This selectivity has been observed in one thermal carbonyl-ene reaction (6-methyl-1,5-heptadiene and the highly electron-deficient diethyl oxomalonate, 180 °C, 24 h): Salomon MF, Pardo SN, Salomon RG. J Am Chem Soc. 1980;102:2473–2475.Salomon MF, Pardo SN, Salomon RG. J Am Chem Soc. 1984;106:3797–3802.
- ix.Ng SS, Jamison TF. J Am Chem Soc. 2005;127:14194–14195. doi: 10.1021/ja055363j. [DOI] [PubMed] [Google Scholar]
- x.See also: Ogoshi S, Oka M-a, Kurosawa H. J Am Chem Soc. 2004;126:11802–11803. doi: 10.1021/ja0460716.Ogoshi S, Ueta M, Arai T, Kurosawa H. J Am Chem Soc. 2005;127:12810–12811. doi: 10.1021/ja0542486.
- xi.See Supporting Information for details.
- xii.Aldehydes bearing one or more α-hydrogens, e.g., isobutyraldehyde, undergo competitive enolsilane formation.
- xiii.Review of the Sakurai reaction: Fleming I, Dunogues J, Smithers R. Org React. 1989;37:57–575.
- xiv.Reactions of p-nitrobenzaldehyde afforded no trace of coupling product.
- xv.Coupling products were not detected when methallylbenzene, cis-4-octene, and trans-4-octene were employed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.