

Abstract
A full account of two recently developed nickel-catalyzed coupling reactions of alkenes, aldehydes and silyl triflates is presented. These reactions provide either allylic alcohol or homoallylic alcohol derivatives selectively, depending on the ligand employed. These processes are believed to be mechanistically distinct from Lewis acid-catalyzed carbonyl-ene reactions, and several lines of evidence supporting this hypothesis are discussed.
Introduction
Alkenes are one of the most versatile, utilized, and readily available classes of functional groups. Simple alpha olefins are produced in megaton scale each year industrially, highlighting the importance of these organic feedstocks.1 Several indispensable transformations utilize olefins, such as Ziegler-Natta polymerization,2 the Heck reaction,3a–3e Wacker oxidation,3a hydroformylation,3a hydrometallation,3a alkene cross-metathesis,4 epoxidation5 and dihydroxylation.5
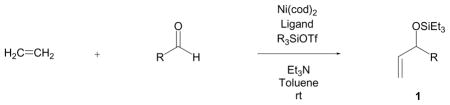
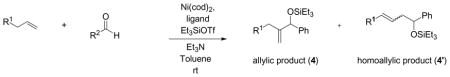
The nickel-catalyzed coupling of alkenes, aldehydes, and silyl triflates that we recently developed is the first example of a transition metal-catalyzed coupling of simple, unactivated olefins and aldehydes that provides allylic alcohol derivatives.6 With careful choice of the supporting ligand on nickel, this coupling reaction can also selectively provide homoallylic alcohol derivatives that are generally not accessible using Lewis acid-catalyzed carbonyl-ene reactions (eq 1).7
| (1) |
Transition metal-catalyzed intermolecular reductive and alkylative coupling reactions have emerged as useful methods for the preparation of alcohol and amine derivatives. Nickel, palladium, rhodium and ruthenium catalysts have been found to be particularly effective in the intermolecular coupling of alkynes, 1,3-enynes, 1,3-dienes, allenes, enoate esters, enones, and enals with aldehydes, ketones, epoxides, glyoxylate esters, and imines.8–11 A variety of reducing agents have been used in these reductive couplings, such as triethylborane, organozinc reagents, organosilanes, and molecular hydrogen. As yet, however, simple, unactivated alkenes such as ethylene and 1-octene have not been reported to undergo analogous catalytic reductive coupling reactions.
As a part of our program directed toward developing C–C bond forming reactions of “off-the-shelf”, simple starting materials, we became very interested in catalytic alkene–aldehyde coupling processes. Intramolecular versions of this transformation have been reported, such as transition metal-catalyzed cyclizations of enals and enones. For example, the titanium-catalyzed intramolecular reductive cyclization of enals and enones was first reported by Buchwald and Crowe.12a,12b Recently Ogoshi has demonstrated a nickel-catalyzed cyclization of enones.12c α,ω-Enals also undergo cyclization by way of a radical process13 and also in a Lewis acid-catalyzed carbonyl-ene reaction.7
Intermolecular coupling of unactivated alkenes and aldehydes is commonly mediated by a transition metal (stoichiometric) or accomplished by way of a carbonyl-ene reaction.7,15 An interesting process developed by Worpel combines an alkene and an aldehyde through a silver-catalyzed silylene transfer reaction.16
Formation of an oxametallacycle through the coupling of simple alkenes and ketones has been observed with several transition metals such as titanium, zirconium and rhodium.14 These studies suggested that transition metal-catalyzed, intermolecular coupling of alkenes and aldehydes would be feasible under the appropriate conditions. Ogoshi recently observed that Lewis acids such as a silyl triflate and trimethylaluminum facilitated the formation of an oxanickellacycle through cyclization of α,ω-enals and α,ω-enones.17 We proposed that if the intermolecular coupling of an alkene and an aldehyde occurred, the nickel alkyl bond could undergo a β-hydride elimination, followed by the removal of triflic acid from nickel to regenerate the nickel catalyst. This mechanistic framework also resembles that in the Heck reaction, a very important example of a catalytic coupling of an alkene and an electrophile. 3a–3e
The carbonyl-ene reaction has historically been the most direct method to combine simple alkenes and carbonyl compounds to provide homoallylic alcohol products. Recent efforts in this area have focused on asymmetric induction through the use of Lewis acids and chiral ligands, such as (bisoxazoline)CuX2, (pybox)ScX3, and (BINAP)TiX2 complexes.18a–18f Typically the alkenes that are employed in intermolecular carbonyl-ene reactions are 1,1-disubstituted and trisubstituted olefins. With respect to the carbonyl component, electron-deficient enophiles such as glyoxylates, glyoxamides, and chloral are generally more efficient than simple aromatic and aliphatic aldehydes. In fact, since the report of the carbonyl-ene reaction in 19437a there have been only a few scattered examples of intermolecular ene reactions between monosubstituted alkenes and simple aromatic and aliphatic aldehydes.19
To summarize, Lewis acid-catalyzed carbonyl-ene reactions are typically not feasible for the most readily available alkene and aldehyde building blocks. One of the nickel-catalyzed coupling of alkenes and aldehydes described herein is thus complementary in scope to the carbonyl-ene reaction; monosubstituted alkenes couple with simple aldehydes to provide a carbonyl-ene-type product in high yield.
Results
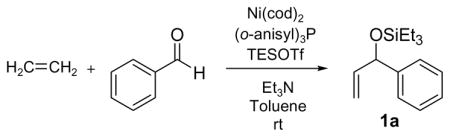
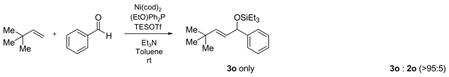
Our investigations commenced with the simplest olefin, ethylene, and benzaldehyde. After a brief examination of phosphorous-based additives, we found that a combination of Ni(cod)2, tris-(o-methoxyphenyl)-phosphine ((o-anisyl)3P), triethylamine, and triethylsilyl triflate (Et3SiOTf) promoted the coupling of ethylene with a variety of aldehydes. In all cases a triethylsilylether of an allylic alcohol is obtained in good to excellent yield (Table 1), providing ready access to a class of allylic alcohol derivatives that have been used in cross metathesis reactions, for example.4
Table 1.
Nickel-Catalyzed Coupling of Ethylene, Aldehydes, and Silyl Triflates
 | ||||
|---|---|---|---|---|
| entry | R (aldehyde) | R3SiOTf | product | isolated yield (%) |
| 1 | Ph | Et3SiOTf |
1a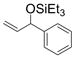
|
82 (65) b |
| 2 | p-tolyl | Et3SiOTf |
1b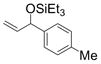
|
88 (65) b |
| 3 | o-tolyl | Et3SiOTf |
1c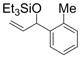
|
93 (64) b |
| 4 | p-anisyl | Et3SiOTf |
1d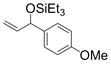
|
95 (65) c |
| 5 | 2-naphthyl | Et3SiOTf |
1e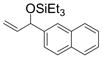
|
95 (83) b |
| 6 | 2-naphthyl | Me3SiOTf |
1f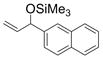
|
60 |
| 7 | 2-naphthyl | t-BuMe2SiOTf |
1g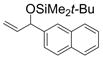
|
67 |
| 8 |
|
Et3SiOTf |
1h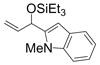
|
80 |
| 9 | 2-furyl | Et3SiOTf |
1i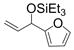
|
38 |
| 10 f |
|
Et3SiOTf |
1j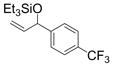
|
25 |
| 11 f |
|
Et3SiOTf |
1k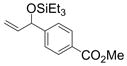
|
34 |
| 12 | piv | Et3SiOTf |
1l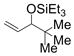
|
70 |
| 13 |
|
Et3SiOTf |
1m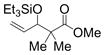
|
81 (40) c, d |
| 14 | cyclohexyl | Et3SiOTf |
1n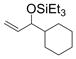
|
25 d (34) d, e |
Standard procedure: Ni(cod)2 (20 mol%) and (o-anisyl)3P (40 mol%) were dissolved in 2.5 mL toluene under argon. Ethylene (balloon, 1atm) was substituted for argon. Triethylamine (600 mol%), the aldehyde (100 mol%, 0.5 mmol), and Et3SiOTf (175 mol%) were added. The reaction mixture was stirred 6–18 h at 23 °C.
(o-anisyl)3P was replaced by Cy2PhP.
(o-anisyl)3P was replaced by Ph3P.
Yields determined by 1H NMR using DMF as a standard.
Conducted under 2 atm of ethylene.
Stirred at room temperature for 30 h.
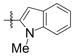
Under 1 atm of ethylene, simple aromatic aldehydes such as benzaldehyde and p-tolylaldehyde undergo efficient coupling (entries 1–2). Ortho substitution on the aromatic aldehyde does not appear to deter the coupling process (entry 3), and notably, acid-sensitive heteroaromatic aldehydes such as 1-methyl-2-indolecarboxaldehyde (entry 8) and 2-furaldehyde (entry 9) are tolerated, even in the presence of Lewis acidic silyl triflates. As an additional advantage, other common silyl triflates can be used in the coupling reaction, providing orthogonal protection of the hydroxyl group when necessary (entries 5–7).
Remarkably, sterically demanding tertiary aliphatic aldehydes such as pivaldehyde and 2,2-dimethyl-3-oxo-propionic acid methyl ester couple with ethylene with the same efficacy as benzaldehyde (entries 12 and 13). Enolizable aldehydes are not appropriate substrates in this system, however, since they react rapidly with the silyl triflate and triethylamine to form alkenyl silyl ethers. The coupling of ethylene with cyclohexanecarboxaldehyde is fast enough, however, that a significant amount of coupling product is observed and can be isolated (entry 14).
Tris-(o-methoxyphenyl)-phosphine is the ligand of choice for the ethylene–aldehyde coupling. Other phosphines such as dicyclohexylphenylphosphine and triphenylphosphine provide lower yield under the same reaction conditions (entries 1–5, 13).
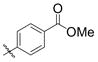
An interesting electronic effect is observed in these coupling reactions. Electron-rich aromatic aldehydes are more efficient substrates than electron-poor aromatic aldehydes. Among the four para-substituted aromatic aldehydes examined, electron-donating para-substituents (–Me and –OMe) improve the yield of the coupling reaction (entries 2 and 4). Electron-withdrawing para-substituents (–CF3 and –CO2Me) suffer from incomplete conversion, even after prolonged reaction time (entries 10–11). In such cases, products resulting from a pinnacol coupling are observed but are not observed in any other example.
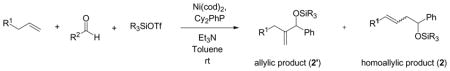
The encouraging results in these ethylene–aldehyde coupling reactions prompted us to examine the scope of the alkenes in detail. Unlike ethylene, 1-octene can afford more than one possible coupling product depending on where the new carbon–carbon bond is formed. The examination of a series of ligands revealed several interesting observations regarding ligand-dependent regioselectivity.
Ligand Effect
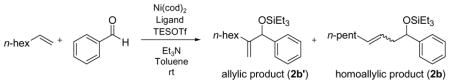
Under similar reaction conditions as the ethylene–aldehyde cases, 1-octene and benzaldehyde undergo coupling in the presence of Ni(cod)2, a ligand, triethylamine, and Et3SiOTf. Two distinct types of coupling products are typically observed, namely a 1,2-disubstituted allylic alcohol product (A) and a homoallylic alcohol product (H). Different classes of phosphine ligands favor one or the other coupling products, as summarized in Tables 2 and 3 and Figure 1.
Table 2.
Ligand-Dependent Regioselectivity — Electron-Rich Phosphines a
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | ligand | cone angle b | νCOb | yield (2b′) c | yield (2b) c | ratio (2b′:2b) d | combined yield (2b′+2b) |
| 1 | (n-Bu)3P | 132 | 1915 | 11% | 27% | 29:71 | 38% |
| 2 | (n-Oct)3P | 107 | 7% | 14% | 23:77 | 21% | |
| 3 | (i-Pr)3P | 160 | 1915 | 11% | 2% | 85:15 | 13% |
| 4 | Cyp3P | 10% | 3% | 78:22 | 13% | ||
| 5 | Cy3P | 170 | 1915 | 13% | 3% | 81:19 | 16% |
| 6 | (t-Bu)3P | 182 | 7% | 2% | 78:22 | 9% | |
|
| |||||||
| 7 | Cy2PhP | 162 | 1917 | 37% | 16% | 70:30 | 53% |
| 8 e | Cy2(o-tol)P | 181 | 30% | 5% | 86:14 | 35% | |
| 9 | Cy2(o-Ph-Ph)P | 32% | 22% | 60:40 | 54% | ||
| 10 f | Cy2FcP | 14% | 2% | 88:12 | 16% | ||
Standard procedure: Ni(cod)2 (20 mol%) and a ligand (40 mol%) were dissolved in 1.5 mL toluene. Alkene (500 mol%), triethylamine (600 mol%), the aldehyde (100 mol%, 0.25 mmol) and Et3SiOTf (175 mol%) were added. The reaction mixture was stirred 18 h at 23 °C.
See ref 20.
Yields determined by 1H NMR using DMF as a standard.
Ratio was determined by 1H NMR of the crude reaction mixture.
48 h reaction time.
1250 mol% alkene was used.
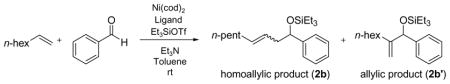
Table 3.
Ligand Dependent Regioselectivity — Electron-Poor Phosphines a
 | ||||||
|---|---|---|---|---|---|---|
| entry | ligand | cone angle b | νCOb | combined yield c (2b+2b′) | ratio (2b:2b′) d | E:Z (2b) d |
| 1 e | Cy2PhP | 162 | 1917 | 73 | 29:71 | n.d. |
| 2 | CyPh2P | 153 | 1917 | 84 | 75:25 | 91:9 |
| 3 | (o-anisyl)3P | 194 | 1919 | 70 | 83:17 | 80:20 |
| 4 | FcPh2P | 173 | 70 | 88:12 | 81:19 | |
| 5 | (p-tol)3P | 145 | 1920 | 78 | 92:8 | 67:33 |
| 6 | Ph3P | 145 | 1922 | 73 | 92:8 | 67:33 |
| 7 | (p-F-Ph)3P | 145 | 1924 | 74 | 92:8 | 57:43 |
| 8 | (EtO)Ph2P | 133 | 1926 | 81 | 95:5 | 75:25 |
| 9 | (p-CF3-Ph)3P | 145 | 1929 | 44 | >95:5 | 69:31 |
| 10 | (EtO)2PhP | 121 | 1932 | 20 | >95:5 | 89:11 |
| 11 | (PhO)3P | 128 | 1951 | <5 | n.d | n.d. |
Standard procedure: Ni(cod)2 (20 mol%) and a ligand (40 mol%) were dissolved in 2.5 mL toluene. Alkene (1 mL), triethylamine (600 mol%), the aldehyde (100 mol%, 0.5 mmol) and Et3SiOTf (175 mol%) were added. The reaction mixture was stirred 18 h at 23 °C.
See ref 20.
Yields were determined by 1H NMR using DMF as a standard.
Ratio was determined by 1H NMR after the products were treated with TBAF.
48 h reaction time.
Figure 1.

Plot of H:A ratio against the σ-electron donating ability of various phosphines.20
The ratio of the allylic to the homoallylic products is opposite for trialkylphosphines in which all alkyl groups are linear (entries 1 and 2), relative to those in which the three alkyl groups are branched (entries 3–5) or tertiary (entry 6). Among six trialkylphosphines with very similar electron donating abilities,20a tri-n-butylphosphine, the smallest of the trialkylphosphines examined, favors the homoallylic alcohol product while tricyclohexylphosphine and tri-t-butylphosphine, the largest among these, favor the 1,2-disubstituted allylic product. However, these sterically demanding ligands are not nearly as effective, affording the coupling products in low yield.
Notably, replacing one of the alkyl substituent of the tricyclohexylphosphine with a phenyl ring dramatically improves the yield (53% vs 16%; Table 2, entries 5 and 7) with a slightly diminished A:H ratio. Other aryldicyclohexylphosphines also display a similar yield enhancement (entries 8 and 9, as compared to entries 4–6). The bulky and electron-rich dicyclohexylferrocenylphosphine, however, seems to be more closely related to tri-t-butylphosphine, (poor yield for both the allylic and homoallylic alcohol products, entry 10).21 All of the sterically demanding dicyclohexylarylderivatives examined favor the allylic alcohol product, and dicyclohexylphenylphosphine is the optimal ligand in terms of yield and selectivity.
The pronounced ligand effects prompted us to examine other organophosphorus ligands (Table 3). Among the four triarylphosphine ligands with a similar cone angle but different para-substituents (entries 5–7 and 9),20b,20c tris-(p-trifluoromethyl-phenyl)-phosphine, the least electron-rich ligand of the four, has the highest H:A ratio (entry 9) whereas tri-p-tolylphosphine, the most σ-electron donating among these four ligands, has the lowest H:A ratio (entry 5).
These data suggest that a higher H:A ratio can be achieved by decreasing the electron donating ability of the phosphine ligand. In accord with this hypothesis, ethyldiphenylphosphinite ((EtO)Ph2P) further improves the H:A ratio in the case of 1-octene and benzaldehyde (entry 8, 95:5). The hypothesis becomes even more convincing when the H:A ratio is plotted against the σ-electron donating ability of various phosphines in Table 3 (Figure 1).20 Very electron deficient phosphonites and phosphites are not effective ligands, however (entries 10 and 11). It should be noted that the cone angle of the ligands also affects the observed H:A ratio. Many of the less electron-rich ligands that favor the homoallylic product in Table 3 are also among the smaller ligands.
Based on the results of this study, we surmised that the coupling product ratio is determined by a combined effect of the electron donating ability and the cone angle of the phosphine ligands. High H:A ratios can be achieved by using less electron-rich phosphines with small cone angle such as (EtO)Ph2P while high A:H ratios can be obtained by using electron-rich phosphines with a large cone angle such as Cy2PhP.22
Effects of the Base
Tertiary amines are the optimal bases for the nickel-catalyzed coupling of alkenes and aldehydes. Among different types of amine bases examined in ethylene couplings, only tertiary amines provide >20% yield of coupling products (Table 4, entries 1 and 3). Amines that likely are able to interact with nickel to a greater degree, such as pyridine (Table 4, entry 5 and Table 6, entry 6), are not effective. No coupling products are detected when inorganic bases are used in place of triethylamine (Table 4, entries 6–8).
Table 4.
Effect of Bases in the Ethylene–Benzaldehyde Coupling a
 | ||
|---|---|---|
| entry | base | yield b |
| 1 | Et3N | 77 |
| 2 | Et2NH | 3 |
| 3 | N-methylpyrrolidine | 36 |
| 4 | proton sponge | 10 |
| 5 | pyridine | 12 |
| 6 c | K3PO4 | <5 |
| 7 | K3CO3 | <5 |
| 8 | Cs2CO3 | <5 |
Standard procedure: Ni(cod)2 (20 mol%) and (o-anisyl)3P (40 mol%) were dissolved in 2.5 mL toluene under argon. Ethylene (balloon, 1atm) was substituted for argon. A base (600 mol%), benzaldehyde (100 mol%, 0.5 mmol), and Et3SiOTf (175 mol%) were added. The reaction mixture was stirred 18 h at 23 °C.
Yields were determined by 1H NMR using DMF as a standard.
Benzaldehyde was replaced by 2-naphthaldehyde and the reaction was run at 0.25 mmol scale.
Table 6.
Effect of Bases in the 1-Octene–Benzaldehyde Coupling (Ph3P)
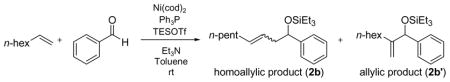 | ||||
|---|---|---|---|---|
| entry | base | combined yield (2b+2b′) b | ratio (2b:2b′) b | ratio (E/Z) c (2b) |
| 1 | Et3N | 64% | 92:8 | 87:13 |
| 2 | Cy2NMe | 35% | >95:5 | 71:29 |
| 3 | N-methylmorpholine | 25% | 94:6 | 69:31 |
| 4 | N-methylpiperidine | <5% | n.d. | n.d. |
| 5 | N-methylpyrrolidine | <5% | n.d. | n.d. |
| 6 | DMAP | <5% | n.d. | n.d. |
Standard procedure: Ni(cod)2 (20 mol%) and Ph3P (40 mol%) were dissolved in 2.5 mL toluene. 1-octene (1 mL), a base (600 mol%), benzaldehyde (100 mol%, 0.5 mmol), and Et3SiOTf (175 mol%) were added. The reaction mixture was stirred 18 h at 23 °C.
Yields and ratios were determined by 1H NMR using DMF as a standard.
Ratio was determined from the desilylated product by 1H NMR.
Tertiary amines were further examined in the 1-octene coupling reaction (Tables 5–6), and triethylamine was consistently superior to other tertiary amines (Table 5, 1–4 and Table 6, 1–5). Tertiary amines smaller or larger than triethylamine compromised the yield of the coupling reaction (Table 5, entries 2–4).
Table 5.
Effect of Bases in the 1-Octene–Benzaldehyde Coupling (Cy2PhP)
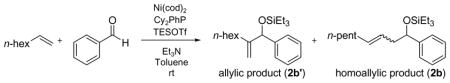 | |||
|---|---|---|---|
| entry | base | combined yield (2b′+2b) b | ratio (2b′:2b) c |
| 1 | Et3N | 61% | 78:22 |
| 2 | Et(i-Pr)2N | 10% | 60:40 |
| 3 | Cy2NMe | 20% | 60:40 |
| 4 | N-methylpyrrolidine | 7% | 71:29 |
| 5 | 2,6-lutadine | 12% | 58:42 |
Standard procedure: Ni(cod)2 (20 mol%) and Cy2PhP (40 mol%) were dissolved in 1.5 mL alkene. A base (600 mol%), benzaldehyde (100 mol%, 0.1 mmol), and Et3SiOTf (100 mol%) were added. The reaction mixture was stirred 18 h at 23 °C.
Yields were determined by 1H NMR using DMF as a standard.
Ratio was determined by 1H NMR of the crude reaction mixture.
The nature of the tertiary amines in determining the yield deserves further comment. It appears that a balance of the nucleophilicity, basicity, and steric bulk of the amine base is required for the coupling reaction to occur efficiently. Amines can compete with the phosphorus ligand, alkene, and aldehyde for a coordination site on nickel. A more nucleophilic (σ-electron donating) or smaller amine might hinder the coordination of any of the other required components to the nickel catalyst. For instance, the less nucleophilic N-methylmorpholine (Table 6, entry 3) provides a better yield than N-methylpiperidine (Table 6, entry 4).
In summary, triethylamine is the best base for the nickel-catalyzed coupling of alkenes and aldehydes, probably because of a combination of low coordinating ability and appropriate basicity.
Source of Nickel
Since the precatalyst Ni(cod)2 has two chelating diene ligands (1,5-cyclo-octadiene), other nickel(II) precatalysts without alkene ligands were examined, including Ni(Ph3P)4, Ni(acac)2/Ph3P/DIBAL-H, Ni(Ph3P)2Cl2/n-BuLi, and Ni(Ph3P)2Br2/n-BuLi (Table 7). Only the Ni(acac)2/Ph3P/DIBAL-H system is as efficient as Ni(cod)2/Ph3P (entry 3). Ni(Ph3P)4 is saturated with phosphine ligand, and perhaps alkene coordination to the nickel catalyst is thus inhibited. Similarly, Ni(cod)2/Cy2PhP is more efficient than Ni(acac)2/Cy2PhP/DIBAL-H and Ni(Cy2PhP)2Cl2/n-BuLi (Table 8).23 Therefore Ni(cod)2 was used in all subsequent investigations.
Table 7.
Examination of Other Nickel Precatalysts a
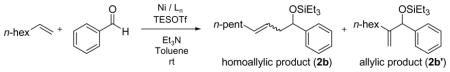 | |||||
|---|---|---|---|---|---|
| entry | catalyst | yield (2b) b | yield (2b′) b | ratio (2b:2b′) c | combined yield b (2b+2b′) |
| 1 | Ni(cod)2/Ph3P | 78% | 6% | 93:7 | 84% |
| 2 | Ni(Ph3P)4 | 41% | 3% | 93:7 | 44% |
| 3 | Ni(acac)2/Ph3P/DIBAL-H | 72% | 5% | 93:7 | 77% |
| 4 | Ni(Ph3P)2Cl2/n-BuLi | 32% | 2% | 94:6 | 34% |
| 5 | Ni(Ph3P)2Br2/n-BuLi | <5% | <5% | n.d. | <5% |
Standard procedure: The nickel precatalyst system (20 mol%) was dissolved in 2.5 mL toluene. The alkene (1 mL), triethylamine (600 mol%), the aldehyde (100 mol%, 0.5 mmol), and Et3SiOTf (175 mol%) were added. The reaction mixture was stirred 48 h at 23 °C.
Yields were determined by 1H NMR using DMF as a standard.
Ratios were determined by 1H NMR of the crude reaction mixture.
Table 8.
Examination of Other Nickel Precatalysts a
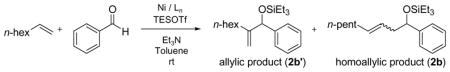 | |||||
|---|---|---|---|---|---|
| entry | catalyst | yield (2b′) b | yield (2b) b | ratio (2b′:2b) c | combined yield b (2b′+2b) |
| 1 | Ni(cod)2/Cy2PhP | 52% | 21% | 71:29 | 73% |
| 2 d | Ni(cod)2/Cy2PhP | 53% | 20% | 73:27 | 73% |
| 3 | Ni(acac)2/Cy2PhP/DIBAL-H | 39% | 16% | 71:29 | 55% |
| 4 | Ni(Cy2PhP)2Cl2/n-BuLi | 21% | 7% | 75:25 | 28% |
Standard procedure: The nickel precatalyst system (20 mol%) was dissolved in toluene. The alkene (500 mol%), triethylamine (600 mol%), the aldehyde (100 mol%), and Et3SiOTf (175 mol%) were added. The reaction mixture was stirred 48 h at 23 °C.
Yields were determined by 1H NMR using DMF as a standard.
Ratios were determined by 1H NMR of the crude reaction mixture.
1 mL 1-octene was used.
Substrate Scope
Applying the results of the studies of ligand and base effects, the substrate scope of the nickel-catalyzed coupling of alkenes, aldehydes, and silyl triflates was next examined (Tables 9–12). In general, ethylene and monosubstituted alkenes are superior substrates in this coupling reaction, while 1,1-disubstituted alkenes and acyclic 1,2-disubstituted (cis or trans) alkenes are significantly less reactive. Trisubstituted alkenes do not react under the standard reaction conditions.
Table 9.
Preparation of Homoallylic Alcohol Products from Nickel-Catalyzed Alkene–Aldehyde Couplings a
| entry | alkene | aldehyde | major product (2) | yield (%)(2:2′) b,c | E:Z (2) b |
|---|---|---|---|---|---|
| 1 d |
|
PhCHO |
2a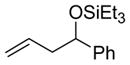
|
73 (89:11) | n.a |
| 2 |
|
PhCHO |
2b |
85 (95:5) | 75:25 |
| 3 e | 72 (>95:5) | 75:25 | |||
| 4 e | p-anisaldehyde |
2c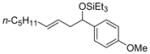
|
85 (>95:5) | 75:25 | |
| 5 e | p- Cl(C6H4)CHO |
2d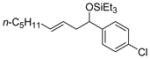
|
37 (>95:5) | 74:26 | |
| 6 f | 2-naphthaldehyde |
2e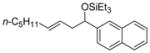
|
88 (>95:5) | 70:30 | |
| 7 | 1-methyl-2-indole- carboxaldehyde |
2f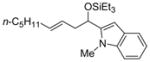
|
56 (>95:5) | 83:17 | |
| 8 f | t-BuCHO |
2g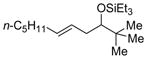
|
64 (>95:5) | 78:22 | |
| 9 |
|
PhCHO |
2h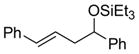
|
86 (92:8) | >95:5 |
| 10 |
o-anisaldehyde m-anisaldehyde p-anisaldehyde |
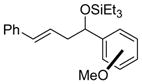 2i (ortho) 2i (meta) 2i (para) |
78 (92:8) 98 (92:8) 99 (92:8) |
>95:5 >95:5 >95:5 |
|
| 11 f,g | p-anisaldehyde | 2i (para) | 98 (92:8) | >95:5 | |
| 12 f | 2-naphthaldehyde |
2j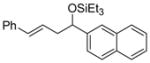
|
88 (95:5) | >95:5 | |
| 13 | 1-methyl-2-indole-carboxaldehyde |
2k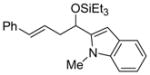
|
57 (>95:5) | >95:5 | |
| 14 | t-BuCHO |
2l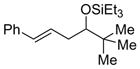
|
65 (>95:5) | 78:22 | |
| 15 |
|
p-anisaldehyde |
2m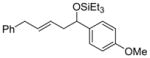
|
91 (92:8) | 69:31 |
| 16 |
|
PhCHO |
2n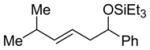
|
82 (>95:5) | 81:19 |
|
| |||||
| 17 |
|
2o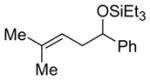
|
95 (86:14) h | n.a. | |
| 18 |
|
2p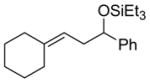
|
99 (75:25) h | n.a. | |
| 19 |
|
3q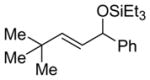
|
14 (>95:5) i | n.a. | |
Standard procedure: (entries 1–8, 15–18): To a solution of Ni(cod)2 (0.1 mmol) and EtOPPh2 (0.2 mmol) in toluene (2.5 mL) at 23 °C under Ar were added the alkene (0.5 mL), triethylamine (3.0 mmol), the aldehyde (0.5 mmol), and Et3SiOTf (0.875 mmol). The mixture was stirred 48 h at room temperature and purified by chromatography (SiO2). Entries 9–14: Ph3P was used in place of EtOPPh2.
Yields were determined by 1H NMR using DMF as a standard.
See Supporting Information for structures of the minor products (2a′-2p′).
Propene (1 atm) was used in place of Ar.
Reaction time 18 h.
Reaction temperature 35 °C.
Fivefold larger reaction scale.
ratio of 2:3.
ratio of 3: (2q+2q′).
Table 12.
Coupling of Oxygen-Containing Alkenes with Aldehydes a
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R1 (alkene) | R2 (aldehyde) | ligand | major product | yield (%) b | ratio (4:4′) b | ratio (E:Z) c |
| 1 |

|
Ph | Cy2PhP |
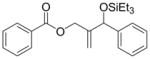 4d |
<5 | n.d. | - |
| (EtO)Ph2P |
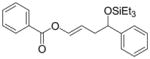 4d′ |
<5 | n.d. | n.d. | |||
| 2 e |
|
Ph | Cy2PhP |
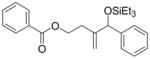 4e |
21 | n.d. | - |
| (EtO)Ph2P |
4e′ |
<5 | n.d. | n.d. | |||
| 3 |
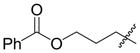
|
o-anisyl | Cy2PhP |
4f |
44 d | 73:27 | - |
| 4 e |
|
o-anisyl | (EtO)Ph2P |
4g |
66 | 7:93 | 50:50 |
Standard procedure: To a solution of Ni(cod)2 (0.1 mmol) and ligand (0.2 mmol) in toluene (2.5 mL) at 23 °C under Ar were added the alkene (2.5 mmol), triethylamine (3.0 mmol), the aldehyde (0.5 mmol), and Et3SiOTf (0.875 mmol). The mixture was stirred 48 h at room temperature and purified by chromatography (SiO2).
Determined by 1H NMR of the crude reaction mixture using DMF as a standard.
The ratio was determined by 1H NMR of the mixture of E and Z homoallylic alcohols after the silyl group of the coupling product was removed by TBAF.
Isolated yield.
1.5 mmol alkene was used.
The Ni–EtOPh2P system efficiently catalyzes the coupling of monosubstituted alkenes and simple aromatic aldehydes such as benzaldehyde (Table 9). While the couplings of ethylene with most aldehydes usually take less than 8 h to reach completion, those involving monosubstituted alkenes typically require more than 18 h (entries 2–3). Nevertheless, with EtOPh2P as the ligand, nickel catalyzes the coupling of several monosubstituted alkenes and aldehydes in excellent yield. The reaction is also highly regioselective and E/Z selective, favoring an E-homoallylic alcohol product.
Aromatic aldehydes (Table 9, entries 2, 6, 9, 12), heteroaromatic aldehydes (entries 7 and 13), and sterically demanding aldehydes (entries 8 and 14) are excellent coupling partners with monosubstituted alkenes, affording an E-homoallylic alcohol derivative as the major product and an allylic alcohol derivative as the minor product, with a selectivity >95:5 in most cases. Monosubstituted aromatic aldehydes of all substitution patterns are tolerated (ortho-, meta-, and para-, entry 10). Aldehydes with an electron donating substituent in the para position (p-MeO–, entry 4) are more reactive than aldehydes with an electron withdrawing group in the same position (Cl–, entry 5), consistent with the observation in the ethylene–aldehyde couplings. The product derived from p-chlorobenzaldehyde can be elaborated further by way of a cross-coupling reaction.
Allylbenzene is an excellent substrate, and the homoallylic products are useful styrene derivatives. The E-isomer is observed exclusively. Oligomerization of the coupling product is not observed, as evidenced by the excellent yield of the coupling reactions (entries 9–14).
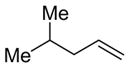
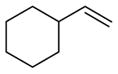
Linear monosubstituted olefins such as propene and 1-octene are not the only terminal olefins that can participate in this nickel-catalyzed reaction (entries 1, 2, 9, 15). Alkenes with substitution at the homoallylic position couple with benzaldehyde in similar regioselectivity and E/Z selectivity as in the case of 1-octene (entry 16, as compared to entry 2).
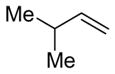
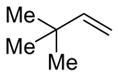
Alkenes with substituents at the allylic position, on the other hand, afford different results. A homoallylic alcohol derivative is still the major coupling product in the coupling of 2-methyl-butene (entry 17) and vinylcyclohexane (entry 18) with benzaldehyde. However, the minor product is an E-1,3-disubstituted allylic alcohol, rather than the usual 1,2-disubstituted allylic alcohol obtained from the coupling of unbranched alkenes (eqs 2–3). The coupling of 3,3-dimethyl-butene and benzaldehyde yields exclusively 1,3-disubstituted allylic alcohol product (eq 4). This observation maybe important in understanding the mechanism of these transformations, which is discussed in more detail in the discussion section below.
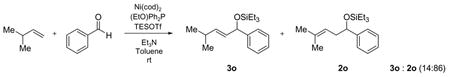 |
(2) |
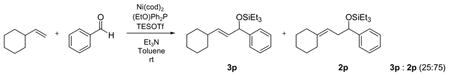 |
(3) |
 |
(4) |
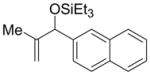
Allylic, rather than homoallylic alcohol derivatives can be prepared by the nickel-catalyzed coupling of alkenes and aldehydes simply by substituting Cy2PhP for EtOPh2P (Table 10). Hence, propene couples with naphthaldehyde to provide the allylic alcohol product in good yield and with the highest selectivity (Table 10, entry 1). In contrast to the Ni–EtOPh2P system, the homoallylic alcohol is the minor product in this case.
Table 10.
Preparation of Allylic Alcohol Products from Nickel-Catalyzed Alkene–Aldehyde Couplingsa
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 (alkene) | R2 (aldehyde) | R3SiOTf | major product | yield (%) b (2+2′) | ratio (2′:2) c |
| 1 d | Me | naphthyl | Et3SiOTf |
 2r′ |
82 | 84:16 |
| 2 | n-hexyl | Ph | Et3SiOTf |
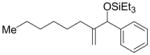 2b′ |
70 | 71:29 |
| 3 d | Me | p-anisyl | Et3SiOTf |
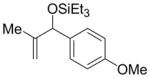 2s′ |
95 | 82:18 |
| 4 | Ph |
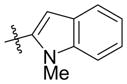
|
Et3SiOTf |
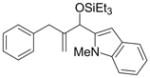 2k′ |
56 e | 80:20 |
| 5 | isobutyl | Ph | Et3SiOTf |
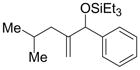 2n′ |
62 | 71:29 |
| 6 |
|
Ph | Et3SiOTf |
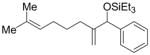 2t′ |
72 | 71:29 |
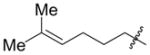
| 7 | c-hexyl | Ph | Et3SiOTf |
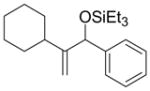 2p′ |
5 f | n.d. |
Standard procedure: Ni(cod)2 (20 mol%), Cy2PhP (40 mol%) were dissolved in 2.5 mL toluene. Excess alkene, triethylamine (600 mol%), the aldehyde (100 mol%, 0.5mmol), and Et3SiOTf (175 mol%) were added. The reaction mixture was stirred 18 h at 23 °C.
Unless specified, isolated yield of all coupling products.
Ratios were determined by 1H NMR of the crude reaction mixture.
1 atm propene (balloon) was used and naphthaldehyde (100 mol%) was mixed with Ni(cod)2 and Cy2PhP before the addition of toluene.
Yields were determined by 1H NMR using DMF as a standard.
Isolated yield of the allylic product 2p′.
Once again, aromatic aldehydes and heteroaromatic aldehydes couple with straight chain monosubstituted alkenes in good yield (entries 1–2, 4). Electron donating p-anisaldehyde is, as before, more reactive than benzaldehyde (entries 1 and 3). Therefore, based on all the data that we gathered so far, it seems to be the trend that generally electron-donating aldehydes are more reactive than electron-poor aldehydes.
There are, however, some differences in the substrate scope of the alkene in the Ni–Cy2PhP system relative to that of the Ni–EtOPh2P system. While branching at the homoallylic position of the alkene does not affect the coupling efficiency (entry 5), branching at the allylic position significantly attenuates the yield of the allylic alcohol product (entry 7). For example, vinylcyclohexane has a dramatically lower A:H ratio (entry 7), and the homoallylic alcohol and a 1,3-disubstituted allylic alcohol are the major products.
In a competition study, benzaldehyde undergoes coupling with a monosubstituted alkene selectively in the presence of a trisubstituted alkene (entry 6); the trisubstituted double bond is stable to the reaction conditions. Carbocyclization is not observed, nor do we observe any isomerization of the trisubstituted double bond in the coupling product. This result enables the use of a trisubstituted double bond as a masked version of other functional groups.
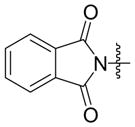
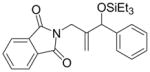
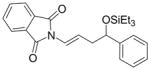
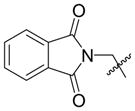
Given that heteraromatic aldehydes are competent substrates in these coupling reactions, we became interested in the effect of heteroatoms on the alkene. N-allylphthalimide, N-homoallylphthalimide and N-homoallyloxazolidinone undergo coupling in both the Ni–Cy2PhP and Ni–EtOPh2P systems (Table 11, entries 1–3). In particular, the coupling of N-allylphthalimide and benzaldehyde in the Ni–EtOPh2P system affords an enamine that appears to be stable to the coupling conditions. In contrast, allylbenzoate and homoallylbenzoate esters are much less efficient (Table 12, entries 1–4). A small amount of the allylic product is detected only with homoallylbenzoate (entry 2). When the benzoate group is further away from the terminal double bond, a better yield of the desired coupling product is observed (entry 3). These findings suggest an interaction of the heteroatoms on the alkenes to the nickel catalyst. We propose that since the oxygen on the phthalimide is less nucleophilic, it does not bind to the nickel as tightly as the benzoate oxygen. Therefore the coupling of N-allylphthalimide occurs more efficiently than allylbenzoate (Table 11, entry 1 and Table 12, entry 1). As the benzoate becomes further away from the double bond, the benzoate is less likely to coordinate to the nickel catalyst, and the reactivity of the alkene is restored (Table 12, entry 3). The silyl ether-tethered alkene (entry 4) does not experience the heteroatom attenuation effect, likely for the same reason as the benzoate ester in entry 3.
Table 11.
Coupling of Nitrogen-Containing Alkenes with Aldehydes a
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R1 (alkene) | R2 (aldehyde) | ligand | major product | yield (%) b | ratio (4:4′) b | ratio (E:Z) c |
| 1 |
|
Ph | Cy2PhP |
 4a |
67 | 74:26 | - |
| (EtO)Ph2P |
 4a′ |
43 | 12:88 | 60:40 | |||
| 2 |
|
o-anisyl | Cy2PhP |
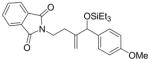 4b |
54 | 71:29 | - |
| Ph3P |
4b′ |
76 | <5:95 | 83:17 | |||
| 3 |
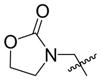
|
Ph | Cy2PhP |
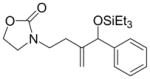 4c |
60 | 83:17 | - |
| (EtO)Ph2P |
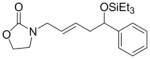 4c′ |
28 | 10:90 | n.d. | |||
Standard procedure: To a solution of Ni(cod)2 (0.1 mmol) and ligand (0.2 mmol) in toluene (2.5 mL) at 23 °C under Ar were added the alkene (1.5 mmol), triethylamine (3.0 mmol), the aldehyde (0.5 mmol), and Et3SiOTf (0.875 mmol). The mixture was stirred 48 h at room temperature and purified by chromatography (SiO2).
Determined by 1H NMR of the crude reaction mixture using DMF as a standard.
The ratio was determined by 1H NMR of the mixture of E and Z homoallylic alcohols after the silyl group of the coupling product was removed by TBAF.
Discussion
General Mechanistic Framework
We believe that the nickel species that catalyzes the alkene–aldehyde coupling reactions above is not functioning simply as a Lewis acid. We propose that the coupling reaction proceeds through the formation of oxanickellacycle from a nickel(0) complex (Scheme 1). A syn β-hydride elimination would afford the coupling product and a nickel–hydride species, analogous to a Heck reaction.3a–c Finally, base-promoted reductive elimination of the nickel–hydride intermediate could regenerate the nickel(0) catalyst. Note that a base-mediated β-elimination of the oxanickellacycle via an E2-like mechanism cannot be completely ruled out. Based on our observations and in analogy to the Heck reaction,3a–c we believe the nickel–hydride pathway is operative (see below).
Scheme 1.

Ligand Effects
The interactions of nickel with the ligand, alkene, and aldehyde govern the assembly of the oxanickellacycle, and the oxanickellacycle in turn determines the product distribution. Scheme 2 summarizes the factors that control the product ratio in the alkene–aldehyde coupling reactions. In general, use of large phosphines favors the allylic alcohol product (A) (e.g., the coupling of 1-octene with benzaldehyde with Cy2PhP as ligand yielded allylic alcohol as the major product.) The use of small phosphines (Bu3P, (EtO)Ph2P, Ph3P, etc) on the other hand affords the homoallylic alcohol (H) as the major product.
Scheme 2.

Size of Coupling Partners
The substituents on the alkene and aldehyde also affect the ratio of the coupling products. The alkene substituents can either be closer to the ligand or the aldehyde substituent in the oxanickellacycle. Allylic alcohol product A is obtained in a significant amount when the alkene has no branching at the allylic position. On the other hand, branching at the allylic position does not affect the coupling process when a small ligand, such as (EtO)Ph2P, is used, and homoallylic allylic alcohol H is formed in good yield. 3,3-dimethyl-1-butene, a sterically demanding monosubstituted alkene with no allylic hydrogen, provides 1,3-disubstituted allylic alcohol A′ as the sole product.
A large substituent on aldehyde favors the production of homoallylic alcohol. Less than 5% allylic alcohol product is observed when propene or 1-octene is coupled with pivaldehyde with Cy2PhP as the ligand. In the following sections, we propose a detailed model consistent with all of these observations.
We begin by examining oxanickellacycle 1 in more detail (Scheme 3). The β-hydrogen of the oxanickellacycle 1 is not aligned with the C–Ni bond. Since β-hydride elimination generally occurs in the syn orientation, The –OSiEt3 group must dissociate from nickel to allow bond rotation such that the β-hydrogen can align with C–Ni bond. At this stage, β-hydride elimination occurs and allylic product A is formed. The larger the phosphine ligand relative to the aldehyde substituent, the more likely oxanickellacycle 1 dominates because the alkene substituent would thus avoid severe steric repulsion with this ligand. The data shown in Table 2 support this proposal; the A:H ratio increases with the cone angle of the trialkylphosphine.
Scheme 3.

Oxanickellacycle 2 accounts for the formation of homoallylic alcohol H and allylic alcohol A′. Examination of oxanickellacycle 2 reveals that although the β-hydrogen in the oxanickellacycle (Hendo) is not aligned with the C–Ni bond, there are β-hydrogens outside the oxanickellacycle (Hexo) that are appropriately poised for β-hydride elimination once a free coordination site is available (Scheme 4).3e The preferred conformation would align R2 of the alkene trans to the C–C bond of the oxanickelacycle 2. Dissociation of one of the ligand on nickel provides a free coordination site for the syn β-hydride elimination to occur and provides the E-homoallylic alcohol H.
Scheme 4.

In order for the unusual allylic alcohol (A′) to form, the β-hydrogens in the oxanickellacycle (Hendo) must be eliminated instead of the exo-β-hydrogen (Hexo). Such a process requires dissociation of –OSiEt3 and maybe favored when the exo-β-hydrogen is not aligned with the C–Ni bond, or when there is no exo-β-hydrogen (Scheme 5).
Scheme 5.

The coupling of vinylcyclohexane and benzaldehyde serves as a good example to illustrate the formation of 1,3-disubstituted allylic alcohol A′ (Scheme 5). Neither R1 nor R2 of vinylcyclohexane is a hydrogen atom, and hence the allylic position is very sterically encumbered. The usual allylic alcohol product A is not favored because the large substituent of vinylcyclohexane will not be accommodated next to the aldehyde substituent (R) in oxanickellacycle 1 (Scheme 3) due to severe steric repulsion.
Experimental data support this theory: The coupling of vinylcyclohexane with benzaldehyde using Cy2PhP as ligand yields only 5% of the allylic alcohol product A (Table 10, entry 7, as compared with other unbranched alkenes in Table 10, entries 1–6). Using a smaller ligand, such as (EtO)Ph2P, the large substituents in vinylcyclohexane can be accommodated by being closer to the ligand than to the aldehyde substituent, favoring oxanickellacycle 2 (Scheme 5). The exo-β-hydrogen of the oxanickellacycle, when aligned to with C–Ni bond, induces an unfavorable steric interaction between the cyclohexyl group and the C–C bond of the oxanickellacycle. Therefore the rate of β-hydride elimination from the exo-β-hydrogen decreases, and that of the endo-β-hydrogen increases, resulting in a greater amount of the unusual E-allylic product A′. The E-double bond geometry of A′ is obtained by minimizing steric repulsion during the β-H elimination step.
Alkenes without an allylic hydrogen cannot afford homoallylic alcohol products in the nickel-catalyzed alkene–aldehyde coupling reaction. For example, 3,3-dimethyl-1-butene, with no allylic hydrogen, couples with benzaldehyde to give exclusively E-1,3-disubstituted allylic alcohol product (A′).24 Also, it appears that the steric bulk of the tert-butyl group renders formation of oxanickellacycle 1 extremely difficult, eliminating the possibility of affording 1,1-disubstituted allylic alcohol product A.
The proposed mechanistic framework is also supported by Ogoshi’s observation that cyclization of an α,ω-enal to form an oxanickellacycle is facilitated by the presence of a silyl triflate.17 A control experiment confirms that without silyl triflate, no coupling product is observed.22
The evidence for the β-hydride elimination as the next step is the observation of isomerization and dimerization (hydrovinylation) of the starting olefins, which suggests the presence of a nickel–hydride (Ni–H) species, likely formed by a β-hydride elimination.3d The requirement of a base in this catalyst system also supports the presence of a Ni–H species. A β-hydride elimination and subsequent base-assisted removal of triflic acid (reductive elimination) from the Ni–H species regenerates the Ni(0) catalyst (Scheme 1) and may also minimize side reactions by suppressing the presence of the Ni–H species.
We do not believe the direct precursor to the oxanickellacycle in this coupling reaction is a cationic nickel (II) species. Ni2+, Pd2+, and Pt2+ catalysts have been reported to be effective Lewis acids for carbonyl-ene reactions.18g–i The nickel-catalyzed coupling of alkenes, aldehydes, and silyl triflates affords carbonyl-ene-type products in good yield, but the substrate scope is entirely different from that of a Lewis acid-catalyzed carbonyl-ene reaction. While the three cationic group 10 transition metal catalysts are effective in the carbonyl-ene reaction of the more nucleophilic alkenes such as 1,1-disubstituted alkenes and the more electrophilic aldehydes such as glyoxylate esters, they do not promote the coupling of monosubstituted alkenes and simple aldehydes.
The nickel catalyst system has the opposite alkene and aldehyde substrate scopes relative to those of the carbonyl-ene reaction. The nickel–phosphine catalyst selectively reacts with monosubstituted olefins, and we observe that electron rich aldehydes, such as p-anisaldehyde, consistently provide better yield than benzaldehyde and electron-deficient aldehydes. Although this coupling reaction readily provides homoallylic alcohol products corresponding to a carbonyl-ene reaction, it is more likely that the oxanickellacycle precursor is a Ni(0) species, and probably not just a Lewis acid catalyst.
To illustrate the difference between the Ni(0)–phosphine system and a Lewis acid system, β-citronellene and benzaldehyde were coupled under two conditions; using a classical Lewis acid and the Ni–EtOPh2P conditions.6b As expected, the Lewis acid-catalyzed reaction reacts at the more nucleophilic trisubstituted double bond. For the Ni–EtOPh2P system, however, the monosubstituted double bond reacts preferentially because it is the kinetically more accessible double bond. These observations are also in accord with many palladium-catalyzed reactions of alkenes (such as Wacker oxidation and alkene hydroamination), in that a monosubstituted double bond is usually more reactive than a more substituted double bond.3a
The difference in substrate scope between the nickel-catalyzed alkene–aldehyde coupling and the carbonyl-ene reaction is further illustrated by competition experiments between a monosubstituted alkene and a 1,1-disubstituted alkene (Scheme 7).25–26 Equal amounts of allylbenzene and methylenecyclohexane were included in the otherwise standard coupling conditions. The coupling reaction was highly selective; 92% of all of the coupling products detected are derived from allylbenzene. The presence of methylenecyclohexane does not change the H:A ratio of the coupling products of allylbenzene (as compared to Table 9, entry 10). A similar trend is observed between 1-octene and methylenecyclohexane (as compare to Table 9, entry 4), but the presence of excess methylenecyclohexane in the reaction mixture seems to lower the yield of the coupling reaction. This lower efficiency might be due to competition for a coordination site on nickel between monosubstituted alkenes and methylenecyclohexane.
Scheme 7.

Competition Experiments Between Monosubstituted and 1,1-Disubstituted Alkenes 25–26
Further evidence that supports the notion that the nickel-catalyzed coupling of alkenes, aldehydes, and silyl triflates does not involve a carbonyl-ene reaction mechanism is that ethylene, with no allylic hydrogen, also participates in this coupling reaction using the same Ni–phosphine catalyst system.
Common side reactions in these nickel-catalyzed reactions are the dimerization (hydrovinylation)27 and isomerization28 of the starting olefin. One explanation for the requirement of excess alkenes in this coupling reaction is that the terminal alkene is isomerized to an internal alkene and that this new internal alkene is not reactive in the coupling process. While isomerization of olefin is common in the coupling reaction, hydrovinylation of olefins is observed in small amounts only when the alkene–aldehyde coupling process is not efficient. The presence of a base in the coupling reaction may keep the Ni-H concentration to a minimum, thus suppressing some of these side reactions.
Summary
The nickel-catalyzed coupling of alkenes, aldehydes, and silyl triflates represents a new alternative to both allylmetal reagents and alkenylmetal reagents (Scheme 8). The parent allylmetal reagent and vinylmetal reagent can now be replaced by propene and ethylene, respectively, using the nickel-catalyzed processes as described herein. The preparation of a terminal, monosubstituted alkene is generally more straightforward than that of the allylmetal species such as those shown in Scheme 8.
Scheme 8.

The transformation in this nickel-catalyzed alkene–aldehyde coupling reaction is, in effect, a C–H functionalization reaction of the alkene, involving addition to an aldehyde. Mechanistically, an entirely different process likely occurs, rather than oxidative addition into a C–H bond that would be expected to have a relatively high energy activation barrier.
Unlike the related transition metal-catalyzed reductive coupling reactions developed by our group and others,8 the nickel-catalyzed coupling of alkenes, aldehydes, and silyl triflates described in this work is not an overall reductive process (Scheme 9). Thus, the coupling of an alkene and an aldehyde, in theory, does not require a third component to form the allylic or homoallylic alcohol derivatives. However, both alkenes and aldehydes are generally unreactive toward each other. Thus, activation of either or both components is necessary. The Lewis acid-catalyzed carbonyl-ene reaction serves as a good example. Intermolecular carbonyl-ene reaction between monosubstituted alkene and unactivated aldehydes such as acetaldehyde is not a practical method under thermal conditions. The presence of a Lewis acid, however, allows the coupling to proceed at room temperature. The Lewis acidic nature of silyl triflate in the nickel-catalyzed alkene–aldehyde coupling reaction likely plays a similar role, providing sufficient activation of the electrophile for the nickel catalyst to promote the coupling reaction.
Scheme 9.

The two classes of the nickel-catalyzed coupling reactions of alkene, aldehyde, and silyl triflate presented here represent unique, non-reductive coupling processes that allow the preparation of derivatives of allylic alcohols or homoallylic alcohols from readily available olefins. The selectivity for these two products is highly ligand dependent, and high selectivity in either direction is possible. These coupling reactions are mechanistically different from Lewis acid-catalyzed carbonyl-ene reactions, and conceptually, alkenes serve as substitutes for both allylmetal reagents and alkenylmetal reagents.
Supplementary Material
Scheme 6.

Acknowledgments
Support for this work was provided by the National Institute of General Medical Sciences (GM-063755). C.-Y. H. thanks The Croucher Foundation for a postdoctoral fellowship. We are grateful to Dr. Li Li for obtaining mass spectrometric data for all compounds (MIT Department of Chemistry Instrumentation Facility, which is supported in part by the NSF (CHE-9809061 and DBI-9729592) and the NIH (1S10RR13886-01)).
Footnotes
Supporting Information Available: Experimental procedures and characterization data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lappin GR, Sauer JD, editors. Alpha Olefins Applications Handbook. Marcel Dekker; New York: 1989. [Google Scholar]
- 2.Blom R, editor. Organometallic Catalysts and Olefin Polymerization. Springer; New York: 2001. [Google Scholar]
- 3.Palladium Reagents and Catalysts: Innovations in Organic Synthesis. Tsuji, J., John Wiley & Sons; New York: 1995. Review of the related Heck reaction and palladium–hydride chemistry: Negishi E-i., editor. Handbook of Organopalladium Chemistry for Organic Synthesis. Wiley-Interscience; New York: 2002. Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048.Detection of a palladium–hydride species in the Heck reaction: Hills ID, Fu GC. J Am Chem Soc. 2004;126:13178–13179. doi: 10.1021/ja0471424.Similar selectivity of the exo hydrogens over the endo hydrogens has been reported in the Heck reaction literature: Ono K, Fugami K, Tanaka S, Tamaru Y. Tetrahedron Lett. 1994;35:4133–4136.
- 4.Grubbs RH, editor. Handbook of Metathesis. John Wiley & Sons; New York: 2003. [Google Scholar]
- 5.Ojima I, editor. Catalytic Asymmetric Synthesis. Wiley-VCH; New York: 2000. [Google Scholar]
- 6.Preliminary communications of this work: Ng SS, Jamison TF. J Am Chem Soc. 2005;127:14194–14195. doi: 10.1021/ja055363j.Ho CY, Ng SS, Jamison TF. J Am Chem Soc. 2006;128:5362–5363. doi: 10.1021/ja061471+.
- 7.Carbonyl-ene reaction was first reported by Alder in 1943. Alder K, Pascher F, Schmitz A. Ber Dtsch Chem Ges. 1943;76:27.For a review of the carbonyl-ene reaction, see: Hoffmann HMR. Angew Chem, Int Ed. 1969;8:556–577.Snider BB. Acc Chem Res. 1980;13:426–432.Snider B. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 2. Pergamon Press; Oxford: 1991. pp. 527–561.Mikami K, Shimizu M. Chem Rev. 1992;92:1021–1050.Dias LC. Curr Org Chem. 2000;4:305–342.
- 8.For a review of the nickel-catalyzed reductive coupling reactions see: Montgomery J. Angew Chem, Int Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634.A general reference of organonickel chemistry: Tamaru Y, editor. Modern Organonickel Chemistry. Wiley-VCH; Weinheim, Germany: 2005. Recent reports of nickel-catalyzed reductive couplings: Alkynes: Patel SJ, Jamison TF. Angew Chem, Int Ed. 2004;43:3941–3944. doi: 10.1002/anie.200460044.Kimura M, Ezoe A, Mori M, Tamaru Y. J Am Chem Soc. 2005;127:201–209. doi: 10.1021/ja0469030.Knapp-Reed B, Mahandru GM, Montgomery J. J Am Chem Soc. 2005;127:13156–13157. doi: 10.1021/ja054590i.Luanphaisarnnont T, Ndubaku CO, Jamison TF. Org Lett. 2005;7:2937–2940. doi: 10.1021/ol050881k.Enyne: Miller KM, Jamison TF. J Am Chem Soc. 2004;126:15342–15343. doi: 10.1021/ja0446799.Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J Am Chem Soc. 2004;126:4130–4131. doi: 10.1021/ja0491735.Miller KM, Colby EA, Woodin KS, Jamison TF. Adv Synth Catal. 2005;347:1533–1536.Miller KM, Jamison TF. Org Lett. 2005;7:3077–3080. doi: 10.1021/ol051075g.Moslin RM, Jamison TF. Org Lett. 2006;8:455–458. doi: 10.1021/ol052719n.Allene: Takimoto M, Kawamura M, Mori M, Sato Y. Synlett. 2005;13:2019–2022.Ng SS, Jamison TF. J Am Chem Soc. 2005;127:7320–7321. doi: 10.1021/ja0521831.Ng SS, Jamison TF. Tetrahedron. 2005;61:11405–11417.Song M, Montgomery J. Tetrahedron. 2005;61:11440–11448.Diene: Takimoto M, Nakamura Y, Kimura K, Mori M. J Am Chem Soc. 2004;126:5956–5957. doi: 10.1021/ja049506y.Sawaki R, Sato Y, Mori M. Org Lett. 2004;6:1131–1133. doi: 10.1021/ol049908a.Takimoto M, Kajima Y, Sato Y, Mori M. J Org Chem. 2005;70:8605–8606. doi: 10.1021/jo051283m.
- 9.Examples of palladium-catalyzed coupling reactions: Anwar U, Grigg R, Rasparini M, Savic V, Sridharan V. Chem Commun. 2000:645–646.Ha YH, Kang SK. Org Lett. 2002;4:1143–1146. doi: 10.1021/ol025557t.Kang SK, Lee SW, Jung J, Lim Y. J Org Chem. 2002;67:4376–4379. doi: 10.1021/jo020017v.Hopkins CD, Malinakova HC. Org Lett. 2004;6:2221–2224. doi: 10.1021/ol0492795.Hopkins CD, Guan L, Malinakova HC. J Org Chem. 2005;70:6848–6862. doi: 10.1021/jo050886v.
- 10.Examples of ruthenium-catalyzed coupling reactions: Trost BM, Pinkerton AB, Seidel M. J Am Chem Soc. 1999;121:10842–10843.Kang SK, Kim KJ, Hong YT. Angew Chem, Int Ed. 2002;41:1584–1586. doi: 10.1002/1521-3773(20020503)41:9<1584::aid-anie1584>3.0.co;2-y.
- 11.Examples of rhodium-catalyzed reductive coupling reactions: Jang HY, Huddleston RR. J Am Chem Soc. 2004;126:4664–4668. doi: 10.1021/ja0316566.Jang HY, Hughes FW, Gong H, Zhang J, Brodbelt JS, Krische MJ. J Am Chem Soc. 2005;127:6174–6175. doi: 10.1021/ja042645v.Kong JR, Cho CW, Krische MJ. J Am Chem Soc. 2005;127:11269–11276. doi: 10.1021/ja051104i.Kong JR, Ngai MY, Krische MJ. J Am Chem Soc. 2006;128:718–719. doi: 10.1021/ja056474l.
- 12.Examples of titanium-catalyzed reductive cyclization of enals: Kablaoui NM, Buchwald SL. J Am Chem Soc. 1995;117:6785–6786.Crowe WE, Rachita MJ. J Am Chem Soc. 1995;117:6787–6788.(c) For a nickel-catalyzed cyclization of enones see ref 17b.
- 13.Examples of radical cyclization: SmI2: Molander GA, McKie JA. J Org Chem. 1994;59:3186–3192.Bu3SnH/PhSiH3: Hays DS, Fu GC. Tetrahedron. 1999;55:8815–8832.Cp2VCl2/Me3SiCl/Zn: Hirao T. Synlett. 1999;2:175–181.t-C12H25SH/AIBN: Yoshikai K, Hayama T, Nishimura K, Yamada KI, Tomioka K. J Org Chem. 2005;70:681–683. doi: 10.1021/jo048275a.
- 14.Observation of oxametallacycle: Titanium: Cohen SA, Bercaw JE. Organometallics. 1985;4:1006–1014.Thorn MG, Hill JE, Waratuke SA, Johnson ES, Fanwick PE, Rothwell IP. J Am Chem Soc. 1997;119:8630–8641.Zirconium: Suzuki N, Rousset CJ, Aoyagi K, Kotora M, Takahashi T, Hasegawa M, Nitto Y, Saburi M. J Organomet Chem. 1994;473:117–128.Rhodium: Godard C, Duckett SB, Parsons S, Perutz RN. Chem Commun. 2003:2332–2333. doi: 10.1039/b306877c.
- 15.Examples of intermolecular coupling of alkenes and aldehydes with stoichiometric transition metals: Titanium: Mizojiri R, Urabe H, Sato F. J Org Chem. 2000;65:6217–6222. doi: 10.1021/jo000925x.Epstein OL, Seo JM, Masalov N, Cha JK. Org Lett. 2005;7:2105–2108. doi: 10.1021/ol050352g.Zirconium: Takahashi T, Suzuki N, Hasegawa M, Nitto Y, Aoyagi KI, Saburi M. Chem Lett. 1992:331–334.
- 16.(a) Cirakovic J, Driver TG, Woerpel KA. J Am Chem Soc. 2002;124:9370–9371. doi: 10.1021/ja020566i. [DOI] [PubMed] [Google Scholar]; (b) Cirakovic J, Driver TG, Woerpel KA. J Org Chem. 2004;69:4007–4012. doi: 10.1021/jo0355505. [DOI] [PubMed] [Google Scholar]
- 17.(a) Ogoshi S, Oka M-a, Kurosawa H. J Am Chem Soc. 2004;126:11802–11803. doi: 10.1021/ja0460716. [DOI] [PubMed] [Google Scholar]; (b) Ogoshi S, Ueta M, Arai T, Kurosawa H. J Am Chem Soc. 2005;127:12810–12811. doi: 10.1021/ja0542486. [DOI] [PubMed] [Google Scholar]
- 18.Examples of highly enantioselective carbonyl-ene reactions: Maruoka K, Hoshino Y, Shirasaka T, Yamamoto H. Tetrahedron Lett. 1988;29:3967–3970.Mikami K, Terada M, Narisawa S, Nakai T. Synlett. 1992:255–265.Evans DA, Burgey CS, Paras NA, Vojkovsky T, Tregay SW. J Am Chem Soc. 1998;120:5824–5825.Evans DA, Tregay SW, Burgey CS, Paras NA, Vojkovsky T. J Am Chem Soc. 2000;122:7936–7943.Yuan Y, Zhang X, Ding K. Angew Chem, Int Ed. 2003;42:5478–5480. doi: 10.1002/anie.200352535.Evans DA, Wu J. J Am Chem Soc. 2005;127:8006–8007. doi: 10.1021/ja0522130.Chiral Pt catalyst: Koh JH, Larsen AO, Gagné MR. Org Lett. 2001;3:1233–1236. doi: 10.1021/ol015702n.Chiral Pd catalyst: Aikawa K, Mikami K. Angew Chem, Int Ed. 2003;42:5458–5461. doi: 10.1002/anie.200352300.Chiral Ni catalyst: Mikami K, Aikawa K. Org Lett. 2002;4:99–101. doi: 10.1021/ol016968x.
- 19.Carbonyl-ene reaction examples that use simple aldehydes: Snider BB, Rodini DJ. Tetrahedron Lett. 1980;21:1815–1818.Snider BB, Rodini DJ, Kirk TC, Cordova R. J Am Chem Soc. 1982;104:555–563.Majewski M, Bantle GW. Synth Comm. 1990;20:2549–2558.Houston TA, Tanaka Y, Koreeda M. J Org Chem. 1993;58:4287–4292.Aggarwal VK, Vennall GP, Davey PN, Newman C. Tetrahedron Lett. 1998;39:1997–2000.Ellis WW, Odenkirk W, Bosnich B. Chem Commun. 1998:1311–1312.Loh T-P, Feng L-C, Yang J-Y. Synthesis. 2002;7:937–940.Pioneering examples with aliphatic aldehydes and monosubstituted alkenes: Snider BB, Phillips GB. J Org Chem. 1983;48:464–469.One isolated example of a carbonyl-ene reaction of an aromatic aldehyde and a monosubstituted alkene has been described (yield not reported): Epifani E, Florio S, Ingrosso G. Tetrahedron. 1988;44:5869–5877.For intramolecular examples of carbonyl-ene reaction between monosubstituted alkenes and sterically demanding aldehydes, see: Andersen NH, Hadley SW, Kelly JD, Bacon ER. J Org Chem. 1985;50:4144–4151.Fujita M, Shindo M, Shishido K. Tetrahedron Lett. 2005;46:1269–1271.
- 20.The stretching frequency (νCO, cm-1) of terminal CO of CpFe(CO)LCOMe (in cyclohexane at room temperature) is a measure of the σ-electron donating ability to a metal center. A less electron donating ligand usually has a higher frequency: Rahman M, Liu H-Y, Eriks K, Prock A, Giering WP. Organometallics. Vol. 8. 1989. pp. 1–7.(b) Tri-p-tolylphosphine (Table 3, entry 5), triphenylphosphine (entry 6), tris-(p-fluoro-phenyl)-phosphine (entry 7) and tris-(p-trifluoromethyl-phenyl)-phosphine (entry 9) have the same cone angle (145°) according to ref 20a.(c) The frequency for (o-anisyl)3P was estimated from (p-anisyl)3P assuming they have similarly electron donating property. Cone angle values and νCO values were obtained from ref. 20a and the following: Tolman CA. Chem Rev. 1977;77:313–348.Otto S. J Chem Crystallogr. 2001;31:185–190.Riihimäki H, Kangas T, Suomalainen P, Reinius HK, Jääskeläinen S, Haukka M, Krause AOI, Pakkanen TA, Pursiainen JT. J Mol Catal A: Chem. 2003;200:81–94.Steinmetz WE. Quant Struct-Act Relat. 1996;15:1–6.
- 21.Application of dicyclohexylferrocenylphosphine as a bulky and electron rich ligand: Ahrendt KA, Bergman RG, Ellman JA. Org Lett. 2003;5:1301–1303. doi: 10.1021/ol034228d.Baillie C, Zhang L, Xiao J. J Org Chem. 2004;69:7779–7782. doi: 10.1021/jo048963u.Thalji RK, Ahrendt KA, Bergman RG, Ellman JA. J Org Chem. 2005;70:6775–6781. doi: 10.1021/jo050757e.Pereira SI, Adrio J, Silva AMS, Carretero JC. J Org Chem. 2005;70:10175–10177. doi: 10.1021/jo051701n.
- 22.A set of four control experiments in which Ni(cod)2, ligand, silyl triflate and the base was each removed from the ethylene–benzaldehyde coupling reaction. No coupling product was detected in any of the four experiments.
- 23.(a) Nickel:phosphine ratio is also important. A 1:2 Ni:phosphine ratio provides a higher yield than a 1:1 Ni:phosphine ratio in the coupling reaction (b) No coupling was observed when Ni(cod)2/Ph3P was replaced with Pd(Ph3P)4.
- 24.Coupling of 3,3-dimethyl-1-butene and benzaldehyde under the standard coupling condition (EtOPh2P, rt, 48h) affording an E-1,2-allylic alcohol product in 14% yield.
- 25.Procedure of the competition experiment: To a solution of Ni(cod)2 (0.1 mmol) and the ligand (Ph3P or (EtO)Ph2P, 0.2 mmol) in toluene (2.5 mL) at 23 °C under Ar were added a monosubstituted alkene (2.5 mmol) , methylenecyclohexane (2.5 mmol), triethylamine (3.0 mmol), p-anisaldehyde (0.5 mmol), and triethylsilyltriflate (0.875 mmol). The mixture was stirred 48 h at room temperature. The yields and ratios were determined by 1H NMR of the crude reaction mixture. Ph3P was the ligand in the reaction between allylbenzene and methylenecyclohexane. (EtO)Ph2P was the ligand in the reaction between 1-octene and methylenecyclohexane.
- 26.As a control experiment, methylenecyclohexane (300 mol%) was coupled with p-anisaldehyde under standard condition (Ni(cod)2, EtOPh2P, Et3SiOTf, Et3N) to give 13% yield of the homoallylic alcohol product. To determine whether the formation of this coupling product requires Ni(cod)2, another control experiment was carried out by stirring methylenecyclohexane, p-anisaldehyde and Et3SiOTf at room temperature. No alkene–aldehyde coupling product was observed.
- 27.A recent review of the nickel-catalyzed hydrovinylation: RajanBabu TV. Chem Rev. 2003;103:2845–2860. doi: 10.1021/cr020040g.Dimerization of ethylene and propylene: Pillai SM, Ravindranathan M, Sivaram S. Chem Rev. 1986;86:353–399.
- 28.Examples of isomerization of olefins by transition-metal hydrides: Nickel: Tolman CA. J Am Chem Soc. 1972;94:2994–2999.Ruthenium: Wakamatsu H, Nishida M, Adachi N, Mori M. J Org Chem. 2000;65:3966–3970. doi: 10.1021/jo9918753.Rhodium: Morrill TC, D’Souza CA. Organometallics. 2003;22:1626–1629.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.