

Abstract
Tauopathies are neurodegenerative diseases in which insoluble fibrillar aggregates of a microtubule-binding protein, Tau, are abnormally accumulated. Pathological Tau fibrils often exhibit structural polymorphisms that differ among phenotypically distinct tauopathies; however, a molecular mechanism to generate polymorphic Tau fibrils remains obscure. Here, we note the formation of a disulfide bond in isoforms of full-length Tau and show that the thiol-disulfide status as well as the isoform composition determines structural and morphological properties of Tau fibrils in vitro. Mainly two regions in a Tau primary sequence are found to act as structural blocks for building a protease-resistant core of Tau fibrils. Interactions among those two blocks for building a core structure depend upon the thiol-disulfide status in each isoform of Tau, which results in the formation of polymorphic fibrils with distinct structural properties. Furthermore, we have found that more diverse structures of Tau fibrils emerge through a cross-seeded fibrillation between heterologous pairs of Tau isoforms. We thus propose that isoform- and disulfide-dependent combinatorial interactions among multiple regions in a Tau sequence endow Tau fibrils with various structures, i.e. polymorphism.
Keywords: Amyloid, Disulfide, Post-translational Modification, Tau, Thiol, Protein Aggregation, Tauopathies
Introduction
In many neurodegenerative diseases, formation of protein fibrillar aggregates has been observed as a major pathological change (1). Although it remains unknown how protein fibrillation is related to degeneration of neurons, there is increasing evidence to show a strong link between the conformation of a protein fibril and the disease phenotype (2–4). In fact, depending upon in vivo pathogenic (5) and in vitro experimental conditions (6), fibrillar structures with various morphologies can be achieved from identical proteins. The molecular basis of the emergence of such a fibril polymorphism will therefore be relevant in understanding a pathomechanism of neurodegenerative diseases.
Tau is one of such proteins that form polymorphic fibrillar structures. Although a physiological function of Tau is to stabilize microtubules (7), formation of insoluble Tau fibrils is a major pathological change in several neurodegenerative diseases called tauopathies (8). Tauopathies include a variety of diseases such as Alzheimer disease (AD),3 Pick disease (PiD), and corticobasal degeneration (CBD). Notably, in each of tauopathies, distinct morphologies of pathological Tau fibrils, such as straight filaments or paired helical filaments (PHFs) of various periodicities and widths, have been observed (9–11); however, a molecular mechanism to generate such polymorphism of insoluble Tau fibrils remains unknown.
Given that there are splicing isoforms in Tau (12), it is possible that each Tau isoform is fibrillized with a distinct morphology. Alternative splicing of Tau mRNA generates two types of “repeat” isoforms, which differ by the presence of either three (3R Tau) or four (4R Tau) tandem pseudo-repeat sequences. A protease-resistant core region in Tau fibrils has been found to include the pseudo-repeat sequences (13); therefore, an additional repeat sequence in 4R Tau that is not present in 3R Tau would alter the protease-resistant core structures and lead to the formation of fibrils with alternative morphology. Indeed, 3R and 4R Tau fibrils prepared in vitro have been reported to exhibit twisted and straight morphologies, respectively (14, 15). In PiD, however, the pathological fibrils composed only of 3R isoforms possess a straight morphology (16). Also, fibrils of 4R Tau in CBD exhibit paired helical as well as straight morphologies (17). Accordingly, we suspect that other structural factors are also involved in diversifying the conformation of Tau fibrils.
One of the factors possibly affecting the fibril structures is a post-translational process. It has been reported that pathological Tau fibrils are often hyperphosphorylated (18) and, in some cases, contain disulfide linkages (19). Although the effects of phosphorylation on the Tau fibrillation still remain controversial (20, 21), formation of a disulfide-linked Tau dimer has been known to facilitate the fibrillation in vitro (14). Despite such extensive studies on fibrillation kinetics of Tau, however, it is yet to be examined whether the structural properties of Tau fibrils are affected by the post-translational modifications. As exemplified by prion diseases, multiple “strains” of neurodegenerative diseases can be distinguished by distinct patterns of neuropathology, each of which seems associated with distinct conformation states of pathogenic proteins (22). A polymorphism in the fibrillar structures of Tau is thus supposed to be relevant in understanding the pathological diversity of tauopathies.
Depending upon the thiol-disulfide status as well as isoform types, we show here that distinct amino acid regions in Tau constitute a protease-resistant core of fibrillar structures. Preformed protein fibrils have been generally known to act as seeds for structural conversion from a soluble protein to the fibrillar state (23), but we have found that such a seeding reaction of Tau proceeds very efficiently only when the isoform type and the thiol-disulfide state are identical between a Tau seed and a seeded soluble Tau. The observed high specificity of a seeding reaction suggests distinct structural properties of Tau fibrils, which would describe the selective aggregation of either 3R or 4R isoform in some tauopathies. Furthermore, albeit inefficiently, a cross-seeding reaction occurred between heterologous states of Tau and produced fibrils with alternative structures. Taken together, we thus propose an isoform- and disulfide-dependent molecular mechanism to generate structural polymorphism in Tau fibrils.
EXPERIMENTAL PROCEDURES
Preparation and Purification of Recombinant Tau Proteins
1N3R and 1N4R Tau cDNA were inserted into a pET15b plasmid vector using NcoI and BamHI, and the six consecutive His residues were directly fused to the C terminus of a protein coding region. Mutations were introduced by a QuikChange mutagenesis. Escherichia coli (Rosetta) transformed with a plasmid was used for overexpression of Tau proteins after induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside at 37 °C for 6 h. After a freeze-thaw cycle, the cells were lysed with a phosphate-buffered saline containing 2% Triton X-100 and a protease inhibitor mixture (Complete, EDTA-free; Roche Diagnostics); then streptomycin sulfate was added to precipitate DNA. After centrifugation, 100 mm NaCl was added to the supernatant, which was heated at 100 °C for 15 min. The precipitate was removed by centrifugation, and the supernatant was loaded on a Proteus Midi IMAC column (Pro-Chem Inc.). The column was washed three times with 50 mm Tris, 500 mm NaCl, 10 mm imidazole, pH 8.0, and Tau proteins were eluted with 50 mm Tris, 100 mm NaCl, 250 mm imidazole, pH 8.0.
Although a disulfide-reduced form of Tau proteins was prepared by treatment with 5 mm DTT and 5 mm EDTA, a disulfide bond was introduced to Tau proteins by incubation with 1 mm H2O2 at room temperature for 20 h. To remove excess chemicals, Tau proteins were then precipitated with 20% trichloroacetic acid, washed with acetone, and redissolved in a pH 7.2 buffer containing 50 mm Tris and 1 mm EDTA. Any insoluble materials were removed by ultracentrifugation at 110,000 × g at 4 °C for 30 min. A Tau protein concentration was determined from the absorption at 280 nm with an extinction coefficient of 7450 cm−1 m−1.
Electrophoresis
To characterize a thiol-disulfide status in Tau proteins, 5 μm Tau was modified with 1 mm (methyl-PEO12)3-PEO4 maleimide (Pierce) in the presence of 2% SDS. After incubation at 37 °C for 30 min, 2 μg of modified Tau was mixed with an SDS-PAGE loading buffer without any reductants, boiled for 5 min, loaded on a 12.5% SDS-PAGE gel, and stained with Coomassie Brilliant Blue.
Tau Fibrillation
Kinetics of Tau fibrillation was monitored by thioflavin T fluorescence using SpectraMax M2 (Molecular Devices). In a 96-well plate, 150 μl of a sample solution containing 10 μm Tau in 50 mm Tris, 1 mm EDTA, 2.5 μm heparin (Sigma; H3393), 16.7 μm thioflavin T, pH 7.2, was set per a well. A fluorescence signal was monitored at 37 °C at intervals of 2 min with 442 and 485 nm of excitation and emission wavelength, respectively. For spontaneous fibrillation, a plate was shaken for 5 s before each fluorescence reading. Instead, when a seeding reaction was examined, a plate was not shaken. Observed fluorescence intensity, F, at time, t, was fit to a sigmoidal function (Equation 1), by which an apparent rate constant, k, a time for formation of half-amounts of aggregates, t½, and a final thioflavin T fluorescence intensity, Ffinal, were estimated.
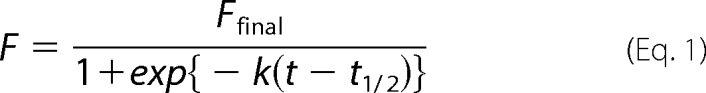 |
After fluorescence intensity reaches a plateau, the pellets were obtained by centrifugation at 110,000 × g for 30 min and used for preparation of seeds by ultrasonication. To estimate a concentration of Tau in seeds, BCA assay (Bio-Rad) as well as SDS-PAGE was performed after seeds were dissolved in 1% SDS.
Morphological observations of Tau fibrils were done using an electron microscope (1200EX; JEOL). The samples were adsorbed on 400-mesh grids coated by a glow-charged supporting membrane and negatively stained with 1% uranyl acetate. The width of a fibrillar structure was measured using Photoshop 7.0 software (Adobe), and 20 different fibrils were examined to estimate errors.
Peptide Mapping Analysis of Tau Fibrils
A fibrillation reaction was performed using 150 μl of 10 μm Tau using SpectraMax M2 as mentioned above. Before and after a fibrillation reaction, sample solutions containing 60 μg of Tau proteins were treated with 1 μg of trypsin (sequencing grade; Promega, V511A) at 37 °C for an hour. After centrifugation at 110,000 × g for 30 min, a pellet was washed once with 50 mm Tris, 1 mm EDTA, pH 7.2, and redissolved in a buffer containing 6 m guanidine hydrochloride and 5 mm DTT. After desalting with a NuTip C-18 (Glygen Co.), α-cyano-4-hydroxycinnamic acid was added as a matrix, and MALDI-MS spectra were acquired using a 4800plus MALDI-TOF/TOF analyzer (Applied Biosystems). Identification of peptides based upon the observed m/z values was performed using a program, MS-Bridge.
RESULTS
A Thiol-disulfide Status in Tau Isoforms Significantly Affects Fibrillation Kinetics
Tau exists as six different isoforms that result from alternative splicing of mRNA (12), and these isoforms are defined by the absence or presence of a 29- or 58-amino acid insert in the N-terminal region (0N, 1N, or 2N, respectively) and the exclusion or inclusion of a 31-amino acid repeat segment in the C-terminal region (3R or 4R, respectively). A longest isoform of Tau (2N4R) possesses two Cys residues, i.e. Cys-291 and Cys-322 (numbering based upon 2N4R isoform), among which Cys-322 exists in all six Tau isoforms, but Cys-291 is present only in four-repeat (4R) isoforms. Accordingly, the three-repeat (1N3R) and four-repeat (1N4R) Tau examined here contain one and two Cys residues, respectively.
Upon oxidation, 1N3R has been shown to form a disulfide-linked dimer, whereas an intra-molecular disulfide between Cys-291 and Cys-322 seems to be favorable in 1N4R Tau (14). To characterize a thiol-disulfide status of our purified Tau proteins, we have utilized the selective reaction of the free thiol groups with (methyl-PEO12)3-PEO4 maleimide (PEG), which adds ∼2.4 kDa per a reactive thiol group and thus retards the mobility of the protein on a denaturing gel. As shown in Fig. 1A (lanes 1 and 5), a DTT-reduced sample of 1N3R Tau (1N3RSH) decreases its electrophoretic mobility upon modification with PEG, although a modification appears not completed, possibly because of the bulkiness of PEG. Similarly, a DTT-reduced sample of 1N4R (1N4RSH) was singly or doubly modified with PEG (Fig. 1A, lanes 3 and 7). These results thus confirm that Cys residues in our 1N3RSH and 1N4RSH proteins are in a free thiol state.
FIGURE 1.

Kinetic effects of disulfide formation on Tau fibrillation. A, an SDS-PAGE analysis on the thiol-disulfide status of Tau proteins. Two μg of Tau proteins were loaded on a 12.5% SDS-PAGE gel (lanes 1–4) with or (lanes 5–8) without a reductant, β-mercaptoethanol. Samples shown in lanes 5–8 were treated with (methyl-PEO12)3-PEO4 maleimide (PEG) before loading on a gel. B and C, kinetics of 1N3R (B) and 1N4R (C) Tau fibrillation monitored by increase in the intensity of ThT fluorescence. A reaction mixture contains 10 μm Tau in a 50 mm Tris buffer, pH 7.2, containing 1 mm EDTA, 16.7 μm ThT, and 2.5 μm heparin. A kinetic trace shown in black represents fibrillation of Tau with disulfide (1N3RS-S in B and 1N4RS-S in C), whereas a fibrillation of Cys-null mutant Tau protein is shown in gray (1N3RCA in B and 1N4RCA in C). The kinetic traces were quantitatively analyzed by a sigmoidal fit (see “Experimental Procedures”), and the fit parameters are summarized in Table 1. Three independent experiments were performed to estimate errors.
When the DTT-reduced Tau samples were treated with hydrogen peroxide (see “Experimental Procedures”), 1N3R efficiently formed a dimer, which disappeared upon treatment with a reductant, β-mercaptoethanol (Fig. 1A, lanes 2 and 6). This result corroborates formation of a disulfide-linked 1N3R dimer (1N3RS-S). In contrast, very small amounts (∼5%, based upon band intensities in an SDS-PAGE gel) of 1N4R formed a β-mercaptoethanol-sensitive disulfide-linked dimer (Fig. 1A, lanes 4 and 8); instead, upon oxidation, 1N4R remains monomeric but becomes no longer modified with PEG, which is consistent with the presence of an intramolecular disulfide bond in oxidized 1N4R (1N4RS-S). Hydrogen peroxide can oxidize several amino acids (e.g. Tyr and His) other than Cys in proteins and also sometimes produce higher oxidation state such as sulfonic acid. We, however, detected no such oxidized products by mass spectrometric analysis on the tryptic fragments of hydrogen peroxide-treated 1N3R and 1N4R (see below).
To test the effects of disulfide formation on a fibrillation process of Tau proteins, heparin-induced Tau aggregation was examined (15). Sulfated glycosaminoglycans such as heparin have been known to stimulate Tau fibrillation in vitro and co-localize with Tau aggregates in the affected neurons of AD brains, supporting a relevance of the heparin/Tau interaction in the pathogenesis of tauopathies. Fibrillation of Tau was monitored by a fluorescence increase of thioflavin T (ThT), which fluoresces upon specific binding with amyloid-like fibrillar aggregates (24). As a model of disulfide-reduced Tau proteins, furthermore, we have used Tau proteins with C291A/C322A mutations (1N3RCA and 1N4RCA) in this study; thereby, possible air oxidation of thiol groups can be ignored during a fibrillation process. As shown in Fig. 1 (B and C), we have found that, in both 1N3R and 1N4R, spontaneous aggregation was significantly accelerated in its disulfide form. Indeed, when these kinetic traces were fit with a sigmoidal function (see “Experimental Procedures”), shorter t½ was obtained in 1N3RS-S and 1N4RS-S than 1N3RCA and 1N4RCA, respectively (Table 1), where t½ is the time required for gaining 50% of the final fluorescence intensity. Although an apparent rate constant (k) in the growth phase of a sigmoidal fit is similar (Table 1), a fluorescence increase occurred after an initial lag time in 1N3RCA and 1N4RCA (until approximately 500 min) but was almost instantaneous in 1N3RS-S and 1N4RS-S (Fig. 1, B and C). We have further confirmed by SDS-PAGE analysis using PEG that the disulfide bond was not reduced in the final aggregates (supplemental Fig. S1). Simple interpretation of these results is that disulfide formation accelerates the conformational conversion of Tau into insoluble fibrillar aggregates. Nonetheless, we have further noted a distinct intensity of ThT fluorescence among four different Tau aggregates examined here (i.e. 1N3RCA, 1N3RS-S, 1N4RCA, and 1N4RS-S). When the fluorescence intensity change reached a plateau, fluorescence intensity of 1N3R aggregates was 1 order of magnitude higher than that of 1N4R aggregates (Ffinal in Table 1 and Fig. 1, B and C). Sample solutions were then separated by ultracentrifugation into supernatant and pellets, and an SDS-PAGE analysis of each fraction has confirmed that all four Tau variants become mostly insoluble upon aggregation (supplemental Fig. S2A). Among the four Tau variants tested here, different amounts of Tau remained soluble, which is, however, not correlated to the intensity of ThT fluorescence. These results imply that the difference in ThT fluorescence intensity is not caused simply by heterogeneity in the protein concentrations. Although the detailed interaction of ThT with protein aggregates remains unclear in general, the four Tau aggregates would possess different structural properties that affect interactions with a ThT molecule.
TABLE 1.
Kinetic parameters of spontaneous Tau fibrillation
Kinetic traces of Tau fibrillation are shown in Fig. 1 (B and C) and fitted to the sigmoidal function (see Equation 1). Errors are estimated by three independent experiments.
| Tau isoform | Ffinal | t½ | k |
|---|---|---|---|
| min | min−1 | ||
| 1N3RCA | (2.23 ± 0.12) × 103 | (7.39 ± 0.92) × 102 | (8.24 ± 0.44) × 10−3 |
| 1N3RS-S | (3.61 ± 0.14) × 103 | (1.17 ± 0.03) × 102 | (1.25 ± 0.09) × 10−2 |
| 1N4RCA | (5.19 ± 1.13) × 102 | (6.55 ± 0.29) × 102 | (1.15 ± 0.06) × 10−2 |
| 1N4RS-S | (4.07 ± 0.31) × 102 | (3.89 ± 0.76) × 102 | (8.14 ± 0.50) × 10−3 |
Thiol-Disulfide Status as Well as Isoform Type Is a Factor Generating Polymorphism of Tau Fibrils
To test whether the thiol-disulfide status in 1N3R and 1N4R Tau impacts structural properties of fibrils, we have first compared morphologies of Tau fibrils by an electron microscopic observation and found distinct morphologies that are dependent upon thiol-disulfide status and isoform composition (Fig. 2). As shown in Fig. 2A, 1N3RCA forms twisted fibrils with a width that varied between 4.8 ± 0.5 and 13.4 ± 1.1 nm and a half-periodicity of 60–70 nm. In contrast, a 1N3RS-S fibril was thicker in its width (16.2 ± 2.0 nm) than that of a 1N3RCA fibril and was also notably shorter (at most 0.3 μm) compared with the other fibrils (well over 1 μm) (Fig. 2B). Because it was of such a fragmented character, it was difficult to tell whether 1N3RS-S fibrils are twisted or straight. Like 1N3RCA fibrils, four-repeat Tau isoforms, 1N4RCA and 1N4RS-S, formed significantly long (>1 μm) fibrils but exhibited less twisted characters; namely, some helical (or zigzag-like) appearance seems to exist, but the cross-over points were difficult to define (Fig. 2, C and D). It is also notable that 1N4RS-S forms a slightly thinner fibril (8.5 ± 0.8 nm) than that of 1N4RCA (9.4 ± 1.2 nm). Thiol-disulfide status is thus relevant in determining the fibril morphologies of each Tau isoform. However, it remains unclear how intramolecular/intermolecular interactions in Tau proteins are realized to produce polymorphism of Tau fibrils. As we have recently shown in Cu,Zn-superoxide dismutase (SOD1) (25), variable regions in a protein molecule can constitute a core of the fibril structure, which further affects the morphologies of fibrils. Depending upon the thiol-disulfide status as well as isoform type, therefore, we suspect that Tau uses different amino acid regions for constructing a core structure upon its fibrillation.
FIGURE 2.

Morphologies of Tau fibrils observed by transmission electron microscopy. A, 1N3RCA fibrils. B, 1N3RS-S fibrils. C, 1N4RCA fibrils. D, 1N4RS-S fibrils. An average value of fibril widths is indicated, and a magnified image (136 × 680 nm) of a representative Tau fibril is also shown on the right side of each panel. White arrowheads indicate cross-over points of Tau fibrils with helical morphologies.
Disulfide Formation Alters Protease-resistant Core Structures of Tau Fibrils
Protein fibrillar aggregates are generally composed of a protease-resistant “core” and the associated “fuzzy coat” that is a susceptible region for proteolysis (13, 26). In Tau, the microtubule-binding region containing three or four pseudo-repeats (R1–R4 in Fig. 3) has been proposed to constitute a structural core of fibrils (13, 27, 28). Furthermore, two hexapeptide motifs, 275VQIINK280 in R2 and 306VQIVYK311 in R3 (Fig. 3), play a critical role in assembly of the core of Tau fibrils by forming a β-sheet structure (29). Given that both Cys-291 and Cys-322 are within the pseudo-repeat regions, therefore, it is possible that the thiol-disulfide status modulates the interaction in the fibril core region. We thus attempted to identify the region(s) of an amino acid sequence that constitutes the structural core of our Tau fibrils. For that purpose, aggregates were first treated with a protease, trypsin. Following ultracentrifugation, the remaining insoluble pellets, which correspond to a trypsin-resistant core of Tau fibrils, were resolubilized in 6 m guanidine hydrochloride and reduced with 5 mm DTT. Mass peaks were then obtained by MALDI-TOF mass spectrometry; however, no mass peaks were observed when the same experimental procedures were applied to soluble Tau proteins (supplemental Fig. S3). Based upon the observed mass, the peptides covering trypsin-resistant cores were identified and mapped on a Tau primary sequence (supplemental Table S1 and Fig. 3). In all four Tau aggregates examined here (i.e. 1N3RCA/S-S and 1N4RCA/S-S), multiple trypsin-resistant peptides were identified that cover either VQIINK in R2, VQIVYK in R3, or both (Fig. 3). R2 and/or R3 regions are therefore considered to be commonly involved as a structural core in all four Tau fibrils, and this result also supports previous findings (29) on the key role of the hexapeptide motifs (VQIINK and VQIVYK) in formation of in vitro Tau fibrils. Furthermore, Wischik and co-workers (13, 27, 28) have previously prepared the protease-resistant fragments from the AD-associated PHFs of Tau, and N-terminal sequencing of some of those fragments has shown the involvement of R2 and/or R3 in the protease-resistant core structures of pathological fibrils. Taken together, therefore, abnormal interactions involving R2 and R3 regions are considered to be essential to the formation of insoluble Tau fibrils both in vitro and in vivo.
FIGURE 3.

Protease-resistant core regions of Tau fibrils. Trypsin treatment of 1N3RCA and 1N3RS-S fibrils (top panel) and 1N4RCA and 1N4RS-S fibrils (bottom panel) produces several peptides, which were identified by MALDI-TOF mass spectrometry (see Fig. S3). The trypsin-resistant regions analyzed from the observed mass peaks (Fig. S3) are shown as bars and mapped on the primary sequence of 1N3R or 1N4R Tau (shown as open bars). Details are also summarized in supplemental Table S1. The numbers indicated above the primary sequence of 1N3R (top panel) or 1N4R (bottom panel) Tau represent the amino residue numbers of the longest Tau isoform. The 1N3R isoform (shown in the top panel) lacks the R2 region (near positions 280–300), and the N-terminal region (near positions 80–100) is also absent in both 1N3R and 1N4R isoforms. The positions of hexapeptide motifs, VQIINK and VQIVYK, as well as two Cys residues are also indicated.
Notably, a C-terminal region (positions 400–447) of both 1N3R and 1N4R Tau was involved in the protease-resistant core when a disulfide is absent (Fig. 3). This is in sharp contrast to the findings that, in the presence of a disulfide linkage, regions outside the pseudo-repeats did not contribute to the core structures of both 1N3R and 1N4R fibrils (except one C-terminal peptide covering the outside of pseudo-repeats in 1N4RS-S; Fig. 3). Depending upon thiol-disulfide status, therefore, each Tau isoform adopts insoluble polymorphic fibrils with a distinct protease-resistant core.
A Cross-seeding Assay Reveals Structural Diversity of Tau Fibrils
Protein fibrils have been well known to function as a structural template or “seed” that facilitates fibrillation of a soluble protein yet unaggregated (23). In general, this seeding reaction occurs very efficiently, when the seed-constituent protein structurally matches with the seeded soluble proteins. We thus expect that the seeding between different pairs of proteins (called “cross-seeding”) measures how distinct the fibril structures are.
To highlight seeding phenomena of Tau fibrils, fibrillation reactions were examined under nonagitating conditions in the presence of heparin, where spontaneous fibrillation was significantly decelerated in 1N3RCA, 1N4RCA, and 1N4RS-S but remained efficient in 1N3RS-S (within at least 1,200 min, a black trace in each panel of Fig. 4). Indeed, without sample agitation, significant amounts of Tau variants except 1N3RS-S remained soluble after incubation for 24 h (supplemental Fig. S2B). Upon addition of the preformed Tau fibrils, in contrast, the corresponding Tau under nonagitating conditions exhibited instantaneous increase in the intensity of ThT fluorescence (Fig. 4) and became almost completely insoluble (supplemental Fig. S2C), consistent with self-seeding reactions (in 1N3RCA, 1N4RCA, and 1N4RS-S). Even in the absence of sample agitation, however, 1N3RS-S instantaneously fibrillized (Fig. 4B) and was found in the pellet fraction (supplemental Fig. S2B), which hampered characterization of self-seeding effects on 1N3RS-S fibrillation. We could inhibit spontaneous fibrillation of Tau proteins by omitting heparin from solutions, but successful seeding reactions were also found to require the presence of heparin (30) (supplemental Fig. S2D). Accordingly, seeding effects on 1N3RS-S fibrillation will be difficult to characterize, and we have thus focused on seeding reactions of 1N3RCA, 1N4RCA, and 1N4RS-S.
FIGURE 4.

A seeding reaction of Tau fibrils monitored by ThT fluorescence intensity. 10 μm Tau of 1N3RCA (A), 1N3RS-S (B), 1N4RCA (C), and 1N4RS-S (D) was prepared in a 50 mm Tris buffer, pH 7.2, containing 1 mm EDTA, 2.5 μm heparin, and 16.7 μm ThT, and fluorescence increase was monitored in the absence of sample agitation. A black trace shown in each panel represents the kinetics in the absence of any added seeds. Seeding reactions were examined by the addition of aggregates of 1N3RCA (blue), 1N3RS-S (red), 1N4RCA (green), and 1N4RS-S (purple) in soluble Tau proteins (A–D). Amounts of added aggregates were 0.5 μm (monomer-base), which corresponds to 5% of the amounts of soluble Tau. The kinetic traces were quantitatively analyzed by a sigmoidal fit (see “Experimental Procedures”), and the fit parameters are summarized in Table 2. Three independent experiments were performed to estimate errors.
It is also important to note that ThT fluorescence intensity of self-seeded 3R fibrils was at most twice higher than that of self-seeded 4R fibrils (Fig. 4, A and B versus C and D). This is somewhat contrasted with the observation in spontaneous fibrils, which exhibited approximately an order of magnitude difference in the ThT fluorescence intensity between 3R and 4R Tau (Fig. 1, B and C). Compared with the spontaneous fibrillation, self-seeding reactions resulted in 1.5-, 3-, and 5-fold increases in the intensity of plateau ThT fluorescence in 1N3RCA, 1N4RCA, and 1N4RS-S Tau, respectively (compare Ffinal values in Tables 1 and 2). We have thus checked the extent of aggregation by an SDS-PAGE analysis after ultracentrifugal fractionation of Tau solutions into supernatant and pellets, and seeding reactions were found to somewhat increase fractions of pelletable Tau (supplemental Fig. S2, A and C); in particular, ∼25% amounts of 1N3RCA and 1N4RS-S remained soluble in the spontaneous fibrillation (supplemental Fig. S2A), but self-seeding reactions of 1N3RCA and 1N4RS-S resulted in almost complete insolubilization (supplemental Fig. S2C). Self-seeding reactions are thus considered to increase amounts of pelletable Tau fibrils, which may contribute to the altered intensity of ThT fluorescence between spontaneously aggregated and seeded Tau solutions. Nonetheless, the difference in fluorescence intensity between spontaneously aggregated and seeded Tau solutions, in particular 1N4RCA and 1N4RS-S, could not be described solely by the difference in extent of aggregation; therefore, such a difference in the ThT fluorescence intensity may indicate that each of the spontaneous and self-seeded fibrils possesses distinct structural properties. Despite this, we would like to emphasize kinetic effects of seeding on the fibrillation of 1N3RCA, 1N4RCA, and 1N4RS-S; namely, rate constants (k in Equation 1) of a growth phase in the seeding reaction (Table 2) will reveal distinct properties of Tau seeds.
TABLE 2.
Kinetic parameters of seeded Tau fibrillation
Kinetic traces of Tau fibrillation are shown in Fig. 4 and fitted to the sigmoidal function (see Equation 1). Errors are estimated by three independent experiments.
| Soluble Tau | Seeds | Ffinal | t½ | k |
|---|---|---|---|---|
| min | min−1 | |||
| 1N3RCA | 1N3RCA | (3.21 ± 0.10) × 103 | (1.26 ± 0.05) × 101 | (9.41 ± 0.51) × 10−2 |
| 1N3RS-S | (2.68 ± 0.06) × 103 | (2.23 ± 0.06) × 102 | (6.09 ± 0.10) × 10−3 | |
| 1N4RCA | N.D.a | N.D.a | N.D.a | |
| 1N4RS-S | N.D.a | N.D.a | N.D.a | |
| 1N4RCA | 1N3RCA | (1.90 ± 0.06) × 103 | (3.09 ± 0.03) × 102 | (3.27 ± 0.02) × 10−3 |
| 1N3RS-S | (5.00 ± 0.09) × 103 | (7.07 ± 0.06) × 102 | (5.50 ± 0.09) × 10−3 | |
| 1N4RCA | (1.51 ± 0.03) × 103 | (1.83 ± 0.09) × 100 | (2.35 ± 0.03) × 10−1 | |
| 1N4RS-S | (2.54 ± 0.11) × 103 | (3.77 ± 0.12) × 102 | (6.82 ± 0.17) × 10−3 | |
| 1N4RS-S | 1N3RCA | (1.01 ± 0.05) × 103 | (2.62 ± 0.29) × 102 | (5.06 ± 0.41) × 10−3 |
| 1N3RS-S | (7.53 ± 0.22) × 102 | (1.76 ± 0.26) × 102 | (3.54 ± 0.30) × 10−3 | |
| 1N4RCA | (5.17 ± 0.22) × 102 | (3.26 ± 0.13) × 102 | (4.05 ± 0.14) × 10−3 | |
| 1N4RS-S | (2.17 ± 0.03) × 103 | (1.17 ± 0.01) × 101 | (1.10 ± 0.01) × 10−1 |
a Not determined. A seeding reaction occurs very slowly within the observed time scale (also see Fig. 4A).
We then tested cross-seeding reactions in a heterologous pair of Tau variants under nonagitating conditions. In the fibrillation of 1N3RCA (Fig. 4A), for example, the addition of 1N3RS-S fibrils accelerated formation of ThT-positive 1N3RCA fibrils (red trace), but such a cross-seeding reaction was not as rapid as a self-seeding reaction by the 1N3RCA fibril seeds (blue trace). Moreover, both 1N4RCA and 1N4RS-S fibrils were very inefficient seeds for facilitating 1N3RCA fibrillation (green and purple traces in Fig. 4A). By fitting kinetic traces of seeded fibrillation with a sigmoidal curve (Equation 1), a self-seeding reaction of 1N3RCA by 1N3RCA fibrils has been quantitatively confirmed to efficiently proceed with shorter t½ and larger k than that by 1N3RS-S fibrils (Table 2). A variable seeding activity among the four types of Tau fibrils was also evident in fibrillation of 1N4RCA and 1N4RS-S (Fig. 4, C and D, and Table 2). A seeding reaction was found to proceed very rapidly (t½ = ∼10 min, k = ∼10−1 min−1; Table 2), when a pair of seed and seeded soluble protein is in the exactly same state including its thiol-disulfide status and isoform type (self-seeding). Between different pairs of a seed and a seeded soluble protein (cross-seeding), in contrast, a seeding reaction was relatively slow (t½ = ∼200 min, k = ∼10−3 min−1; Table 2). Accordingly, a seeding assay again supports our proposal that distinct properties are realized among four types of Tau fibrils with different thiol-disulfide status and different isoforms. In Fig. 4, it should also be noted that a cross-seeding reaction did occur between different states of Tau proteins, albeit inefficiently. A cross-seeding between a heterologous set of Tau may therefore produce alternative properties of seeded Tau fibrils that are adapted to the seed structure.
Alternative Fibril Species Emerges through a Cross-seeding Reaction
As shown in Fig. 4A (green trace), a 1N4RCA fibril functions less efficiently as a seed for 1N3RCA fibrillation. Once 1N4RCA acquires a fibril structure adapted to that of a 1N3RCA fibril through a cross-seeding reaction (Fig. 4C, blue trace), however, we expected that, compared with an original 1N4RCA fibril, such a 1N3RCA-seeded 1N4RCA fibril exhibits higher seeding efficiency for 1N3RCA fibrillation. To test this idea, 1N4RCA fibrillation was first cross-seeded by adding 5% amounts of 1N3RCA fibrils (Fig. 4C, blue trace), and the resultant insoluble fibrils were obtained by ultracentrifugation. To reduce amounts of original 1N3RCA fibril in seeds, we then repeated a cross-seeding reaction; namely, 1N4RCA fibrillation was again cross-seeded by adding 5% amounts of 1N3RCA-seeded 1N4RCA fibrils. The resultant insoluble fibrils were recovered by ultracentrifugation and called 1N4RCA(3RCA) fibrils here. Then 5% amounts of 1N4RCA(3RCA) fibrils were added to soluble 1N3RCA Tau as seeds. As shown in Fig. 5A (red trace), 1N4RCA(3RCA) fibrils now function very efficiently as seeds for 1N3RCA fibrillation to the extent that cross-seeded 1N3RCA fibrillation occurs instantaneously. This was not observed by adding 1N4RCA seeds (Fig. 5A, green trace, which is the same trace shown in Fig. 4A) and also not caused by the residual 1N3RCA seeds, given that 1.25/100% amounts of 1N3RCA seeds (theoretical amounts of residual 1N3RCA in 1N4RCA(3RCA) seeds) were too small to trigger 1N3RCA fibrillation within the observed time (Fig. 5A, purple trace). Similarly, a 1N4RCA-seeded 1N3RCA fibril (1N3RCA(4RCA) fibril) exhibited higher seeding efficiency for 1N4RCA fibrillation than a 1N3RCA fibril (Fig. 5B). These results thus suggest that structural adaptation occurs during a cross-seeding between heterologous pairs of Tau proteins.
FIGURE 5.

Tau fibrils with alternative structural properties are emerged by a seeding reaction. A, a seeding reaction of 10 μm 1N3RCA in a 50 mm Tris buffer, pH 7.2, containing 1 mm EDTA, 2.5 μm heparin, and 16.7 μm ThT is monitored by a fluorescence increase of ThT in the absence of sample agitation. A kinetic trace in the absence of added seeds was also shown (black). As also shown in Fig. 4A, the addition of 0.5 μm 1N3RCA aggregates to soluble 1N3RCA significantly accelerates the fibrillation (blue trace). Although 1N4RCA aggregates (0.5 μm, green trace) did not efficiently function as seeds, 0.5 μm 1N4RCA(3RCA) aggregates significantly accelerated the fibrillation of 1N3RCA (red trace). This is not due to the residual 1N3RCA aggregates in the 1N4RCA(3RCA) aggregates, which was evidenced by an inefficient seeding of 1N3RCA fibrillation by adding 1.25 × 10−3 μm (1.25/100% amounts of soluble 1N3RCA) 1N3RCA aggregates (purple trace). B, a seeding reaction of 10 μm 1N4RCA in the conditions that are the same with those in A. Seeds added are as follows: 1N3RCA aggregates (0.5 μm, blue trace), 1N4RCA (0.5 μm, green trace; 1.25 × 10−3 μm, purple trace) and 1N3RCA(4RCA) aggregates (0.5 μm, red trace). A spontaneous fibrillation of 1N4RCA in the absence of agitation is also shown as a black trace. C and D, electron micrographs of cross-seeded Tau fibrils, 1N4RCA(3RCA) (C) and 1N3RCA(4RCA) (D). An average value of fibril widths is indicated, and a magnified image (136 × 680 nm) of a representative Tau fibril is also shown on the right side of each panel. White arrowheads indicate cross-over points of Tau fibrils with helical morphologies.
Indeed, we have found that 1N4RCA(3RCA) fibrils exhibit alternative twisted morphologies with a width that varied between 7.1 ± 1.1 and 15.6 ± 1.3 nm (Fig. 5C). This is in contrast to the less twisted morphology of a 1N4RCA fibril spontaneously formed without any seeds (Fig. 2C). Given the clearly twisted morphologies of 1N3RCA fibrils (Fig. 2A), a cross-seeding reaction is considered to transmit the structural properties of 1N3RCA seeds to 1N4RCA, which results in the formation of twisted 1N4RCA(3RCA) fibrils. It is also notable that 1N3RCA forms alternative straight fibrils when cross-seeded with 1N4RCA seeds (Fig. 5D). Based upon these results, therefore, we have shown that Tau fibrils display diverse sets of structures dependent upon thiol-disulfide status and isoform types and further proposed that more diverse fibril structures can be realized through a cross-seeding reaction.
DISCUSSION
Tauopathies are neurodegenerative diseases in which insoluble Tau fibrils are abnormally accumulated (8). In phenotypically distinct tauopathies, a different ratio of three- and four-repeat Tau isoforms has been observed in the pathological inclusions; more interestingly, insoluble Tau fibrils constituting those inclusions exhibit a variety of morphologies (9–11). Given that structural diversity of protein fibrils appears to regulate phenotypes in yeast prions (3) and also in several neurodegenerative diseases (2, 5), polymorphism of Tau fibrils may have important pathological roles in tauopathies. However, it remains to be established how such a polymorphism emerges in insoluble Tau fibrils. Here, we have performed morphological, structural, and biochemical analysis on fibrils of full-length Tau proteins and proposed a disulfide- and isoform-dependent mechanism to produce Tau fibrils with distinct molecular properties.
A Thiol-disulfide Status in Both Repeat Isoforms of Full-length Tau Impacts Fibrillation Kinetics
From the 1990s, kinetic effects of disulfide formation on Tau fibrillation in vitro have been studied by using several truncated Tau peptides comprised of pseudo-repeats and also using several full-length Tau isoforms such as 0N3R (also called htau23) and 2N4R (also called htau40) (14, 31–33). Based upon those previous studies, formation of a disulfide-linked three-repeat Tau dimer has been shown to significantly accelerate fibrillation kinetics, whereas a disulfide-linked monomer of four-repeat Tau (often called a “compact monomer”) has been regarded as a fibrillation-incompetent species. To our knowledge, however, limited numbers of studies have systematically examined important structural as well as kinetic roles of thiol-disulfide status in the fibrillation process of Tau.
We have found here that introduction of a disulfide linkage accelerates fibrillation kinetics in both 1N3R and 1N4R isoforms (Fig. 1, B and C). Acceleration of 1N3R fibrillation by intermolecular disulfide formation is completely consistent with the previous results (14), whereas apparent contradiction on 1N4R fibrillation kinetics between previous (14) and our current studies remains to be solved. Unfortunately, however, the disulfide formation in four-repeat isoforms has not been fully characterized in previous studies. Our thiol-disulfide assay using SDS-PAGE has revealed that oxidation of 1N4R Tau largely produces an intramolecular disulfide linkage but also forms a disulfide-linked dimer in small amounts (Fig. 1A). Possible formation of an intermolecular disulfide in a four-repeat isoform appears unnoticed in previous studies; however, even small amounts of such a disulfide-linked 1N4R dimer would rapidly form aggregates (31), which could then act as seeds to facilitate fibrillation of soluble 1N4RS-S. By using gel filtration chromatography, we attempted to separate a compact monomer from disulfide-linked dimer of 1N4R Tau, which, however, failed because of insufficient separation of monomer and dimer elution peaks. As we have shown in Fig. 1, B and C, and Table 1, alternatively, the relatively low intensity of ThT fluorescence may hamper the detection of 1N4RS-S fibrillation in the previous studies. Because the intramolecular disulfide bond remains intact in our 1N4RS-S fibrils (supplemental Fig. S1), one of our findings here is that a four-repeat Tau with an intramolecular disulfide linkage is not a fibrillation-incompetent species but is able to form insoluble fibrils; in other words, both repeat isoforms of Tau are capable of adopting insoluble fibrillar structures regardless of its thiol-disulfide status.
A Molecular Mechanism to Produce Tau Fibrils with Distinct Structural Properties
In addition to the propensity for fibrillation, we have also successfully reproduced distinct morphologies of Tau fibrils that are dependent upon both isoform compositions and thiol-disulfide status (Fig. 2). Several molecular mechanisms have so far attempted to describe morphological differences in in vitro Tau fibrils, in which variations of the interactions among hexapeptide motifs (VQIINK and VQIVYK) play important roles (34, 35). It is also notable that R2 and R3 regions containing those motifs constitute a protease-resistant core in pathological Tau fibrils, PHFs, purified from AD brain (13, 27, 28). As expected, we have also identified the repeat regions including those hexapeptide motifs as being protease-resistant in all Tau fibrils examined here (Fig. 3); therefore, the R2 region containing VQIINK motif that is specific to the four-repeat isoform may add alternative interactions in a protease-resistant core structure, resulting in different morphologies between 1N3R and 1N4R fibrils.
In addition to the hexapeptide motifs in the repeat region, our results further suggest the relevance of a C-terminal region in the formation of a protease-resistant core of disulfide-reduced Tau fibrils (Fig. 3). Given that the protease-resistant peptides of PHFs were analyzed by an N-terminal sequencing in previous studies (28), it remains obscure whether a C-terminal region of Tau is involved in the core structures of pathological PHFs. It is, however, notable that the C-terminal peptide spanning from Ser-409 to Lys-438 has previously been shown to possess a fibril-forming propensity (36). In a soluble state of Tau, furthermore, the C-terminal region (Ser-422 to Leu-441) has been proposed to assume an α-helical structure and interact with a part of the repeat region (Lys-321 to Lys-375) (37). Because Cys-322, which exists in all Tau isoforms, is in the repeat region, we speculate that inter- and intramolecular disulfide formation somehow hampers the interaction of the C-terminal region with the repeats and favors such conformations that preclude the C-terminal region from the protease-resistant core upon fibrillation (Fig. 6, left panel). When both Cys residues are in a free thiol state, the C-terminal region would be allowed to interact with the repeat region and thereby recruited to the protease-resistant core of fibrils (Fig. 6, right panel). The mechanism proposed here is still speculative and needs more fine-tuning; for example, an exact arrangement of Cys residues in a fibril core remains totally unknown. Despite this, our experimental results here would support our proposals that the two regions within a Tau sequence (i.e. a repeat region and a C-terminal region) function as main building blocks for the formation of a fibril core and that the thiol-disulfide status in Tau affects the interactions among those building blocks in the core structures.
FIGURE 6.

A proposed mechanism to generate structural polymorphism of Tau fibrils. Three- and four-repeat Tau isoforms are schematically represented together with pseudo-repeats (R1, R2, R3, and R4) and a C-terminal region highlighted as boxes. N and C indicate the N and C termini, respectively. In both 1N3R and 1N4R isoforms, a disulfide-reduced state is assumed to allow the C-terminal region to interact with the repeat region (1N3RSH and 1N4RSH), although such interactions may be precluded by formation of either intermolecular (1N3R) or intramolecular (1N4R) disulfide (1N3RS-S and 1N4RS-S). An exact alignment of Tau molecules in a fibril remains unknown; therefore, alignment/orientation of boxes and Cys residues in the schematic representation is still speculative. Despite this, our results support that a C-terminal region is involved in the core of disulfide-reduced Tau (right side) but not in that of disulfide-bonded Tau (left side). Protease-resistant core regions (Fig. 3) are colored gray and also highlighted by dotted lines.
Implications for Tauopathies
Depending upon subtypes of tauopathies, the observed Tau fibrils have been known to exhibit distinct ultrastructural characteristics (8). For example, AD is pathologically characterized by the formation of PHFs, which are composed of two strands of the 3R/4R Tau filament twisted around one another with a periodicity of 80 nm and a width varying from 8 to 20 nm (8). In PiD, insoluble inclusions called Pick bodies are composed of straight 3R Tau fibrils with 12 nm of the width and PHF-like twisted 3R Tau fibrils with 16 nm of the maximum width (10). Straight and PHF-like twisted fibrils are also observed in CBD with (maximum) widths of 15 and 29 nm, respectively; however, unlike PiD, the fibril component is selectively a 4R Tau isoform (10). Although morphologies of our in vitro Tau fibrils were found to depend upon the isoform-type and the thiol-disulfide status of Tau, it remains elusive which state(s) of Tau is responsible for the formation of pathological fibrils in each subtype of tauopathies. Despite this, it is notable that the widths of our in vitro fibrils range from 8.5 to 16.2 nm, which are comparable with those of pathological fibrils and also those of in vitro fibrils previously reported (14).
We have further supposed that isoform- and disulfide-dependent core structures of Tau fibrils play key roles in describing the pathological diversity of tauopathies. As mentioned above, selective aggregation of either three- or four-repeat Tau has been observed in some tauopathies such as PiD and CBD (8). It still remains an open question why a specific isoform(s) of Tau constitutes pathological inclusions in a disease-dependent manner. Based upon our seeding assay, nonetheless, a high specificity in a seeding fibrillation between homologous states of Tau (Fig. 4) would result in selective propagation of fibrils of a specific Tau isoform. Interestingly, a seeding reaction with high specificity has been proposed to describe a selective deposition of a pathogenic P301L mutant Tau in vivo (30). Fibrils of P301L mutant Tau would be structurally different from those of wild-type Tau and therefore did not act as a seed to facilitate the fibrillation of wild-type Tau (30). Furthermore, a seeded aggregation of Tau in cultured cells has been shown to efficiently occur only between a pair of the same repeat isoforms (38). In our current study, a seeding reaction proceeds very efficiently only when both the isoform composition and the thiol-disulfide status of the soluble Tau are identical to those of the added seeds (Fig. 4). Although it needs to be further characterized whether pathogenic conditions disturb the thiol-disulfide equilibrium in Tau (19), core structures of Tau fibrils in vitro are discriminated by mutations, isoform types, and thiol-disulfide status, all of which would be relevant in describing selective aggregation of specific Tau species in vivo. It is also important to note that a conformation of soluble Tau can be adapted to that of heterologous seeds through a cross-seeding reaction to produce fibrils with alternative structural and morphological properties (Fig. 5). Based upon these in vitro data, therefore, such conformational plasticity of Tau would contribute to diversify the pathologies of tauopathies.
Recently, we have reported using another protein, SOD1, that a mutation-dependent polymorphism of SOD1 fibrils is described by distinct combinations of interactions among three independent regions in an SOD1 sequence (25). A distinct core structure of Tau fibrils would also be realized by pathogenic mutations (e.g. P301L) in Tau (30, 39), and importantly, as we have found here, formation of a disulfide bond in both Tau isoforms produces an alternative fibril core structure. Taken together, therefore, we propose a general mechanism producing fibril polymorphism, in which multiple regions in a protein sequence act as blocks for building a core structure of fibril; and then, mutation- and modification-dependent combinatorial diversity of the interactions among those building blocks introduces polymorphism in protein fibrils.
Supplementary Material
Acknowledgments
We are grateful to Prof. Yasuo Ihara (Doshisha University) for the gift of cDNAs of human Tau isoforms. We also thank the Support Unit for Bio-material Analysis, RIKEN BSI Research Resources Center, especially Masaya Usui and Kaori Otsuki for mass analysis and Yuriko Sakamaki for electron micrographic observations.
This work was supported by Grants-in-Aid 22240037 (to N. N.), 22110004 for Scientific Research on Innovative Areas (to N. N.), and 22770162 (to Y. F.), and the Strategic Research Program for Brain Science (to N. N.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by funds from the Uehara Memorial Foundation (to Y. F.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S3.
- AD
- Alzheimer disease
- PiD
- Pick disease
- CBD
- corticobasal degeneration
- PHF
- paired helical filament
- ThT
- thioflavin T
- SOD1
- Cu,Zn-superoxide dismutase.
REFERENCES
- 1. Chiti F., Dobson C. M. (2006) Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 2. Angers R. C., Kang H. E., Napier D., Browning S., Seward T., Mathiason C., Balachandran A., McKenzie D., Castilla J., Soto C., Jewell J., Graham C., Hoover E. A., Telling G. C. (2010) Science 328, 1154–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanaka M., Chien P., Naber N., Cooke R., Weissman J. S. (2004) Nature 428, 323–328 [DOI] [PubMed] [Google Scholar]
- 4. Westermark G. T., Westermark P. (2009) FEBS Lett. 583, 2685–2690 [DOI] [PubMed] [Google Scholar]
- 5. Nekooki-Machida Y., Kurosawa M., Nukina N., Ito K., Oda T., Tanaka M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 9679–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Petkova A. T., Leapman R. D., Guo Z., Yau W. M., Mattson M. P., Tycko R. (2005) Science 307, 262–265 [DOI] [PubMed] [Google Scholar]
- 7. Weingarten M. D., Lockwood A. H., Hwo S. Y., Kirschner M. W. (1975) Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee V. M., Goedert M., Trojanowski J. Q. (2001) Annu. Rev. Neurosci. 24, 1121–1159 [DOI] [PubMed] [Google Scholar]
- 9. Arima K. (2006) Neuropathology 26, 475–483 [DOI] [PubMed] [Google Scholar]
- 10. Ksiezak-Reding H., Wall J. S. (2005) Microsc. Res. Tech. 67, 126–140 [DOI] [PubMed] [Google Scholar]
- 11. Spillantini M. G., Goedert M. (1998) Trends Neurosci. 21, 428–433 [DOI] [PubMed] [Google Scholar]
- 12. Andreadis A. (2005) Biochim. Biophys. Acta 1739, 91–103 [DOI] [PubMed] [Google Scholar]
- 13. Wischik C. M., Novak M., Thøgersen H. C., Edwards P. C., Runswick M. J., Jakes R., Walker J. E., Milstein C., Roth M., Klug A. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 4506–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barghorn S., Mandelkow E. (2002) Biochemistry 41, 14885–14896 [DOI] [PubMed] [Google Scholar]
- 15. Goedert M., Jakes R., Spillantini M. G., Hasegawa M., Smith M. J., Crowther R. A. (1996) Nature 383, 550–553 [DOI] [PubMed] [Google Scholar]
- 16. Munoz-Garcia D., Ludwin S. K. (1984) Ann. Neurol. 16, 467–480 [DOI] [PubMed] [Google Scholar]
- 17. Ksiezak-Reding H., Morgan K., Mattiace L. A., Davies P., Liu W. K., Yen S. H., Weidenheim K., Dickson D. W. (1994) Am. J. Pathol. 145, 1496–1508 [PMC free article] [PubMed] [Google Scholar]
- 18. Grundke-Iqbal I., Iqbal K., Tung Y. C., Quinlan M., Wisniewski H. M., Binder L. I. (1986) Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sahara N., Maeda S., Murayama M., Suzuki T., Dohmae N., Yen S. H., Takashima A. (2007) Eur. J. Neurosci. 25, 3020–3029 [DOI] [PubMed] [Google Scholar]
- 20. Abraha A., Ghoshal N., Gamblin T. C., Cryns V., Berry R. W., Kuret J., Binder L. I. (2000) J. Cell Sci. 113, 3737–3745 [DOI] [PubMed] [Google Scholar]
- 21. Schneider A., Biernat J., von Bergen M., Mandelkow E., Mandelkow E. M. (1999) Biochemistry 38, 3549–3558 [DOI] [PubMed] [Google Scholar]
- 22. Parchi P., Strammiello R., Giese A., Kretzschmar H. (2011) Acta Neuropathol. 121, 91–112 [DOI] [PubMed] [Google Scholar]
- 23. Harper J. D., Lansbury P. T., Jr. (1997) Annu. Rev. Biochem. 66, 385–407 [DOI] [PubMed] [Google Scholar]
- 24. Naiki H., Higuchi K., Hosokawa M., Takeda T. (1989) Anal. Biochem. 177, 244–249 [DOI] [PubMed] [Google Scholar]
- 25. Furukawa Y., Kaneko K., Yamanaka K., Nukina N. (2010) J. Biol. Chem. 285, 22221–22231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nukina N., Ihara Y. (1985) J. Biochem. 98, 1715–1718 [DOI] [PubMed] [Google Scholar]
- 27. Jakes R., Novak M., Davison M., Wischik C. M. (1991) EMBO J. 10, 2725–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Novak M., Kabat J., Wischik C. M. (1993) EMBO J. 12, 365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. von Bergen M., Friedhoff P., Biernat J., Heberle J., Mandelkow E. M., Mandelkow E. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aoyagi H., Hasegawa M., Tamaoka A. (2007) J. Biol. Chem. 282, 20309–20318 [DOI] [PubMed] [Google Scholar]
- 31. Bhattacharya K., Rank K. B., Evans D. B., Sharma S. K. (2001) Biochem. Biophys. Res. Commun. 285, 20–26 [DOI] [PubMed] [Google Scholar]
- 32. Friedhoff P., von Bergen M., Mandelkow E. M., Davies P., Mandelkow E. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 15712–15717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schweers O., Mandelkow E. M., Biernat J., Mandelkow E. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 8463–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goux W. J., Kopplin L., Nguyen A. D., Leak K., Rutkofsky M., Shanmuganandam V. D., Sharma D., Inouye H., Kirschner D. A. (2004) J. Biol. Chem. 279, 26868–26875 [DOI] [PubMed] [Google Scholar]
- 35. Margittai M., Langen R. (2006) J. Biol. Chem. 281, 37820–37827 [DOI] [PubMed] [Google Scholar]
- 36. Yanagawa H., Chung S. H., Ogawa Y., Sato K., Shibata-Seki T., Masai J., Ishiguro K. (1998) Biochemistry 37, 1979–1988 [DOI] [PubMed] [Google Scholar]
- 37. Berry R. W., Abraha A., Lagalwar S., LaPointe N., Gamblin T. C., Cryns V. L., Binder L. I. (2003) Biochemistry 42, 8325–8331 [DOI] [PubMed] [Google Scholar]
- 38. Nonaka T., Watanabe S. T., Iwatsubo T., Hasegawa M. (2010) J. Biol. Chem. 285, 34885–34898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barghorn S., Zheng-Fischhöfer Q., Ackmann M., Biernat J., von Bergen M., Mandelkow E. M., Mandelkow E. (2000) Biochemistry 39, 11714–11721 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.