

Abstract
To date, CCR5 variants remain the only human genetic factors to be confirmed to impact HIV-1 acquisition. However, protective CCR5 variants are largely absent in African populations, in which sporadic resistance to HIV-1 infection is still unexplained. Here we perform a genome-wide association study (GWAS) in a population of 1,532 individuals from Malawi, a country with high prevalence of HIV-1 infection, to investigate whether common genetic variants associate with HIV-1 susceptibility in Africans. Using single nucleotide polymorphisms (SNPs) present on the genome-wide chip, we also investigated previously reported associations with HIV-1 susceptibility or acquisition. Recruitment was coordinated by the Center for HIV/AIDS Vaccine Immunology at two sexually transmitted infection clinics. HIV status was determined by HIV rapid tests and nucleic acid testing.
After quality control, the population consisted of 848 high-risk seronegative and 531 HIV-1 seropositive individuals. Logistic regression testing in an additive genetic model was performed for SNPs that passed quality control. No single SNP yielded a significant P-value after correction for multiple testing. The study was sufficiently powered to detect markers with genotype relative risk ≥ 2.0 and minor allele frequencies ≥12%. This is the first GWAS of host determinants of HIV-1 susceptibility, performed in an African population. The absence of any significant association can have many possible explanations: rarer genetic variants or common variants with weaker effect could be responsible for the resistance phenotype; alternatively, resistance to HIV-1 infection might be due to non-genetic parameters or to complex interactions between genes, immunity and environment.
Keywords: Human immunodeficiency virus (HIV-1), acquisition, resistance, Genome Wide Association Study (GWAS), Africa
INTRODUCTION
Throughout the history of the AIDS epidemic, subsets of individuals have appeared to resistHIV-1 infection despite multiple exposures to the virus [1, 2]. However, almost 30 years after the first description of AIDS, variants in the CCR5 gene remain the only human genetic variants that have been proven to significantly impact HIV-1 acquisition [3, 4]. When present in homozygous or combined heterozygous form, the Δ32 deletion and the much rarer m303T>A point mutation confer complete resistance to infection by viruses that use CCR5 as co-receptor. Nevertheless, these mutations only explain a fraction of the apparently HIV-1 exposed, yet uninfected cases. Importantly, they are only found in individuals with a northern European or central Asian heritage and thus are not responsible for resistance observed in African populations [5]. The identification of additional human genetic factors influencing HIV-1 susceptibility would shed new light on transmission mechanisms and pathogenesis, and potentially suggest novel preventive or therapeutic approaches.
Genome-wide association studies (GWAS) are a widely accepted approach for the investigation of common genetic variation in the human genome [6]. Not relying on candidate gene selection, these hypothesis-free studies have the potential to implicate new genomic regions and pathways affecting complex human traits and diseases. Several recent GWAS have provided a detailed description of how common variation influences control of HIV-1 in infected individuals from European and African America ancestry [7 – 10]. To date, however, there have been no reported GWAS studies of HIV-1 resistance/susceptibility. A major reason for this has been the difficulty in recruiting enough well-characterized, highly exposed, yet seronegative individuals for an adequately powered study [11]. We here describe the first GWAS of host determinants of HIV-1 susceptibility, performed in a homogeneous African population.
METHODS
STUDY POPULATION
To identify common gene variants influencing HIV-1 acquisition in the highly affected Sub-Saharan African region, we performed a genome-wide association study in a population of over 1,500 individuals recruited from two Sexually Transmitted Infections (STI) clinics in Blantyre and Lilongwe, Malawi. These clinics are integrated in the Center for HIV/AIDS Vaccine Immunology (CHAVI) Clinical Core. The prevalence of HIV-1 infection in Malawi is one of the highest in the world, with an estimated 12% of adults infected [12]. An even higher prevalence (around 30%) was observed among the patients screened for this study. We therefore assume that sexually active individuals recruited at these sites were likely to have been exposed to the HIV virus. We did not, however, collect information about individual exposure level, sexual orientation, or intravenous drug use.
This study was approved by all local and by the sponsoring institution’s ethics committees. All participants consented to a blood sample collection and genetic testing. HIV status was determined by HIV rapid tests and nucleic acid testing (NAT): a positive HIV-1 diagnosis required a confirmed positive rapid test, and a negative HIV-1 diagnosis was based on two negative rapid tests followed by a negative NAT, or discordant results from rapid tests with a negative NAT.
GWAS GENOTYPING AND QUALITY CONTROL
DNA samples were genotyped using either the Illumina Human1M or 1M-Duo DNA Analysis BeadChips. Genotype clustering was performed using the Infinium BeadStudio program. Samples that obtained a very low intensity or call rate (<99%) were excluded. Further quality control was performed using PLINK [13], by checking the genetic gender and removing the gender misclassified individuals. Then, cryptic relatedness was assessed using pair-wise identity-by-descent (IBD). All pairs of DNA samples showing ≥0.125 (estimated proportion of alleles IBD) were individually inspected, and one sample in each pair was excluded from further analyses.
To account for the possibility of spurious associations resulting from residual population stratification, we used a modified EIGENSTRAT method to correct for population ancestry within the remaining case and control data [14].
GENOME-WIDE ASSOCIATION ANALYSIS
We searched for an association between HIV infection status and each of the single-marker genotypes by logistic regression in an additive genetic model using PLINK, correcting for age, gender and the significant principal component analysis axes identified with EIGENSTRAT. Bonferroni correction was applied to correct for multiple testing; however, we first used a linkage disequilibrium pruning procedure to remove entirely dependent markers, defined as r2=1, and then used the Bonferroni adjustment based on this reduced set of SNPs. This allowed for improved control of multiple marker testing.
Power calculations for association analysis were performed using the genetic power calculator (GPC) (available at http://pngu.mgh.harvard.edu/~purcell/gpc/)[15].
Previous studies have reported variants that might influence HIV-1 susceptibility in other populations. We investigated whether these previously reported associations could be replicated in a genome-wide context within the Malawi study by looking at the SNP variants that have previously been published with a p<0.05. If the originally reported SNP was not genotyped, we report the best available proxy SNP based on the HapMap YRI data, also reporting the r2 value. Moreover, we report the SNP with the lowest p value for each of the previously reported candidate genes.
RESULTS
A total of 1,532 Chichewa-speaking individuals recruited from Malawi STI clinics between December 2006 and August 2008 were genotyped. Of these, n=922 (60.2%) were HIV negative cases and n=610 (39.8%) were HIV positive controls.
DNA samples from 86 individuals (6%) did not pass initial quality control filtering. An additional 19 individuals were removed due to gender misclassification, and 37 individuals were removed due to cryptic relatedness. In addition, to assess population stratification, PCA was performed on a subset of 191,212 SNPs not in Linkage Disequilibrium. In the first iteration, 11 outliers were identified and excluded. Following the above quality control steps, the population adopted in the association testing consisted of 848 HIV-negative cases, of which 52% were females, and531 HIV-positive controls, of which 62% were females. Age distribution significantly differed between the HIV-1 seropositive and seronegative samples (median 29 [range, 18–62] vs. 29 [range, 20–66], p=0.002).
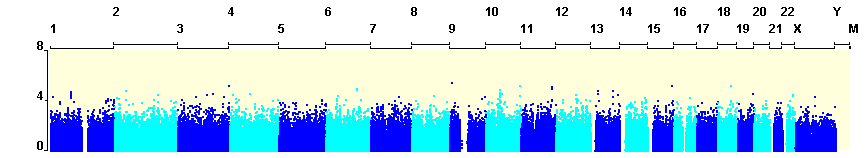
Logistic regression testing in an additive genetic model was performed for the 844,489single markers that passed quality control. No single SNP yielded a P-value below the Pcutoff = 6.03×10−8 (Supplementary Figure 1, Manhattan Plot). An annotated list of all markers obtaining a P-value less than 2×10−4 was generated using WGA viewer software[16](Supplementary Table1).
The Q-Q plot of the GWAS P-value distribution shows that the distributions of the observed and expected P-values are very similar with a lambda value of 0.9982 that suggests no inflation of association signals after correction for population stratification.
As an additional subset analysis, based on previously published reports of variants associated with HIV-1 susceptibility, we checked the p values across 22candidate genes of 36previously reported candidate SNPs or their closest proxies within 100kb. For each gene, we report the candidate SNPs when possible, and otherwise the best available proxy. We also report the lowest p-value identified within each candidate gene, first uncorrected and then corrected for the number of SNPs analysed in that gene (Table 1). However, failure to find a significant association within a candidate gene where the originally reported SNP was not examined does not translate to a failure to replicate the original association. We were able to directly test 17 of the 36 SNPs previously reported. For those SNPs not present on the chips, none have good proxies r2>0.8. Of these17SNPs, only rs1946518–IL18, originally reported to increase susceptibility to HIV-1 infection in a pediatric Brazilian population (p=0.02) [17], was significant in our Malawi study at the p<0.05 level where the C allele is significantly more represented in the HIV positive Malawi group than the high-risk seronegative group (67 vs. 62%, p=0.004). The meta p-value remains non-significant at the genome-wide level, p=0.001, Stouffer’s z trend.
Table 1. Candidate study of previously reported variants associated with HIV-1 susceptibility.
Column 1) Reported candidate gene; Column 2) The identical candidate SNPs originally identified and available after passing QC on the 1M-Duo chip; Column 3) The identical candidate SNPs originally identified and available via a proxy found on the 1M-Duo chip; Column 4) The proxy SNP and the corresponding r2 value as identified in the YRI population for the candidate SNP identified in column 3; Column 5) the lowest p-value for the SNPs identified in Column 2, or Column 4, if a proxy SNP; Column 6) the lowest p-value for SNPs linked to the candidate gene; Column 7) The number of SNPs tested in each candidate gene; Column 8) gene-wide corrected p-value for the lowest p-value identified within the candidate gene; Column 9) set-wide corrected p-value for the lowest p-value identified per candidate gene and corrected for all candidate genes investigated; Column 10) genome-wide corrected p-value for the lowest p-value identified per candidate gene; and Column 11) Studies reporting corresponding candidate SNPs and genes
| Gene | previously associated SNPs | previously associated SNPs represented by an LD proxy | Closest proxy (r2 in YRI) | SNP replication P | lowest P in gene | Number of SNPs tested in gene | Gene-Wide correction | Set-wide correction | Genome- wide correction | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| ABCB1 | rs1045642 | 0.87 | 0.004 | 105 | 0.42 | 1 | 1 | Fellay Jet al.,2002; Lancet, 359:30–36 | ||
| APOBEC3G | 0.26 | 4 | 1 | 1 | 1 | Valcke et al., 2006; AIDS, 20:1984–1986 | ||||
| CCL2 | rs1024610 rs1024611 |
0.54 | 0.0008 | 175 | 0.14 | 0.96 | 1 | Modi et al., 2003; AIDS, 17:2357–2365 | ||
| CCL3 | rs1719134 | rs1719126 (0.43) | 0.41 | 0.1 | 3 | 0.3 | 1 | 1 | Gonzalez et al., 2001; Proc Natl Acad Sci USA, 98: 5199–5204 | |
| CCL4 | 0.15 | 6 | 0.9 | 1 | 1 | Colobran et al., 2005; J Immunol, 174:5655–5664 | ||||
| CCL5 | rs2107538 rs2280789 |
0.68 | 0.04 | 10 | 0.4 | 1 | 1 | McDermott et al., 2000; AIDS, 14:2671–2678 Gonzalez et al., 2001; Proc Natl Acad Sci USA, 98: 5199–5204 An et al., 2002; Proc Natl Acad Sci USA, 99:10002–10007 Fernandez et al., 2003; AIDS Res Hum Retroviruses, 19: 349–352 Liu et al., 2004; J Infect Dis, 190:1055–1058 |
||
| CCL7 | 0.11 | 3 | 0.33 | 1 | 1 | Modi et al., 2003; AIDS, 17:2357–2365 | ||||
| CCL11 | 0.2 | 13 | 1 | 1 | 1 | Modi et al., 2003; AIDS, 17:2357–2365 | ||||
| CCR5 | 0.19 | 3 | 1 | 1 | 1 | [3] Dean et al., 1996 [4] Samson et al., 1996 Huang et al.,1996; Nat Med, 2:1240–1243 |
||||
| CD209 | 0.07 | 19 | 1 | 1 | 1 | Martin et al., 2004; J Virol, 78:14053–14056 | ||||
| CD4 | rs28919570 | 0.21 | 0.007 | 33 | 0.23 | 1 | 1 | Oyugi et al, 2009; J Infect Dis, 199:1327–1334 | ||
| CX3CR1 | rs3732378 rs3732379 |
0.56 | 0.03 | 32 | 0.96 | 1 | 1 | Faure et al., 2000; Science, 287:2274–2277 | ||
| CXCL12 | rs1801157 | rs10900029 (0.22) | 0.8 | 7.07E-05 | 179 | 0.01 | 0.08 | 0.6 | [15] Petersen et al., 2005 Modi et al., 2005; Genes Immun, 6:691–698 |
|
| DARC | rs2814778 | 1 | 0.1 | 8 | 0.8 | 1 | 1 | He et al., 2008; Cell Host Microbe, 4:52–62 | ||
| DEFB1 | 0.03 | 51 | 1 | 1 | 1 | Braida et al., 2004; AIDS, 18:1598–1600 Milanese et al., 2006, AIDS, 20:1673–1675 |
||||
| IL10 | rs1800872 rs1800896 |
0.2 | 0.007 | 18 | 0.13 | 1 | 1 | Shin et al., 2000; Proc Natl Acad Sci USA, 97:14467–144721 Naicker et al,2009; J Infect Dis, 200:448–452 |
||
| IL18 | rs1946518 | 0.004 | 0.004 | 17 | 0.07 | 1 | 1 | [14] Segat et al., 2006 | ||
| IRF1 | rs17848424 | 0.34 | 0.34 | 12 | 1 | 1 | 1 | Ball et al., 2007; AIDS, 21:1091–1101 | ||
| MBL2 | rs5030737 rs1800450 rs1800451 |
0.45 | 0.001 | 297 | 0.3 | 1 | 1 | Garred et al., 1997; Scand J Immunol, 46:204–208 Pastinen et al., 1998; AIDS Res Hum Retroviruses, 14:695–698 Boniotto et al., 2003; AIDS, 17:779–780 Vallinoto et al., 2005; Mol Immunol, 43:1358–1362 |
||
| PPIA | 0.03 | 9 | 0.27 | 1 | 1 | An et al., 2007; PLoS Pathog, 3:e88 Rits et al., 2008; PLoS One, 3:e3975 |
||||
| PTPRC | 0.03 | 188 | 1 | 1 | 1 | Tchilian et al, 2001; AIDS, 15:1892–1894 | ||||
| TRIM5 | rs3740996 | rs10838525 | rs10769175 (0.19) | 0.67 | 0.005 | 23 | 0.12 | 1 | 1 | Javanbakht et al, 2006; Virology, 354:15–27 |
Of the 22 candidate genes tested, 13 genes were found to have at least one SNP significant at the p<0.05 level, with the most strongly associated gene, CXCL12 [18], represented by 8 SNPs below the p<0.001 level. After correcting for the number of SNPs per gene, only rs2437935–CXCL12 remained significant with a p-value of 0.01 (corrected for 179 SNPs). However, that p-value increased to 0.085 when correcting for all 1,208 SNPs tested across the 22candidate genes.
DISCUSSION
Here we performed a GWAS of determinants of resistance to HIV-1 infection by testing for associations at over 800,000 SNPs. We failed to detect significant signals for differences in HIV-1 susceptibility in this study of samples collected from Malawi STI clinics. Our lowest p-value was 3.97×10−6, which is substantially higher than the Pcutoff estimated to be 6.03×10−8 on this dataset.
This is, to our knowledge, the first report of a genome-wide search for genetic variants associated with differences in susceptibility to HIV-1 infection. We studied a homogenous Sub-Saharan African population, comparing genotypes of HIV-infected and non-infected subjects that attended the same STI clinics in Malawi. Due to the high prevalence of HIV-1 in this region, it is believed that HIV negative individuals attending these STI clinics are in a high-risk category and are likely to have been exposed to the virus. However, clinical data on exposure details was not collected, and as such, we had no information on the number and type of sexual contacts, the number of partners, sexual orientation, co-infections, or discordance in long-term relationship with a known HIV-1 infected partner.
Failing to detect a GWAS signal in this study can have many possible explanations, including the hypothesis that resistance or reduced susceptibility to HIV-1 infection might be due to complex interactions between innate and acquired immunity, modulated by epistasis and environment. Non-genetic factors might include mode of transmission; concurrent STI infections; viral load of infected partner; and multiple viral strain exposures. However, it is still possible that resistance or reduced susceptibility to HIV-1 infection is due to common human genetic variants not identified in the present study, either because they are not represented, directly or indirectly, on the genome-wide genotyping chip that we used (the Illumina 1M-Duo chip is the best currently available platform for investigating African population [19], but it still has suboptimal coverage), or because they have relatively weak effects, undetected with our sample size. Even when there is incomplete exposure in the high-risk seronegative group, there is still an expectation of allele frequency imbalance since the HIV-positive individuals are infectable, and the power depends on the precise degree of exposure. Under the assumption that all seronegative individuals recruited at the Malawi STI clinics have some protection, given MAFs of 5% and 20%, under a additive disease model with a type I error rate of 6.03×10−8, our Malawi study provides at least 80% power to detect an association for HIV-1 reduced susceptibility, with genotype relative risks (GRR) of ≥2.65 and >1.7, respectively. Moreover, the power to detect markers with GRRs ≥2.0 was 13%, 68%, and 99% for markers with 5%, 10%, and 20% MAFs, respectively. Considering a larger population and thus increasing power might detect a signal from African populations. An alternative hypothesis to common causation would be that rare variants are causal, and therefore, resequencing efforts on highly exposed, HIV-1 uninfected individuals might return informative data on the genetic determinants of HIV-1 resistance.
Supplementary Material
{kind=link}
Acknowledgments
Funding:
Funding was provided by the NIAID Center for HIV-1/AIDS Vaccine Immunology grant AI067854.
We thank all the individuals that agreed to participate in the study and the staff at the two STI clinics (Blantyre and Lilongwe) in Malawi that recruited the participants. Funding was provided by the NIAID Center for HIV-1/AIDS Vaccine Immunology grant AI067854. SP acknowledges a Fellowship from the American Australian Association.
Footnotes
Conflicts of interest:
We have no conflicts of interest.
JF, KVS, NLL, AJM, BFH, MSC and DBG contributed to the design of the study. SP and JF analyzed the data. SP and JF wrote the paper and all coauthors reviewed the manuscript. NC, JK, GK, DDK and members of the CHAVI team designed, established and maintained the study cohort and provided the samples. All authors contributed to interpreting the data, revising the manuscript, and reading and approving the final version.
References
- 1.Detels R, Liu Z, Hennessey K, et al. Resistance to HIV-1 infection. Multicenter AIDS Cohort Study. J Acquir Immune Defic Syndr. 1994;7:1263–1269. [PubMed] [Google Scholar]
- 2.Lederman MM, Alter G, Daskalakis DC, et al. Determinants of Protection among HIV-Exposed Seronegative Persons: An Overview. J Infect Dis. 2010;202(S3):S333–S338. doi: 10.1086/655967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dean M, Carrington M, Winkler C, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study. San Francisco City Cohort, ALIVE Study. Science. 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 4.Samson M, Libert F, Doranz BJ, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 5.Fowke KR, Nagelkerke NJ, Kimani J, et al. Resistance to HIV-1 infection among persistently seronegative prostitutes in Nairobi, Kenya. Lancet. 1996;348(9038):1347–1351. doi: 10.1016/S0140-6736(95)12269-2. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 7.Fellay J, Shianna KV, Ge D, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Limou S, Le Clerc S, Coulonges C, et al. Genomewide Association Study of an AIDS-Nonprogression Cohort Emphasizes the Role Played by HLA Genes (ANRS Genomewide Association Study 02) J Infect Dis. 2009;199:419–426. doi: 10.1086/596067. [DOI] [PubMed] [Google Scholar]
- 9.Fellay J, Ge D, Shianna KV, et al. Common Genetic Variation and the Control of HIV-1 in Humans. PLoS Genet. 2009;5:e1000791. doi: 10.1371/journal.pgen.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pelak K, Goldstein DB, Walley NM, et al. Host Determinants of HIV-1 Control in African Americans. J Infect Dis. 2010;201(8):1141–1149. doi: 10.1086/651382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horton RE, McClaren PJ, Fowke K, Kimani J, Ball TB. Cohorts for the Study of HIV-1 Exposed but Uninfected Individuals: Benefits and Limitations. J Infect Dis. 2010;202(S3):S377–S381. doi: 10.1086/655971. [DOI] [PubMed] [Google Scholar]
- 12.UNAIDS. 2008 Report on the global AIDS epidemic. UNAIDS World Health Organization (WHO); 2008. viewed April 16–2010, < http://www.unaids.org/en/KnowledgeCentre/HIVData/GlobalReport/2008/2008_Global_report.asp>. [Google Scholar]
- 13.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am Journal Hum Genet. 2007;81(3):557–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal Components Analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 15.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19(1):149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 16.Ge D, Zhang D, Need AC, et al. WGA Viewer: Software for Genomic Annotation of Whole Genome Association Studies. Genome Res. 2008;18(4):640–643. doi: 10.1101/gr.071571.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segat L, Bevilacqua D, Boniotto M, et al. IL-18 gene promoter polymorphism is involved in HIV-1 infection in a Brazilian pediatric population. Immunogenetics. 2006;58:471–473. doi: 10.1007/s00251-006-0104-7. [DOI] [PubMed] [Google Scholar]
- 18.Petersen DC, Glashoff RH, Shrestha S, et al. Risk for HIV-1 infection associated with a common CXCL12 (SDF1) polymorphism and CXCR4 variation in an African population. J Acquir Immune Defic Syndr. 2005;40(5):521–526. doi: 10.1097/01.qai.0000186360.42834.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhangale TR, Rieder MJ, Nickerson DA. Estimating coverage and power for genetic association studies using near-complete variation data. Nature Genetics. 2008;40:841–843. doi: 10.1038/ng.180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.