

Abstract
Biological variation exists across a nested set of hierarchical levels from nucleotides within genes to populations within species to lineages within the tree of life. How selection acts across this hierarchy is a long-standing question in evolutionary biology. Recent studies have suggested that genome size is influenced largely by the balance of selection, mutation and drift in lineages with different population sizes. Here we use population cage and maternal transmission experiments to identify the relative strength of selection at an individual and cytoplasmic level. No significant trends were observed in the frequency of large (L) and small (S) mtDNAs across 14 generations in population cages. In all replicate cages, new length variants were observed in heteroplasmic states indicating that spontaneous length mutations occurred in these experimental populations. Heteroplasmic flies carrying L genomes were more frequent than those carrying S genomes suggesting an asymmetric mutation dynamic from larger to smaller mtDNAs. Mother-offspring transmission of heteroplasmy showed that the L mtDNA increased in frequency within flies both between and within generations despite sampling drift of the same intensity as occurred in population cages. These results suggest that selection for mtDNA size is stronger at the cytoplasmic than at the organismal level. The fixation of novel mtDNAs within and between species requires a transient intracellular heteroplasmic stage. The balance of population genetic forces at the cytoplasmic and individual levels governs the units of selection on mtDNA, and has implications for evolutionary inference as well as for the effects of mtDNA mutations on fitness, disease and aging.
Keywords: mtDNA, Selection, Heteroplasmy, Genetic drift, Insertion-deletion, Population cage
Introduction
Life is organized into a hierarchy of populations. Nucleotides exist within genes, genes within chromosomes, chromosomes within individuals, individuals within populations, all of which are nested within species and higher units of biodiversity. How the forces of population genetics act across this hierarchy to produce patterns of variation in nature is an old and thorny problem in evolutionary biology (Wynne-Edwards 1962; Williams 1966; Dawkins 1976). When nonrandom distributions of molecular polymorphisms are observed in natural populations, natural selection is often invoked to account for the pattern. In many cases, however, unequal mutation pressures or genetic drift could have generated an identical pattern of variation. In some cases it may be unclear whether selection is acting at the level of the gene, the individual or the population (Lewontin and Dunn 1960). As the power of genomic analysis continues to increase, large scale surveys of molecular variation promise to resolve many issues in population and evolutionary genetics. However, we ultimately want to associate molecular variation in nature with some mechanistic explanations for static patterns, so there is a growing interest in coupling functional studies with patterns of static variation to broaden inference on how selection can act.
Several studies have shown that distribution of mitochondrial DNA (mtDNA) length variants in natural populations is skewed towards smaller genomes (see Fig. 1a; Hale and Singh 1986; Hale and Singh 1991; Rand and Harrison 1989; Brown et al. 1992; Arnason and Rand 1992; Rand 1993; Townsend and Rand 2004). The shapes of these distributions have implicated selection for small mitochondrial genomes in a race for replication in the germline cytoplasm (see Fig. 1b; ops. cit.). Support for this notion comes from genetic transmission studies that show an increase of the small mtDNA genomes from mother to offspring (Solignac et al. 1984; Rand and Harrison 1986; Solignac et al. 1987). Following the transition from RFLP to PCR based analyses of mtDNA polymorphism, mtDNA length variation has been largely ignored in studies of variation because individual genes are targeted by PCR. At the same time, there has been a growing interest in the population genetic phenomena in the germ line cytoplasm motivated by studies of the inheritance of mitochondrial disease (Cree et al. 2008; Van Leeuwen et al. 2008; Wai et al. 2008). Because the mtDNAs in the germ line are inherited as a population of molecules, the fundamental forces of population genetics (mutation, section and genetic drift) are primary determinants of how novel mtDNA variants are transmitted from one generation to the next. Indeed, all of the mtDNA sequence variants that are used to quantify population variation, differentiation and phylogenetic affiliation must first pass through a polymorphic phase in the germ line cytoplasm (heteroplasmy). For this reason, studies quantifying the fate of mtDNA variants in the germline could uncover a microcosm of population genetic forces that have implications for the use of mtDNA as a marker in population and evolutionary genetics, as well as contributing understanding to the impact of cytoplasmic drift on the etiology of mitochondrial disease and aging.
Fig. 1.
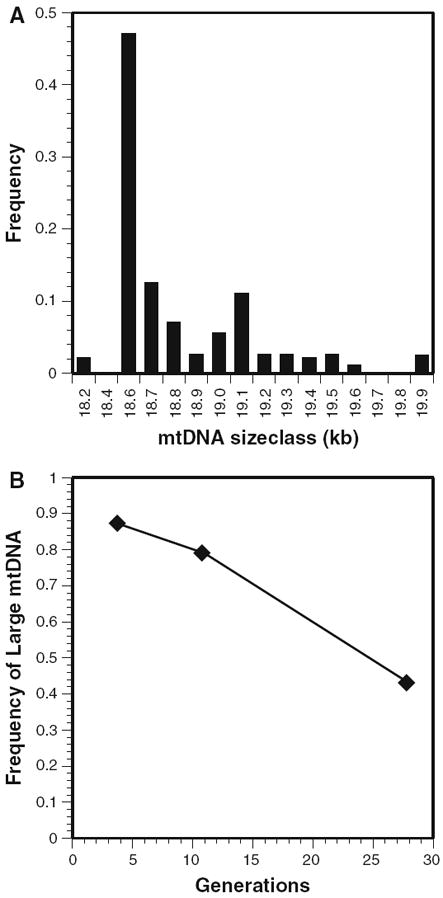
MtDNA length variation in Drosophila melanogaster. a A skewed distribution towards smaller mtDNAs (data from Hale and Singh 1991). b Smaller mtDNA length variants accumulate in heteroplasmic lines maintained in the laboratory (data from Solignac et al. 1984, 1987)
It is now widely recognized that mtDNA is not necessarily a neutral genetic marker that provides an accurate tracer of population and evolutionary history (Whittam et al. 1986; Clark and Lyckegaard 1988; Ballard and Kreitman 1994; Rand et al. 1994; Rand 1994; Ballard and Rand 2005; Bazin et al. 2006; Nabholz et al. 2008; Meiklejohn et al. 2007; Dowling et al. 2008). Functional studies from population cages (MacRae and Anderson 1988; Fos et al. 1990; Hutter and Rand 1995; Kilpatrick and Rand 1995a; Ballard and James 2004) and direct measurements of fitness traits among mtDNA haplotypes have demonstrated phenotypic effects (Clark and Lyckegaard 1988; Rand et al. 2001; James and Ballard 2003). None of these studies, however, were designed in a manner that would allow mitochondrial genome size to be isolated as an independent experimental variable. Considering the observation that smaller mtDNAs can be favored in the cytoplasm during transmission from mother to offspring (Rand and Harrison 1986) but see (Solignac et al. 1987), and that mtDNA D-loop variants have been associated with longevity in humans (Michikawa et al. 1999; Coskun et al. 2004), mtDNA length variation provides a simple system with which to address questions regarding the units of selection (Lewontin 1970; Rand 2001; Taylor et al. 2002).
Here I report the results of a population cage study using Drosophila melanogaster where mtDNAs of two different lengths (the S and L genomes) were competed against one another to determine fitness effects at the organismal level. The S and L mtDNAs were collected from the same Texas locality and were identical in restriction patterns for 10 restriction enzymes, but differed in length by 1.4 kilobases (kb; 7.5% of genome length; details in Methods). These mtDNAs were chosen for fitness studies because they provided an opportunity to separate coding sequence variation from mtDNA length variation. No significant fitness effects were observed in the population cage experiments, but spontaneous length mutations generating heteroplasmic flies were detected in replicate cages. Analyses of mother-offspring transmission of heteroplasmy for these novel length variants showed that the L variant increased in frequency within individuals. Because mtDNA length variants do not maintain their size long enough for selection to act on them at the level of the individual, mtDNA size variation can be considered effectively neutral at the organismal level, but not neutral in the germ line cytoplasm in D. melanogaster.
Materials and methods
Fly strains
Two lines of Drosophila melanogaster collected in Brownsville, Texas were obtained from Dr. Larry Hale. The lines had mtDNAs with identical restriction patterns for 10 enzymes: 4 cutters (DdeI, HaelII, Hinfl, HpaII, MboI, TaqI); 4.5 cutter (AvaII); 6 cutters (EcoRI, HindIII, XbaI) as described in Hale and Singh (1991). The only known difference in the mtDNAs of these two lines was the presence of an additional 1.4 kilobases (kb) of DNA inserted in the A + T-rich region of the longer molecule (20.0 kb Hale and Singh 1986; Hale and Singh 1991; hereafter the L genome). The smaller genome (hereafter S) was 18.6 kb in length and is the most frequent length variant found in natural populations. The Brownsville S line was identical to nine lines from California for >1,500 bp of the ND5 gene (Rand et al. 1994), consistent with low levels of mtDNA polymorphism in North American D. melanogaster. While complete sequencing would be required to establish the identity of the coding region of the L and S mtDNAs, we assumed that they were similar, if not identical, at the nucleotide level in the 13 protein coding genes, the 22 transfer RNA (tRNA) genes and the two ribosomal RNA (rRNA) genes of the mitochondrial genome. The A + T-rich region of D. melanogaster mtDNA is about 4 kb in length, is at least 95% A + T in nucleotide composition and contains the origin of replication (Fauron and Wolstenholme 1976, 1980; Lewis et al. 1994). Restriction map studies of the A + T region in the melanogaster subgroup of species indicate that this region is composed of tandemly repeated sequences (Solignac et al. 1986), and this has been confirmed from direct sequencing in D. melanogaster (Lewis et al. 1994). Prior to initiation of the population cages, the size of the mtDNA was confirmed using the molecular analyses described below.
Initiation and maintenance of population cages
The experimental population cages were initiated by isolating virgin females from the S and L lines and mating 250 of these virgins to an equal number of males from the opposite line. These flies were allowed to mate for 24 h in bottles after which they were transferred to two different laying cages: one with S females mated to L males and a second cage with L females mated to S males. Each laying cage was provided with 12 plastic laying cups containing instant Drosophila food (10 grams of 4–24 medium, Carolina Biological Supply Company, plus 30 milliliters of distilled water). After each 24-h laying period in the cages, three laying cups were moved from each of the laying cages to each replicate experimental cage. This was done for five successive days so that each cage contained five sets of three cups carrying F1 eggs from the forced crosses between S and L flies. This established a continuous population of flies that were maintained at 24°C in overlapping generations by adding three fresh cups every 3 days, thus imposing a generation time of a maximum of 15 days. Six population cages were initiated: three cages at high frequency of L (~90 L%; originally called cages 1–3 and hereafter referred to as Lhigh1, Lhigh2, Lhigh3) and three cages at intermediate frequencies (~50 L%; originally called cages 4–6 and hereafter referred to as Lmid1, Lmid2, Lmid3). These frequencies were manipulated by allowing different number of adults to lay eggs in the laying cups that were transferred to the initial cages.
Sampling and estimates of frequencies
The starting frequencies of the L and S mtDNAs in each cage were estimated from the eggs laid by the F1 adults that emerged from the original laying cages. A second sample was taken for each cage one generation (15 days) later. Subsequent estimates of L and S mtDNA frequencies were made approximately once each month. One day after a fresh set of three cups was placed in each cage, a small spoon was used to scoop out a sample of the new food approximately 2 cm in diameter that was covered with eggs. Each sample was placed in a separate half-pint Drosophila culture bottle with 30 mL of standard cornmeal and molasses medium. All of the adults that hatched from these egg samples were frozen at −80°C for later DNA analysis. Frequencies of the L and S genomes in each cage were estimated from a sample of 64 flies using restriction digests and Southern blots.
DNA preparations
Total genomic DNA was prepared by grinding either one or two adult flies in 25 μl of Lifton Buffer (0.1 M Tris, 50 mM EDTA, 0.2 M Sucrose, 0.5% SDS, ph 9.0; (Bender et al. 1983) with 0.5 μl of DEPC using a plexiglass pestle shaped to fit the inside of a 1.5 milliliter microcentrifuge tube. After grinding, an additional 25 μl of Lifton buffer was added to the homogenate and the tube placed at 65°C for 20–30 min. Seven μl of 8.0 M potassium acetate was then added and the tubes incubated on ice for 15–30 min. The samples were spun in a microcentrifuge for 10 min, the supernatant recovered and precipitated with two volumes of cold ethanol at −80°C for 10 min. Crude nucleic acids were recovered by centrifugation in a microcentrifuge for 10 min, resuspended in 100 μl TE (10 mM Tris, 1 mM EDTA) and extracted once with a 25:24:1 mixture of Phenol:Chloroform:Isoamyl alcohol. The aqueous phase was precipitated with 1/10 volume of 3 M Sodium Acetate and two volumes of cold ethanol at −80°C for 10 min. Total nucleic acids were pelleted by centrifugation for 10 min, rinsed once with 70% EtOH and dried in a vacuum desiccator. The dried pellet was resuspended in 20 μl TE for single fly preps and 50 μl for two-fly preps.
Southern blot analysis
MtDNA haplotype frequencies were scored from autoradiographs of Southern blots of total genomic DNA. An aliquot of one third to one half of the total genomic DNA (prepared as described above) was digested with Hae III or its less expensive isoschizimer Pal I, according to the manufacturers specifications (New England Biolabs for Hae III; Stratagene for Pal I). The digested DNA was electrophoresed in 0.7% Agarose gels using Tris Acetate buffer. The resulting gels were blotted to nitrocellulose membranes by capillary transfer or a pressure blotter (Stratagene). Membranes were prehybridized in 5× SSPE with 0.1% SDS for a minimum of 30 min at 65°C. Hybridization to probes was performed in the same solution for a minimum of 8 h at 65°C.
As a probe, a clone of the 4.8 kb Hind III fragment of Drosophila melanogaster mtDNA was used because it hybridizes to the large Hae III fragment containing the L-S length variation, and to the adjacent 6.2 kb Hae III fragment which provides an internal size standard for comparison to the length-variable band (see Fig. 2).
Fig. 2.
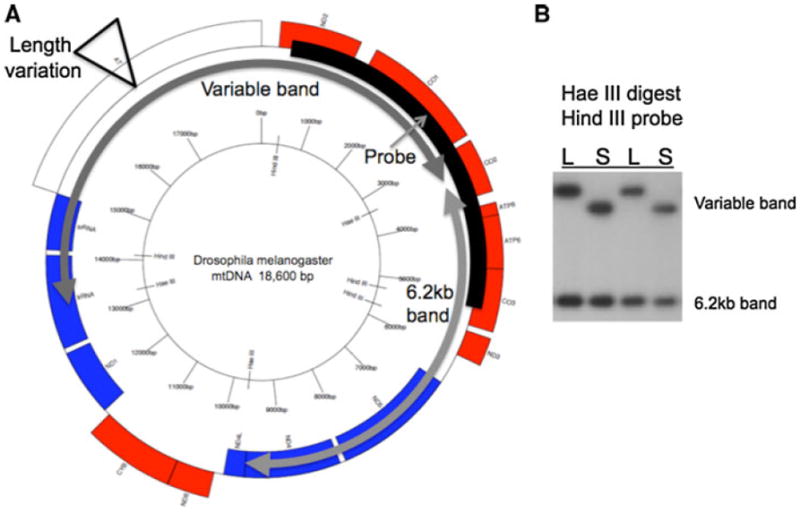
Map of D. melanogaster mtDNA. a Circular genome map showing location of two Hae III fragments, one containing the length variation and a smaller 6.2 kb band that is constant in size (gray arrows). The Hind III band used as a probe overlaps both Hae III bands (black arc labeled ‘Probe’ with thin gray arrow). b Radiograph of a Southern blot from a Hae III digest probed with the Hind III band probe, showing individual flies with Large or Small (L, S) length variation in the larger band and no variation in the 6.2 kb band. The L and S bands differ by ~1.5 kb
Initially, gels were scored using two-fly DNA preps since two data points were obtained from each lane of a gel: a single large band indicated both flies carried L mtDNA, a large and a small band indicated that one L and one S fly was present and a single small band indicated that both flies carried S mtDNA. Since novel length variants were detected during the course of the experiment, single-fly preps were used to repeat the first samples showing novel length variants, and all samples thereafter, to allow for precise identification of heteroplasmic individuals.
Transmission of L-S heteroplasmy
The transmission of the heteroplasmic mtDNA population through the female germ line was analyzed by comparing the frequency of L and S mtDNAs in a focal heteroplasmic mother to the frequency of these variants in a sample of individual offspring from each mother. The heteroplasmic mothers for these analyses were derived from the spontaneous heteroplasmic lines that emerged in the cage experiment. Analyses of heteroplasmic transmission were done manipulating two conditions: (1) the age of the mother (early vs. late broods of offspring), and (2) the culture temperatures (18̇C vs. 25̇C). The maternal age effect was motivated by the findings of (Solignac et al. 1987) who showed an effect of maternal age on mean and variance of offspring heteroplasmies. Temperature was manipulated to determine if the delayed development at 18̇C would alter heteroplasmic transmission.
Mother—offspring transmission was scored in independent heteroplasmic lines with samples of offspring taken from early broods (eggs collected 1–3 days after mating) and late broods (eggs collected 11–20 days after mating). These early and late broods were each collected from cultures maintained at two temperatures (18 and 25°C). By necessity, collection of early and late broods from heteroplasmic mothers maintained at different temperatures could not be the same individuals. To minimize the effects of different L and S frequencies in heteroplasmic mothers allowed to lay eggs at 18 and 25°C, sibling females from the same heteroplasmic culture were split between the two temperatures for collection of early and late broods. As such, the sets of offspring emerging from early vs. late broods, or 18 vs. 25̇C were descended from the same grandmother, and had sibling mothers for each condition. Nine mother-offspring families were quantified for early- and late-brood mtDNA frequencies from an average of 13 offspring per mother per time point (total of 470 individuals). The goal of this experiment was to determine the effect of female age and temperature on the transmission dynamics of heteroplasmy through the female germline.
The frequency of L and S mtDNA within each fly was quantified using densitometry of autoradiographs from Southern blots where the frequency of the two length variants was estimated from the intensity of signal corresponding to each band. The densitometer trace generates peaks of varying height and breadth, and the area under the trace for each band was taken as an estimate for frequency, following removal of autoradiograph background. Replicate gel runs and autoradiographs were scored for each mother-offspring sample, and the average value across these replicates was taken as the frequency of L or S mtDNA within an entire fly. Saturation of autoradiographs was avoided by ignoring densitometer traces that had flat peaks, suggesting over-exposure of the film in the middle of a band. Details of this procedure are reported in Kann et al. (1998).
Estimates of selection coefficients and effective population sizes
Selection coefficients were estimated for haploid alleles from the slope of the regression of natural logarithm of the ratio of the frequencies of alternative alleles on time, as described by Dykhuisen and Hartl (1980, 1983):
| (1) |
where xLarge and xSmall are the frequencies (or counts) of the L and S mtDNA, s is the selection coefficient and t is time. Because the population cages were maintained with continuous overlapping generations, time is measured in days but the cycle of food forced a generation time of 15 days. The significance of this regression is tested by analysis of variance. This was done for each cage individually and, where appropriate, for data pooled among replicate cages. For comparison, selection can be inferred from the change in frequency of mtDNAs and the product of the frequencies of the alternative haplotypes at generation t + 1 and t (Δp = spt+1qt).
Allele frequency changes are also caused by genetic drift, and since effective neutrality occurs when s ≤1/Ne, the estimates of selection above need to be put in context with the strength of genetic drift. The population cage data are samples of mtDNA frequencies over multiple generations, and the change in these frequencies within each population can be used to estimate effective population size (Ne) based on the principle that the change in frequency across generations should be inversely proportional to Ne, under neutrality. To arrive at an estimate of Ne, we compared two methods using population cage data: (1) estimates from allele frequencies across time in individual cages, and (2) estimates from the variance in allele frequency among replicate population cages. Approaches for estimating Ne from samples takes across time points have been described previously (Waples 1989; Nei and Tajima 1981). The simplest approach is to estimate the standardized variance (F) in allele frequency (P) between time points 0 and t, as
| (2) |
and estimate Ne as
| (3) |
This holds for true allele frequencies, but when allele frequencies are estimated from samples, F and Ne should be estimated as follows (Nei and Tajima 1981):
| (4) |
where K = number of alleles, xi = frequency of allele i at time 1 and yi = frequency of allele i at time 2. Ne can be estimated from F̂c after adjusting for the empirical sampling variance (Waples 1989; Nei and Tajima 1981):
| (5) |
where t = the time interval in generations, S0 and St = the sample sizes of individuals scored at generations 0 and t, respectively. Expression (3) assumes that N = Ne, and we know from population cage studies that Ne is roughly between 25 and 75% of N (Crow and Morton 1955; Hutter and Rand 1995; Kilpatrick and Rand 1995b). Waples (1989) offers a modified expression using a correction factor of r = N/Ne, and suggests trying different values of r that may give realistic estimates of Ne.
| (6) |
In the current study, using a realistic value of r = N/Ne = 2 gave many estimates of Ne that were negative, indicating that the experimental sampling variance from S0 and St can mask the evolutionary sampling variance due to the true Ne (i.e., the sum of 1/2S0 and 1/2St is greater than the estimate of Fc, in (4), giving an estimate of Ne < 0). Since census sizes in population cages were at least 2,000, estimates of Ne less than 0 were ignored, and values > 0 were retained for comparison, accepting that an order-of-magnitude error is possible. Using expressions (3) or (6), estimates of Ne could be made by comparing the allele frequency estimates at time 0 to estimates at each successive temporal sample, or across the full 14–15 generation time interval that the population cages were maintained (see Table 1).
Table 1.
Effective neutrality of mtDNA length variants in laboratory populations
| Cage | s | Lower 95% C.I. | Upper 95% C.I. | Ne (adjacent mean) | Ne (harmonic) | Ne (cumul. mean) | Ne (variance) |
|---|---|---|---|---|---|---|---|
| Lhigh1 | −0.0018 | −0.007 | 0.004 | 343 | 49 | – | |
| Lhigh2 | 0.0013 | −0.007 | 0.010 | 125 | 51 | 83 | |
| Lhigh3 | 0.0015 | −0.011 | 0.014 | 365 | 24 | 21 | 1,077 |
| Lmid1 | −0.0029 | −0.006 | 0.001 | 87,117 | 26 | 2,233 | |
| Lmid2 | 0.0019 | −0.002 | 0.005 | 95 | 49 | 88 | |
| Lmid3 | 0.0018 | −0.004 | 0.008 | 102 | 21 | 2,589 | 87 |
s = Selection coefficient estimated from expression (1)—see “Methods”
95% C.I. are confidence limits from regression slopes used to estimate s
Ne = effective population size, as the mean of estimates from different time intervals
Ne (adjacent mean) is the arithmetic mean of Ne estimates from adjacent time samples
Ne (harmonic) is the harmonic mean of Ne estimates from adjacent time samples
Ne (cumulative) is the mean of Ne estimates from time = 0 to t = 1, 2, 3, etc
See “Methods” and “Results” for further details
Ne was also be estimated from the variance in allele frequency among replicate populations descended from an initial founding population after g generations of drift, assuming the variants are neutral (Wright 1968). This approach can be used for the experimental populations, or for heteroplasmy transmitted through the female germline (Solignac et al. 1984; Rand and Harrison 1986):
| (7) |
In expression (7), Vg is the variance among populations, p is the frequency of the L mtDNA, Ne is the haploid effective population size of mitochondria, and g is the number of organismal generations. This same expression can be used to estimate the effective number of mitochondria in the germ line cytoplasm, in which case Vg is the variance among offspring in the frequency of L mtDNAs, g expresses the number of cellular divisions in the germ line cytoplasm from mother to offspring, assuming one organismal generation of transmission. The population cage experiment and the transmission experiment both provide empirical estimates of p and the variance of p at generation g (Vg), allowing the estimate of the average effective population size across replicate cages, or the effective number of sampled mitochondria in the female germline in the transmission study, as follows:
| (8) |
Novel mtDNA length variants were observed in the cages and the change in frequency can provide an estimation of mutation rates:
| (9) |
where p0 = 1.0, pt = 0.90 or 0.80 assuming the novel length variant = 0.1 or 0.2 and t = 15 generations. Solving for μ gives
| (10) |
Results
Selection at the level of individuals
Two sets of replicate population cages were established at different initial frequencies: cages Lhigh1, Lhigh2, Lhigh3 at ~90% L mtDNA and cages Lmid1, Lmid2, Lmid3 at ~50% L mtDNA. Cages Lhigh1–3 were maintained for 223 days and cages Lmid1–3 were maintained for 213 days (approximately 15 and 14 generations, respectively, assuming 15 days per generation—see “Methods”). There was no apparent selective advantage of flies carrying either the S or L mtDNA in either set of replicate cages (see Fig. 3). Selection coefficients were estimated for each cage using expression (1; see “Methods”), and in each case the confidence limits included zero (see Table 1). The two sets of cages with different initial frequencies were intended to test the possibility that a stable frequency at one initial frequency could be the result of frequency-dependent selection. The observation that both sets of cages show no apparent change in frequency from the initial state does not support this hypothesis. It remains possible that two separate frequency-dependent equilibria are being maintained in these cages, but this seems unlikely and unparsimonious in light of the evidence. The simplest conclusion is that mtDNA length variants are effectively neutral at the level of the whole fly in these cages.
Fig. 3.
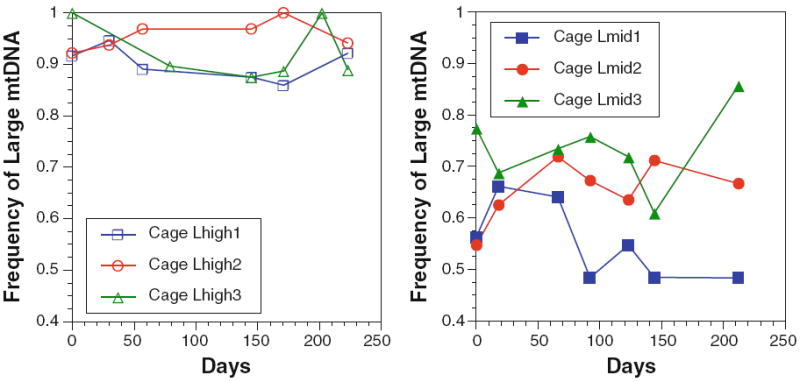
Frequency trajectories of mtDNA length variants in experimental population cages. Left panel cages Lhigh1–3 were initiated with ~90% LmtDNA; right panel cages Lmid1–3 were initiated with ~50% L mtDNA. See “Methods” and “Results” for details
Estimates of Ne in population cages
Effective neutrality occurs when the selection coefficient (s) < 1/Ne. The population cage data can be used to estimate Ne in the cages, based on changes in haplotype frequency between time points (Waples 1989). Several different variations on this methods were done, and the results are presented in Table 1. First, each sample interval was considered and the change in mtDNA haplotype frequency was taken as the true frequency without considering sampling error (Ne = t/2F; using expression (3) above). Each point estimate between adjacent samples was then averaged from the samples across the 15 generations of each cage (‘Ne adjacent mean’ in Table 1). For comparison, the harmonic mean of these adjacent values is also shown in Table 1 (‘Ne harmonic’). Next, the error variance due to population sampling was incorporated (expressions 4–6), and Ne was estimated in successive cumulative comparisons from the first sample to the nth sample (1 vs. 2, 1 vs. 3, 1 vs. 4, etc.). Some of these comparisons produced negative estimates of Ne because the sampling variance was greater than F̂c. These cases were ignored, and only those intervals that gave positive estimates of Ne were used, and then averaged across all time intervals (‘Ne cumulative mean’ in Table 1). Finally, expression (8) was used to estimate Ne from the variance in haplotype frequency across the three final samples, separately for cages Lhigh1–3 and cages Lmid1–3 (‘Ne variance’ in Table 1). The initial frequency was taken as the average of the three initial frequencies for each cage.
As expected, estimates of Ne correspond roughly to the reciprocal of observed changes in frequency, with some notable outliers (see Table S1). The values using the ‘cumulative’ method that incorporates sampling variance (expression 6) are Ne = 83–21 for cages Lhigh1–3 and Ne = 88–2,589 for cages Lmid1–3. These individual estimates contrast those based on the variance among individual cages at the final time point (Lhigh1–3 Ne = 1,077, Lmid1–3 = 87). Despite this noise, Ne falls between ~100 and ~2,000. This means that a selection coefficient on the order of 1/100–1/2,000 = 0.01 to 0.0005 will be neutral in these experimental cages. These observations further support the effective neutrality of the mtDNA length variants (see Table 1).
Novel mutations and heteroplasmy
In all six cages, samples taken after day 200 showed clear evidence of novel length variants on Southern blots of two-fly samples (see “Methods”). Band sizes suggested that these length variants were also present in heteroplasmic states. To confirm heteroplasmy in individual flies, replicate samples of single flies were prepared and Southern blots hybridized as described. In each cage novel mtDNA length variants were detected as were flies heteroplasmic for two different length variants. To confirm that the novel mtDNA length variants were heritable, 30 female flies from cages Lmid1–3 were collected and allowed to lay eggs singly in vials. After the offspring of these single females had hatched and themselves laid eggs for the maintenance of new isofemale lines, DNA was extracted from 5 adult offspring of the original females taken from the cages. These lines showed clear evidence of novel mtDNA length variants and heteroplasmy (see Fig. 4). Subsequent single-fly Southern blot analysis of later generation adults of the heteroplasmic lines (e.g., 4–19, 5–27, Fig. 4) revealed flies heteroplasmic for the two mtDNA length variants that were originally detected in the isofemale lines shown in Fig. 4 (within the size resolution of these gels). Samples from the cages revealed that novel variants approach 10–20% in cages 4–6 (data not shown).
Fig. 4.
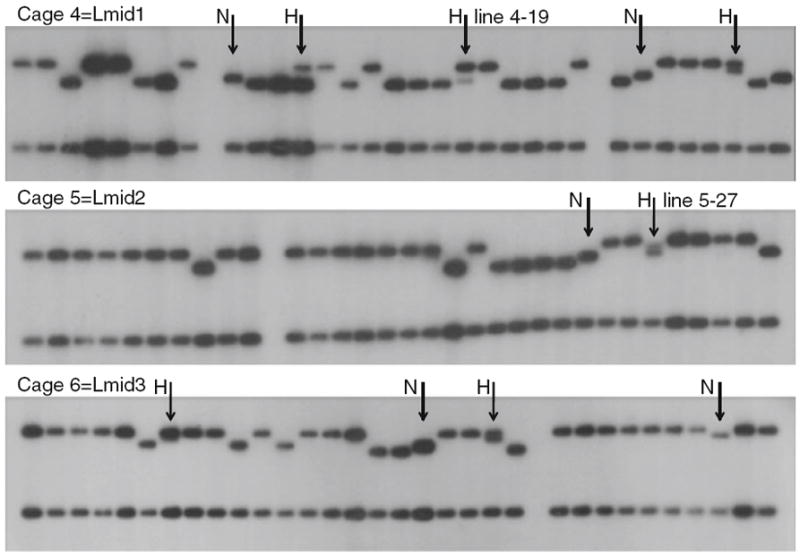
MtDNA length variation and novel heteroplasmy in experimental population cages. Each lane is the Hae III digest pattern of five offspring from a single female. Novel (N) and heteroplasmic (H) lineages are identified, in each cage. Note that more heteroplasmies include an L band as one of the length variants suggesting deletion events. Lines 4–19 and 5–27 were used for subsequent mtDNA transmission studies. Sizes of bands are shown in Fig. 2
If mutation is the only force increasing the frequency of these variants, we can estimate that mutation rate from the decrease in frequency of L mtDNA that is associated with an increase in the novel length variant (see expressions 9, 10 in “Methods”). From cage frequency data, p0 = 1.0, pt = 0.90 or 0.80 assuming the novel length variant = 0.1 or 0.2 and t = 15 generations. Solving for μ (equation 7) gives: μ = 0.007 when the novel length variants are at 10% and μ = 0.015 when novel length variants are at 20%. These estimates indicate that the mutation pressure changing the length of an mtDNA can be an order of magnitude stronger than the selection acting on these variants at the level of the individual (see Table 1). These findings underscore the effective neutrality of mtDNA length variants at the individual level because an mtDNA length variant does not maintain its size long enough for individual selection to achieve a change in frequency.
Selection and drift within germ lines and individuals
The novel heteroplasmic individuals were used to quantify selection and drift in the female germline by measuring the frequency of mtDNA length variants within mothers and samples of their offspring. The proportions of the two length variants were estimated using densitometry of Southern blots of mothers, and early and late broods of offspring descended from these mothers (see “Methods”). In general, the frequency of the L mtDNA increased in frequency between mothers and offspring, with further increases between the early and late broods (see Fig. 5; Table 2). On average the L mtDNA increased by 12% between mother and early brood offspring, and increases a further 6% when the late brood offspring are considered. The consistent pattern of an increase of L mtDNA across multiple lines indicates that drift is an unlikely explanation for this change in heteroplasmy. Note that the frequency of L mtDNA in the mother was that measured after she produced the late brood, so any change in frequency of the mtDNA inside the cells of the mother during her lifetime could alter this measurement. But since the estimates indicate an increase in L mtDNA with age (Table 2; see also (Kann et al. 1998), this will only underestimate the increases in the L mtDNA shown in Table 2.
Fig. 5.
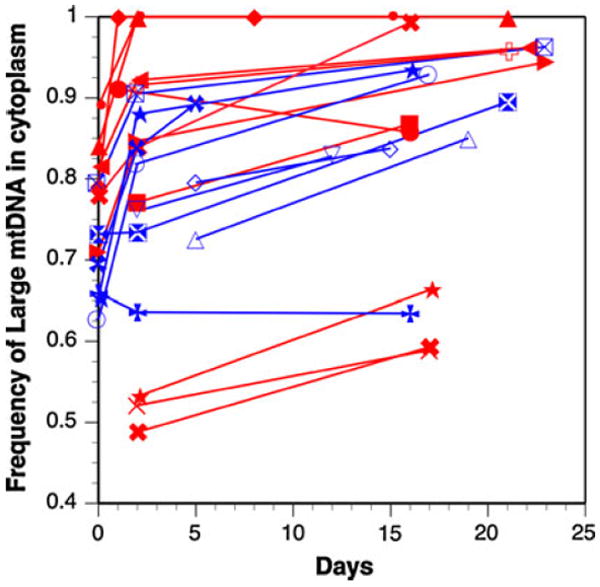
Increase of L mtDNA in transmission from mother to offspring. Day zero is the frequency of L mtDNA in heteroplasmic mothers, and subsequent days are the frequencies of L mtDNA in early or late brood offspring from those mothers. Not all families had data from mothers, so some lines start after day 0. Red lines are 25°C and blue lines are 18°C cultures. There is no significant effect of temperature on frequency shifts (t = 0.46, df = 10, P = 0.65). (Color figure online)
Table 2.
Selection and drift of mtDNA length variants within and between generations
| Line-subline | Temp. (°C) | Days d2-d1 | %L mother | %L early | V(L) early | Ne–mt early | %L late | V(L) late | Ne–mt late | Δ%L early-mother | Δ%L late-mother | Δ%L late-early |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4-19D4-D1 | 25 | 14 | NA | 0.771 | 0.00384 | 318 | 0.868 | 0.00397 | 199 | 0.097 | ||
| D2 | 25 | 15 | NA | 0.911 | 0.00360 | 155 | 0.858 | 0.00740 | 112 | −0.053 | ||
| B3 | 25 | 19 | 0.842 | 1.000 | 1.000 | 0.158 | 0.158 | 0.000 | ||||
| B4 | 25 | 7 | 0.791 | 1.000 | 1.000 | 0.209 | 0.209 | 0.000 | ||||
| G4 | 25 | 20 | 0.815 | 0.922 | 0.00176 | 283 | 0.962 | 0.00020 | 1,312 | 0.107 | 0.147 | 0.040 |
| H4 | 25 | 21 | 0.710 | 0.847 | 0.00314 | 287 | 0.944 | 0.00044 | 833 | 0.137 | 0.234 | 0.098 |
| D3 | 18 | 14 | NA | 0.726 | 0.00490 | 281 | 0.850 | 0.00578 | 151 | 0.125 | ||
| D4 | 18 | 10 | NA | 0.796 | 0.00384 | 293 | 0.838 | 0.00221 | 428 | 0.042 | ||
| B2 | 18 | 19 | 0.732 | 0.734 | 0.00476 | 284 | 0.895 | 0.00672 | 95 | 0.002 | 0.163 | 0.161 |
| G2 | 18 | 21 | 0.795 | 0.905 | 0.00723 | 80 | 0.963 | 0.00137 | 178 | 0.110 | 0.168 | 0.058 |
| H1 | 18 | 15 | 0.627 | 0.819 | 0.00250 | 412 | 0.929 | 0.00116 | 397 | 0.192 | 0.302 | 0.110 |
| D1F | 18 | 10 | NA | 0.760 | 0.01145 | 108 | 0.828 | 0.00518 | 189 | 0.068 | ||
| 5-27H3-D | 25 | 11 | NA | 0.488 | 0.00410 | 424 | 0.593 | 0.00903 | 184 | 0.105 | ||
| O | 25 | 11 | NA | 0.521 | 0.00240 | 725 | 0.589 | 0.00384 | 438 | 0.068 | ||
| J | 25 | 11 | NA | 0.532 | 0.00102 | 1699 | 0.663 | 0.00314 | 496 | 0.131 | ||
| Held virgins (10 days) | ||||||||||||
| 4-19D4-D5 | 25 | 19 | NA | 0.916 | 0.00372 | 142 | 0.958 | 0.00194 | 144 | 0.042 | ||
| B6 | 25 | 15 | 0.779 | 0.839 | 0.00865 | 106 | 0.993 | 0.00008 | 598 | 0.060 | 0.214 | 0.154 |
| C5 | 25 | 13 | 0.890 | 1.000 | 1.000 | 0.110 | 0.110 | 0.000 | ||||
| B8 | 18 | 15 | 0.659 | 0.636 | 0.00563 | 285 | 0.635 | 0.00740 | 216 | −0.023 | −0.024 | −0.002 |
| G8 | 18 | 4 | 0.695 | 0.839 | 0.00578 | 161 | 0.893 | 0.01124 | 56 | 0.144 | 0.198 | 0.055 |
| H7 | 18 | 14 | 0.653 | 0.880 | 0.00348 | 209 | 0.934 | 0.00102 | 419 | 0.227 | 0.281 | 0.054 |
| Averages | 0.0045 | 347 | 0.0040 | 358 | 0.119 | 0.180 | 0.064 | |||||
The action of drift was quantified by measuring the variation among individual offspring from individual mothers, and applying this variance to estimate the effective population size of ‘segregating’ mitochondrial genomes (Ne-mt) during maternal transmission, using expressions (7) and (8). The early and late broods produced similar results with variances of 0.0045 and 0.0040, respectively, leading to an estimate of approximately 350 segregating units (Ne-mt; see Table 2). This number is based on an assumption of seven germ cell generations per animal generation (Solignac et al. 1987); with 10 germ cell generations the estimate of Ne increases to Ne ~ 500 segregating units. There were no significant effects of temperature on the change in relative frequencies across generations or between early and late broods (see Table 2; Fig. 5).
Discussion
Mitochondrial DNA continues to be the most widely used marker in population and evolutionary genetics. The high mutation rate, maternal inheritance, haploid transmission, and low rates of recombination make it ideal for inferring the evolutionary history of organisms (Avise 1986, 2000; Harrison 1989). A common assumption is that the variation in the molecule is neutral with respect to fitness and that the patterns of nucleotide variation among individuals, populations and species can be used to estimate effective population sizes or ages of common ancestry in recent history. This view has been challenged by an increasing number of studies (Ballard and Rand 2005; Bazin et al. 2006; Meiklejohn et al. 2007; Dowling et al. 2008). At the same time, a broad literature has emerged documenting correlations between mtDNA mutations and a variety of human diseases including age related decline (Wallace 2005, 2007). The variety of ways that mtDNA variants can be associated with fitness and diseases provide motivation for functional studies of mtDNA polymorphism. Some late-onset mtDNA diseases in humans exist as benign heteroplasmies in early life that lead to disease only after the mutant mtDNA increases in frequency in certain tissues (Castagna et al. 2007, 2010). Thus, an intracellular population genetics exists that may be critical for understanding the functional effects of mtDNA mutants at the level of the whole organism. As such, there can be selection at different levels, and the cytoplasmic population genetics of mtDNA presents a fascinating problem in the units of selection (Birky 2001; Rand 2001; Taylor et al. 2002).
The mtDNA length variation described in this paper presents such a problem. A strongly skewed frequency distribution of mtDNA length variants in natural populations of Drosophila melanogaster suggests selection favoring smaller mtDNA length variants (Hale and Singh 1991); Fig. 1). The relevant unit of selection is not clear from this static pattern. An experimental approach was taken to dissect the relative contributions of population genetic forces at two levels: among individuals within populations, or among mtDNAs within the female germ line. The population cage experiments revealed no evidence for fitness differences among flies carrying mtDNAs of different length. Estimates of effective population sizes suggest that selection coefficients in the range of 0.01–0.0005 could be detected in these cages (Table 1). Moreover, novel heteroplasmic mtDNA length variants appeared in experimental time indicating that the size of an mtDNA is not stable for sufficient time that selection at the individual level can mediate significant change in frequency. Within the organism, however, a strong and repeatable bias was observed favoring the transmission of L (long) mtDNA variant into the offspring generation. Estimates of effective population size in the female germ line indicate that the strength of drift at the cytoplasmic level was about the same order of magnitude as at the level of the whole organism, yet a consistent selective transmission was observed. Thus selection is measurably stronger within the cytoplasm than among individual flies.
A race for replication?
How can we accommodate the transmission bias favoring longer mtDNAs with the skewed frequency distribution in natural populations (Hale and Singh 1991) that shows a bias towards shorter mtDNAs? Earlier studies with heteroplasmic mtDNA length variants in the D-loop region have shown that the smaller mtDNA length variants increase in frequency during transmission (Solignac et al. 1984; Rand and Harrison 1986), or in laboratory mass culture (Solignac et al. 1987). This has been attributed to a race for replication in the cytoplasm favoring mtDNAs with less DNA (Rand and Harrison 1986). In Drosophila the increase in short (S) mtDNA within heteroplasmic lines was attributed to the rapid turnover of lab cultures that favor reproduction at early age (Solignac et al. 1987). Subsequent analyses showed that early reproducing heteroplasmic females showed an increase in S mtDNA, while late reproducing females showed an increase in L mtDNA (op. cit.). The skewed frequency distribution reported in Hale and Singh (1991) may have resulted from rapid turnover of cultures since the lines for that study had been in lab culture for some time before scoring. However, the skewed distribution is repeatable in younger collections (Townsend and Rand 2004). The current study confirms the increase in L mtDNA in later-age broods, but provides little evidence for an increase in S mtDNAs in the early broods. It could be that this ‘race for replication’ effect was missed, but most of the early broods were sampled from mothers aged 1–2 days, so it would have to be a very early effect to have been missed.
Scramble competition for nuclear encoded mtDNA polymerase?
An earlier study using the lines described here showed that when mated females are allowed to continue producing offspring throughout their life there is a steeper increase in the L mtDNA within individual females than within virgin females that are not allowed to reproduce (Kann et al. 1998). This suggests that continued turnover of the germ line cytoplasm is an important force in the change of mtDNA variants in heteroplasmic cells. The A + T-rich region of Drosophila mtDNA contains the origin of replication where the nuclear-encoded polymerase γ binds to initiate DNA synthesis (Oliveira and Kaguni 2009). This region contains tandem repeats with likely secondary structures (Lewis et al. 1994) that may facilitate the binding of polymerase. As such, a larger mtDNA may have an advantage over a smaller mtDNA in attracting or maintaining mtDNA polymerase at the site of replication. The additional cost of replicating mtDNA may be a small trade-off compared with the advantage of ensuring the engagement of polymerase γ.
This is an appealing model from an mtDNA’s perspective, since nuclear encoded proteins are limiting resources that are not readily available under local genetic control. When conditions are not ideal for a mitochondrion, retrograde signaling is thought to be initiated that sends signals from the mitochondrion back to the nucleus to alter nuclear gene expression for mitochondrial biogenesis and other biochemical functions (Liu and Butow 2006). It is not clear if individual mitochondria can be targeted under these conditions, or whether a general mitochondrial malaise is needed before retrograde signaling begins to rescue mitochondrial function. As such, an individual mtDNA could out compete other mtDNAs by sequestering more of the limiting import products that are needed to maintain function. This selfish, units of selection argument (Koonin and Wolf 2009) is similar to the logic of smaller mtDNAs gaining representation in the next generation, but requires that the metabolic cost of extra mtDNA replication is smaller than that of acquiring additional nuclear proteins to maintain individual mitochondrial function. While certain D-loop mutations appear to have replication advantages in mammalian systems (Michikawa et al. 1999; Coskun et al. 2004), the length variation is not sufficient to be considered a differential cost (a few base pairs). In Drosophila, the A + T rich region is poorly understood and difficult to work with for mtDNA replication assays, but modification of polymerase γ levels is a feasible means of testing this hypothesis.
Alternatively, the skewed frequency distribution could be due to a mutation bias that is length-dependent. If the A + T-rich region has a lower and an upper size limit, and the insertion/deletion rate is dependent on the number of tandem repeats units, a frequency distribution skewed towards smaller genomes can result even if the insertion rate equals the deletion rate (Townsend and Rand 2004). Under this model, a transmission bias that leads to an increase in the L genome will be counteracted by a mutational effect that tends to push the distribution back towards smaller genomes. Interestingly, more of the novel heteroplasmic lines discovered in the population cage experiment involved a heteroplasmy with the L genome, suggesting deletions from this state are more common (see Fig. 4). As with many static distributions in natural populations, there are opposing forces at work that may maintain the distribution at some equilibrium. In this particular system the population genetic forces at work below the level of the individual, and most likely in the meta-population of the female germ line, are most critical in explaining this pattern of genetic variation.
Supplementary Material
Acknowledgments
This work was motivated by the fortuitous discovery of mtDNA length heteroplasmy in crickets while I was learning how to use mtDNA to study the Gryllus hybrid zone in Rick Harrisons’ lab (Harrison et al. 1985). The microcosm of competing mtDNAs in the cytoplasm seemed like too interesting a problem to pass up, even if it was far afield from the ecological genetics that was the focus of my PhD thesis. It was an exciting time to be in Rick’s lab, and I owe much to Rick in making graduate school seem like summer science camp. The cricket heteroplasmy raised many questions and it seemed logical to do a population cage experiment in Drosophila, which was initiated as a postdoc in Dick Lewontin’s lab in 1988. From there, flies seemed like the best system to study how mtDNA variation is related to organismal fitness, and thus another person was sucked into the Drosophila model. I would like to thank W. Anderson, E. Arnason, D. Dykhuisen, A. MacRae, T. Prout for helpful comments and K. Zvonar for help with the frequency estimates. An anonymous reviewer provided many helpful comments that significantly improved the manuscript. Supported by National Research Service Award GM12357 to DMR and Grant GM21179 to R. C. Lewontin from the NIH, and grants DEB-9120293 from the NSF, grant GM067862 from the NIH and grant AG027849 from the NIA to DMR.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10709-011-9576-y) contains supplementary material, which is available to authorized users.
References
- Arnason E, Rand DM. Heteroplasmy of short tandem repeats in mitochondrial DNA of Atlantic cod, Gadus morhua. Genetics. 1992;132:211–220. doi: 10.1093/genetics/132.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avise JC. Mitochondrial DNA and the evolutionary genetics of higher animals. Philos Trans R Soc Lond B Biol Sci. 1986;312(1154):325–342. doi: 10.1098/rstb.1986.0011. [DOI] [PubMed] [Google Scholar]
- Avise JC. Phylogeography: the history and formation of species. Harvard University Press; Harvard: 2000. [Google Scholar]
- Ballard JW, James AC. Differential fitness of mitochondrial DNA in perturbation cage studies correlates with global abundance and population history in Drosophila simulans. Proc Biol Sci. 2004;271(1544):1197–1201. doi: 10.1098/rspb.2004.27096QVXE53Q0031KLN0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard JWO, Kreitman M. Unraveling selection in the mitochondrial genome of Drosophila. Genetics. 1994;138(3):757–772. doi: 10.1093/genetics/138.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard JWO, Rand DM. The population biology of mitochondrial DNA and its phylogenetic implications. Annu Rev Ecol Evol Syst. 2005;36:621–642. doi: 10.1146/Annurev.Ecolsys.36.091704.175513. [DOI] [Google Scholar]
- Bazin E, Glemin S, Galtier N. Population size does not influence mitochondrial genetic diversity in animals. Science. 2006;312(5773):570–572. doi: 10.1126/science.1122033. [DOI] [PubMed] [Google Scholar]
- Bender W, Spierer P, Hogness DS. Chromosomal walking and jumping to isolate DNA from the Ace and rosy loci and the bithorax complex in Drosophila melanogaster. J Mol Biol. 1983;168(1):17–33. doi: 10.1016/s0022-2836(83)80320-9. [DOI] [PubMed] [Google Scholar]
- Birky CW., Jr The inheritance of genes in mitochondria and chloroplasts: laws, mechanisms, and models. Annu Rev Genet. 2001;35:125–148. doi: 10.1146/annurev.genet.35.102401.090231. [DOI] [PubMed] [Google Scholar]
- Brown JR, Beckenbach AT, Smith MJ. Mitochondrial DNA length variation and hetroplasmy in populations of white sturgeon (Acipenser transmontanus) Genetics. 1992;132:221–228. doi: 10.1093/genetics/132.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagna AE, Addis J, McInnes RR, Clarke JT, Ashby P, Blaser S, Robinson BH. Late onset Leigh syndrome and ataxia due to a T to C mutation at bp 9, 185 of mitochondrial DNA. Am J Med Genet A. 2007;143A(8):808–816. doi: 10.1002/ajmg.a.31637. [DOI] [PubMed] [Google Scholar]
- Clark AG, Lyckegaard EM. Natural selection with nuclear and cytoplasmic transmission. III. Joint analysis of segregation and mtDNA in Drosophila melanogaster. Genetics. 1988;118(3):471–481. doi: 10.1093/genetics/118.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun PE, Beal MF, Wallace DC. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci USA. 2004;101(29):10726–10731. doi: 10.1073/pnas.04036491010403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun PE, Wyrembak J, Derbereva O, Melkonian G, Doran E, Lott IT, Head E, Cotman CW, Wallace DC. Systemic mitochondrial dysfunction and the etiology of alzheimer’s disease and down syndrome dementia. J Alzheimers Dis. 2010;20(Suppl 2):S293–S310. doi: 10.3233/JAD-2010-100351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree LM, Samuels DC, de Sousa Lopes SC, Rajasimha HK, Wonnapinij P, Mann JR, Dahl HH, Chinnery PF. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet. 2008;40(2):249–254. doi: 10.1038/ng.2007.63. [DOI] [PubMed] [Google Scholar]
- Crow JF, Morton NE. Measurement of gene frequency drift in small populations. Evolution. 1955;9:202–214. [Google Scholar]
- Dawkins R. The selfish gene. Oxford University Press; Oxford: 1976. [Google Scholar]
- Dowling DK, Friberg U, Lindell J. Evolutionary implications of non-neutral mitochondrial genetic variation. Trends Ecol Evol. 2008;23(10):546–554. doi: 10.1016/j.tree.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Dykhuizen D, Hartl DL. Selective neutrality of 6PGD allozymes in E. coli and the effects of genetic background. Genetics. 1980;96(4):801–817. doi: 10.1093/genetics/96.4.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykhuizen DE, Hartl DL. Selection in chemostats. Microbiol Rev. 1983;47(2):150–168. doi: 10.1128/mr.47.2.150-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauron CM, Wolstenholme DR. Structural heterogeneity of mitochondrial DNA molecules within the genus Drosophila. Proc Natl Acad Sci USA. 1976;73(10):3623–3627. doi: 10.1073/pnas.73.10.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauron CM, Wolstenholme DR. Intraspecific diversity of nucleotide sequences within the adenine + thymine-rich region of mitochondrial DNA molecules of Drosophila mauritiana, Drosophila melanogaster and Drosophila simulans. Nucleic Acids Res. 1980;8(22):5391–5410. doi: 10.1093/nar/8.22.5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fos M, Dominguez MA, Latorre A, Moya A. Mitochondrial DNA evolution in experimental populations of Drosophila subobscura. Proc Natl Acad Sci USA. 1990;87(11):4198–4201. doi: 10.1073/pnas.87.11.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale LR, Singh RS. Extensive variation and heteroplasmy in size of mitochondrial DNA among geographic populations of Drosophila melanogaster. Proc Nat Acad Sci USA. 1986;78:8813–8817. doi: 10.1073/pnas.83.22.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale LR, Singh RS. A comprehensive study of genic variation in natural populations of Drosophila melanogaster. IV. Mitochondrial DNA variation and the role of history vs selection in the genetic structure of geographic populations. Genetics. 1991;129(1):103–117. doi: 10.1093/genetics/129.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RG. Mitochondrial DNA as a genetic marker in population and evolutionary genetics. Trends Ecol Evol. 1989;4:6–11. doi: 10.1016/0169-5347(89)90006-2. [DOI] [PubMed] [Google Scholar]
- Harrison RG, Rand DM, Wheeler WC. Mitochondrial DNA size variation within individual crickets. Science. 1985;228(4706):1446–1448. doi: 10.1126/science.228.4706.1446. [DOI] [PubMed] [Google Scholar]
- Hutter CM, Rand DM. Competition between mitochondrial haplotypes in distinct nuclear genetic environments: Drosophila pseudoobscura vs. D. persimilis. Genetics. 1995;140(2):537–548. doi: 10.1093/genetics/140.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AC, Ballard JWO. Mitochondrial genotype affects fitness in D. simulans. 2003 doi: 10.1093/genetics/164.1.187. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann LM, Rosenblum EB, Rand DM. Aging, mating, and the evolution of mtDNA heteroplasmy in Drosophila melanogaster. Proc Natl Acad Sci USA. 1998;95(5):2372–2377. doi: 10.1073/pnas.95.5.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick ST, Rand DM. Conditional hitchhiking of mitochondrial DNA: Frequency shifts of Drosophila melanogaster mtDNA variants depend on nuclear genetic background. Genetics. 1995a;141:1113–1124. doi: 10.1093/genetics/141.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick ST, Rand DM. Fitness of Drosophila melanogaster mitochondrial DNA variants depends on nuclear genetic background. Genetics. 1995b doi: 10.1093/genetics/141.3.1113. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI. The fundamental units, processes and patterns of evolution, and the tree of life conundrum. Biol Direct. 2009;4:33. doi: 10.1186/1745-6150-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DL, Farr CL, Farquhar AL, Kaguni LS. Sequence, organization, and evolution of the A + T region of Drosophila melanogaster mitochondrial DNA. Mol Biol Evol. 1994;11:523–538. doi: 10.1093/oxfordjournals.molbev.a040132. [DOI] [PubMed] [Google Scholar]
- Lewontin RC. The units of selection. Annu Rev Ecol Syst. 1970;1:1–18. [Google Scholar]
- Lewontin RC, Dunn LC. The evolutionary dynamics of a polymorphism in the house mouse. Genetics. 1960;45(6):705–722. doi: 10.1093/genetics/45.6.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Butow RA. Mitochondrial retrograde signaling. Annu Rev Genet. 2006;40:159–185. doi: 10.1146/annurev.genet.40.110405.090613. [DOI] [PubMed] [Google Scholar]
- MacRae AF, Anderson WW. Evidence for non-neutrality of mitochondrial DNA haplotypes in Drosophila pseudoobscura. Genetics. 1988;120(2):485–494. doi: 10.1093/genetics/120.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Montooth KL, Rand DM. Positive and negative selection on the mitochondrial genome. Trends Genet. 2007;23(6):259–263. doi: 10.1016/j.tig.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science. 1999;286(5440):774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- Nabholz B, Mauffrey JF, Bazin E, Galtier N, Glemin S. Determination of mitochondrial genetic diversity in mammals. Genetics. 2008;178(1):351–361. doi: 10.1534/genetics.107.073346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Tajima F. Genetic drift and estimation of effective population size. Genetics. 1981;98(3):625–640. doi: 10.1093/genetics/98.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira MT, Kaguni LS. Comparative purification strategies for Drosophila and human mitochondrial DNA replication proteins: DNA polymerase gamma and mitochondrial single-stranded DNA-binding protein. Methods Mol Biol. 2009;554:37–58. doi: 10.1007/978-1-59745-521-3_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM. Endotherms, ectotherms, and mitochondrial genome-size variation. J Mol Evol. 1993;37:281–295. doi: 10.1007/BF00175505. [DOI] [PubMed] [Google Scholar]
- Rand DM. Thermal habit, metabolic rate and the evolution of mitochondrial DNA. Trends Ecol Evol. 1994;9(4):125–131. doi: 10.1016/0169-5347(94)90176-7. [DOI] [PubMed] [Google Scholar]
- Rand DM. The units of selection on mitochondrial DNA. Annu Rev Ecol Syst. 2001;32:415–448. [Google Scholar]
- Rand DM, Harrison RG. Mitochondrial DNA transmission genetics in crickets. Genetics. 1986;114(3):955–970. doi: 10.1093/genetics/114.3.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Harrison RG. Molecular population genetics of mtDNA size variation in crickets. Genetics. 1989;121:551–569. doi: 10.1093/genetics/121.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Dorfsman ML, Kann LM. Neutral and non-neutral evolution of Drosophila mitochondrial DNA. Genetics. 1994;138:741–756. doi: 10.1093/genetics/138.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Clark AG, Kann LM. Sexually antagonistic cytonuclear fitness effects in Drosophila melanogaster. Genetics. 2001;159:173–187. doi: 10.1093/genetics/159.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solignac M, Genermont J, Monnerot M, Mounolou J-C. Genetics of mitochondria in Drosophila: mtDNA inheritance in heteroplasmic strains of D. mauritiana. Mol Gen Genet. 1984;197:183–188. [Google Scholar]
- Solignac M, Monnerot M, Mounolou J-C. Mitochondrial DNA evolution in the melanogaster species subgroup of Drosophilia. J Mol Evol. 1986;23:31–40. doi: 10.1007/BF02100996. [DOI] [PubMed] [Google Scholar]
- Solignac M, Genermont J, Monnerot M, Mounolou J-C. Drosophila mitochondrial genetics: evolution of heteroplasmy through germ line cell divisions. Genetics. 1987;117:687–696. doi: 10.1093/genetics/117.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DR, Zeyl C, Cooke E. Conflicting levels of selection in the accumulation of mitochondrial defects in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2002;99(6):3690–3694. doi: 10.1073/pnas.072660299072660299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend JP, Rand DM. Mitochondrial genome size variation in new world and old world populations of Drosophila melanogaster. Heredity. 2004;93(1):98–103. doi: 10.1038/sj.hdy.68004846800484. [DOI] [PubMed] [Google Scholar]
- Van Leeuwen T, Vanholme B, Van Pottelberge S, Van Nieuwenhuyse P, Nauen R, Tirry L, Denholm I. Mitochondrial heteroplasmy and the evolution of insecticide resistance: non-Mendelian inheritance in action. Proc Natl Acad Sci USA. 2008;105(16):5980–5985. doi: 10.1073/pnas.0802224105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Teoli D, Shoubridge EA. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat Genet. 2008;40(12):1484–1488. doi: 10.1038/ng.258. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem. 2007;76:781–821. doi: 10.1146/annurev.biochem.76.081205.150955. [DOI] [PubMed] [Google Scholar]
- Waples RS. A generalized approach for estimating effective population size from temporal changes in allele frequency. Genetics. 1989;121(2):379–391. doi: 10.1093/genetics/121.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittam TS, Clark AG, Stoneking M, Cann RL, Wilson AC. Allelic variation in human mitochondrial genes based on patterns of restriction site polymorphism. Proc Natl Acad Sci USA. 1986;83(24):9611–9615. doi: 10.1073/pnas.83.24.9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC. Adaptation and natural selection. Princeton University Press; Princeton, New Jersey: 1966. [Google Scholar]
- Wright S. The theory of gene frequencies. Vol. 2. University of Chicago Press; Chicago: 1968. Evolution and the genetics of populations. [Google Scholar]
- Wynne-Edwards VC. Animal dispersion in relation to social behavior. Oliver and Boyd; Edinburgh: 1962. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.