

Abstract
As the fastest folding protein, the villin headpiece (HP35) serves as an important bridge between simulation and experimental studies of protein folding. Despite the simplicity of this system, experiments continue to reveal a number of surprises, including structure in the unfolded state and complex equilibrium dynamics near the native state. Using 2.5 ms of molecular dynamics and Markov state models, we connect to current experimental results in three ways. First, we present and validate a novel method for the quantitative prediction of triplet–triplet energy transfer experiments. Second, we construct a many-state model for HP35 that is consistent with previous experiments. Finally, we predict contact-formation time traces for all 1,225 possible triplet–triplet energy transfer experiments on HP35.
Keywords: protein dynamics, near-native dynamics
A detailed understanding of how proteins fold remains a grand challenge in biophysics, whose solution would yield both satisfying physical insight and useful biological applications. To approach this problem, many have carried out detailed studies of model systems such as the 35-residue villin headpiece protein (HP35) (1). Sometimes called "the hydrogen atom of protein folding," this small protein folds in only microseconds (2), yet retains the clear secondary structure and well-packed core characteristic of larger proteins. These features make HP35 an ideal model; in particular, the fast-folding timescale allows for a detailed connection between experiments and molecular dynamics simulations. The allure of in silico protein folding has led to several computational studies of this small protein. Early landmark simulations showed partial folding (3). Later simulations achieved a small number of complete folding trajectories (4) and suggested residue-level interactions involved in the rate limiting steps of folding. Today’s computers permit the study of multiple folding trajectories, permitting a statistically sound approach to questions of states, rates, and pathways (5, 6). With myriad computational and experimental studies of this tiny system, one may expect the story of HP35 to be complete.
However, recent experimental research highlights a number of questions and even a few surprises. Laser temperature jump studies showed deviations of observed relaxations from single-exponential behavior (7). Further studies suggested that such relaxations might be fit using several exponentials, indicating the possibility of high barriers within the unfolded or partially folded states (8, 9). NMR and IR measurements revealed residual structure in the unfolded state (10), particularly in the two N-terminal helices (11). Finally, triplet–triplet energy transfer (TTET) experiments have observed fluctuations between native and several nonnative conformations on multiple timescales (12). This flurry of results suggests that our understanding of this “simple” system remains incomplete.
Recent advances in experimental methods can resolve ever-finer details in the protein folding process (12, 13). Likewise, molecular mechanics force fields have steadily improved (14–16), with accompanying enhancements in computer performance (17, 18). Interpreting molecular dynamics simulations is facilitated by analysis methods such as Markov state models (MSMs) (19–22), which provide a probabilistic framework for describing the conformational transitions observed in simulations. Progress on these fronts suggests that simulation and experiment should be more tightly coupled than ever before, with computation giving specific, falsifiable predictions and experiments quickly testing them (4, 9, 23).
Here, we use molecular dynamics simulations and MSMs to investigate the dynamics of the fast-folding, double-norleucine HP35 mutant (7). By modeling TTET in an MSM framework, we show near-quantitative agreement with recent experiments. Our analysis identifies states consistent with the observations in ref. 12 and elucidates these states in atomic detail, suggesting a set of specific nonnative contacts that frequently occur in the N terminus and a fluctuation in the C terminus that leaves a helix partly but not completely unraveled. These phenomena are strikingly apparent in a comprehensive set of simulated TTET traces, whose fine structure is surprisingly robust to modeling and force field errors. We present these calculations as a challenge to the experimental community. Further validation of these predictions will critically test our understanding of HP35 and demarcate the limitations of state-of-the-art simulation.
Results
Comparing TTET Experiments to Simulations.
The recent application of TTET experiments to submicrosecond fluctuations of HP35 (12) raised the possibility of testing simulations at an unprecedented level of detail. We therefore sought to reproduce four experiments that probed the rate of contact formation between a xanthone donor and napthylalanine acceptor attached at residue pairs (0, 23), (7, 23), (35, 23), and (0, 35) (Fig. 1). (The experiments of ref. 12 attached 9-oxoxanthene-2-carboxylic acid to the N terminus via a peptide bond and called this position 0.) To make direct comparisons to TTET measurements, we developed a method for simulating contact-formation time traces that expands the MSM formalism to simultaneously model both conformational and triplet state dynamics (see Methods). We created MSMs based on molecular dynamics simulations of a fast-folding double-norleucine HP35 mutant (7), extending prior calculations (5) to include more than 1 ms of simulation and requiring more than 10 million CPU hours of computation (see Methods and SI Methods).
Fig. 1.

Probe locations For TTET experiments. The TTET experiments in ref. 12 monitored contact formation between a (donor, acceptor) pair inserted at positions (7, 23), (0, 23), (35, 23), and (0, 35). Residues 1 (blue), 7 (cyan), 23 (yellow), and 35 (red) and the tightly packed phenylalanine core (gray) are shown as sticks (PDB ID code 2F4K). The N terminus (residue 1) is located on the left and is colored in blue. TTET interaction radii are shown as 5-Å spheres on beta carbons.
Systematic error in comparing simulations to TTET experiments results from six primary sources: MSM error, uncertainty in modeling the TTET observable, differences between simulated and experimental constructs, differences in solution conditions between simulation and experiment, simulation force field error, and sampling limitations. First, we estimate the systematic error from MSM construction by examining several MSMs for a given simulation dataset (see Fig. S1). Second, simulated TTET depends on the relationship between TTET rate and donor-acceptor distance; the error associated with this relationship is minimized by fitting a single parameter (see Methods, SI Methods, and Fig. S2). Third, the Lys24Nle/Lys29Nle construct used in molecular dynamics simulations is different from the Met12Nle/double-fluorophore constructs used in the experiments of ref. 12. To address this error, we analyzed short simulations of a fluorophore-containing construct (see SI Methods and Fig. S2). Fourth, experiments were performed primarily at 278 K, but fixed charge force fields are optimized for 300 K; we performed most calculations at 300 K but repeated one set at 278 K for comparison. Fifth, we control the error due to force field uncertainty by using datasets simulated with different force field/solvation model combinations. In total, we analyzed four datasets (ff03-TIP3P-300K, ff03-GBSA-300K, ff99SB-ILDN-TIP3P-300K, and ff99SB-ILDN-TIP3P-278K). Finally, limited sampling introduces uncertainties in folding stability; this destabilizing effect can be mitigated by examining experimental data under denaturing conditions, as is discussed below (see Table S1). In the following, we report data from the ff99SB-ILDN-TIP3P-300K dataset, with errors bracketed by the differences between the four datasets, which dominate most other sources of error.
The first comparison (Fig. 2A) measures contact formation between residues 0 and 23. This process is thought to require disrupting the entire core of the protein, and is experimentally measured to be slower than 8 μs—TTET measurement is limited by the 10-μs xanthone lifetime (12). The simulation also finds a slow timescale (5.5 ± 2.0 μs; 1/e time) for this contact formation. The second comparison (Fig. 2B) involves another slow (7.8 ± 2.0 μs) unfolding process that is required for contact between 7 (the N terminus) and 23. The multiexponential MSM traces and the measurements overlay within systematic uncertainties. Furthermore, unlike experimental measurements of TTET, the simulation-based method can observe timescales beyond 10 μs by disabling the intrinsic xanthone decay (Fig. S3). Simulations predict 20–100-μs timescales for these processes, consistent with experimentally measured unfolding under mildly denaturing conditions (7).
Fig. 2.

Comparing simulated TTET to experiment. Simulated TTET traces are compared to experiments. The experimental data are calculated using the exponential fits in ref. 12. GdmCl concentrations are shown as color gradients, with the darkest representing 0 M. Simulated TTET is shown for all four datasets. A displays the TTET between residues (0, 23). B shows (7, 23). C shows (23, 35). D shows (0, 35). C also contains experimental data at (0 M GdmCl, 300 K). Simulations assume 10-μs xanthone decay.
A third comparison involves excitation transfer between probes at residues 23 and 35 and is sensitive to fluctuations of the C-terminal helix (Fig. 2C). Simulated and experimental TTET traces agree, suggesting a timescale of 400 ( ± 300) and 380 ns, respectively—an order of magnitude faster than global unfolding. For this probe location, experimental data were available at multiple temperatures. With the exception of the implicit solvent data, simulation predictions rest between the experimental values for 278 and 300 K. The experimental observation of a C-terminal unraveled state is not limited to TTET studies; previous NMR work has observed a strong temperature dependence of slow motion order parameters in this helix (24).
The fourth comparison (Fig. 2D) measures contact formation between residues 0 and 35. Fits of experimental TTET traces yielded very fast kinetics: a dominant nanosecond (< 50 ns) phase and an additional submicrosecond (330 ns) phase. Simulations predict somewhat slower TTET (150 ± 100 ns) than is observed experimentally, which may be attributed to two factors. First, the nonpolar interaction between fluorophores in the experimental construct may accelerate the kinetics of 0–35 contact formation. Control simulations including fluorophores suggest rate enhancement by a factor of three (see Fig. S2); similarly, CD experiments found the 0–35 construct up to 0.4 kcal/mol more stable than the other three constructs. Second, the present simulations (at 300 K) may favor the near-native state rather than the closely packed native state probed by experiments at 278 K, a point further discussed below. Indeed, all four TTET predictions agree with experiments at moderately denaturing (1–3 M GdmCl) conditions.
Simulation of Denaturant Dependence.
As a final comparison to TTET experiments, we carried out studies of GdmCl denaturation. Additional molecular dynamics simulations at 300 K using available GdmCl parameters (25) gave insignificant destabilization of HP35 by denaturant; the available parameterization may not be sufficiently transferable for use with the ff99SB-ILDN force field. We achieved a more robust and predictive analysis by combining MSM perturbation theory (SI Methods) with two separate models for GdmCl effects: Myer’s solvent accessible surface area (SASA) m-value correlation (26) and a group transfer free energy (GTFE) model (27). In terms of thermodynamics, these calculations yield m-values of 0.53 ± 0.2 kcal·mol-1·M-1 (SASA) and 0.46 ± 0.2 kcal·mol-1·M-1 (GTFE), in agreement with the measured value of 0.69 ± 0.1 kcal·mol-1·M-1 (12) (Fig. S4A). In terms of kinetics, simulated TTET traces show weakly decreasing or constant rates of 0–23, 7–23, and 23–35 contact formation with increasing GdmCl, and strongly increasing rates of 0–35 contact formation (Fig. S4 B–E). The 0 M GdmCl calculations are in quantitative agreement with experimental TTET measurements at 2 M GdCl (Fig. S4B), further confirming that simulations give an accurate picture of HP35 dynamics in moderately denaturing conditions.
Comparison to Prior Models.
The quantitative comparison between simulation and experiment described above requires a high-resolution MSM with 10,000 states or more. In contrast, prior models for HP35 folding typically use a small number of states to concisely explain existing experimental data. Thus, a key question is to what extent do simulations capture the behavior of previous empirical models.
Simulation gives reasonable agreement with prior experiment-based models of HP35’s structure and stability. Grouping states into folded and unfolded macrostates based on the similarity (rmsd) to the crystal structure [Protein Data Bank (PDB) ID code 2F4K] suggests an apparent two-state ΔG of -0.5 ± 0.5 kcal/mol (Table S1). Circular dichroism studies of the same mutant report a value of -4.0 kcal/mol (7). This thermodynamic comparison suggests that the native state is understabilized in the simulation, and is consistent with the agreement of simulations and TTET experiments in moderate concentrations of chemical denaturant.
To compare our model with previous experiments and to gain a qualitative picture of folding dynamics, we applied the Perron Cluster Cluster Analysis (28) model reduction procedure to the ff99SB-ILDN-TIP3P-300K dataset (see SI Methods). Reduced MSMs permit comparison to the recent four-state model based on TTET data (12). The four-state model included native (N), unlocked (N′), and intermediate (I) states to account for the experimentally observed near-native fluctuations, as well as a globally unfolded state U (U not shown in Fig. 3). In the previous model, the native state N was shown to be associated with close contact between the terminal residues 0 and 35. The near-native state (N′) was shown to be native-like, but lacking close contact between the terminal residues. Finally, the intermediate I was characterized by unfolding of the C-terminal helix, as measured by TTET between residues 23 and 35. The simulation-based reduced MSM is consistent with this picture (Fig. 3, Fig. S5, Fig. S6, and Table S2).
Fig. 3.

Two models for HP35 dynamics. The model from ref. 12 is shown in A and has three distinct states: native (N), near-native globule (N′), and C-terminal unraveled intermediate (I). (Reproduced with permission from ref. 12.) The ten most populated states (B) from a 1,000-state reduced MSM show clear similarity to the previous model for HP35.
From the 1,000-state reduced MSM, we focus on the ten most populated states, which are labeled 1 through 10 in order of population, with 1 being the most populated state (state properties are given in Table S2). We identify N with states 1 and 5; both of these states bring together the N and C termini, permitting 0–35 TTET transfer. State 1 is also the most similar to the crystal structure (PDB ID code 2F4K), as indicated by its 2.2-Å rmsd. The states 2, 6, 7, 8, and 9 are characterized by native-like secondary structure and topology, but with structural differences as compared to the crystallographic structure. We identify this ensemble of near-native states with the unlocked state (N′). States 3, 4, and 10 show visible unraveling of the C-terminal helix, suggesting a connection to the intermediate I. Indeed, state 3 puts residues 23 and 35 in contact, providing an explanation for the submicrosecond 23–35 TTET previously assigned to I. The relative populations of the states (N′, N, and I in decreasing population) in the (300 K) simulation are in qualitative agreement with experimental estimates at (278 K, 2 M GdmCl). Simulations thus capture HP35 dynamics under moderately denaturing conditions in which the native state (N) is sampled but the unlocked (N′) state is more frequently populated.
Structural Predictions from Simulations.
Despite the similarities above, the reduced MSM is distinct from the previous four-state model (12). We describe two differences here.
The previous model for the observed intermediate (I in ref. 12; states 3, 4, and 10 herein) proposed complete unfolding of the 11-residue C-terminal helix (residues 22–32) to allow contact formation between residues 23 and 35. Simulations suggest that this state involves only partial unraveling of the C-terminal helix, rather than a complete disordering. In particular, the segment 22–25 is helical; the next turn (residues 26–29) is partly unstructured, and the C-terminal residues (30–35) are fully disordered. Although the differences between the MSM description and the previous description are subtle, hydrogen/deuterium exchange measurements (1) support the molecular dynamics picture in finding a monotonic decrease in protection factors from residues 22–35 in the C-terminal helix; this monotonic decrease is in contrast to the approximately constant protection factors one would expect of a cooperatively unfolded helix. Additional experiments could further discriminate between these models, including measurements that monitor the position dependence of the rate of contact formation, as described below.
Second, simulations provide a structural model for a near-native, compact state (N′ in ref. 12; states 2, 6, 7, 8, and 9 herein) that accounts for rapid fluctuations measured in end-to-end contacts (12). This state, previously called an “unlocked” or “dry molten globule” ensemble, was hypothesized to display large fluctuations in the C-terminal helix, making it a natural stepping stone to the more disordered intermediate I (see above). In contrast, molecular dynamics simulations display strong structural heterogeneity in both the N-terminal and C-terminal helices (Figs. S6 and S7) (29). The MSM predicts not just fluctuations but also specific nonnative contacts that have not been previously described. Most notably, residues Asp3 and Ser15, which are well separated in the crystallographic model, are predicted to hydrogen bond via the Ser hydroxyl and Asp carboxylate groups at a frequency of up to 50% (Fig. S7). This contact should be testable experimentally, through NMR nuclear Overhauser effect measurements (of protons on Cβ).
A Comprehensive Set of TTET Predictions.
Rigorous validation of simulations requires not only consistency checks but also nontrivial predictions that can be quantitatively compared to experiments. We therefore extended our contact-formation analysis from four residue pairs to all pairwise combinations.
We present these predictions as a 35 × 35 matrix of timescales (1/e times in Fig. 4; full time-traces in SI Methods). Although the uncertainties in predicted contact timescales are in the range of 2–4 fold, these errors are much smaller than the overall span of timescales, which cover a 105-fold range. The fastest rates are subnanosecond and involve residue pairs that are in close contact in the most populated states. The slowest timescales extend beyond 100 μs and involve the proline-containing turn at residues 18–25. There are small differences in datasets with different force fields, solvation models, and temperatures, but, strikingly, the same overall pattern is observed in all four datasets (Fig. 4). On average, the 278 K data is 1.5 times slower than its 300 K counterpart; however, temperature comparisons at individual probe positions are limited by systematic uncertainty.
Fig. 4.

Simulated TTET for all residue pairs provide a single-residue resolution map of contact-formation timescales. The heat map shows the time required for TTET to drop to 1/e of its starting amplitude; minimum and maximum represent 1 ns and 100 μs, respectively. (A) Using the ff99SB-ILDN-TIP3P-300K dataset. (B) Using the ff99SB-ILDN-TIP3P-278K dataset. (C) Using the ff03-TIP3P-300K dataset. (D) Using the ff03-GBSA-300K dataset. No xanthone decay is assumed.
The most interesting features of the contact timescale matrix are sharp variations as the probe positions are shifted by single residues. For example, consider contact formation between residue 23 and residues 34 and 35. A previous model (12) suggested that contact formation for these pairs involves full disordering of the C-terminal helix (state I); under this model, the residue pair (23, 34) would have slightly faster timescales than the (23, 35) pair. In contrast, simulations give a different picture of I, with decreasing disorder from residue 35 to lower numbered residues. This picture gives a distinct prediction that TTET timescales for the (23, 34) pair will be nearly 10-fold slower than for (23, 35), although the magnitude of the change is near the limit of the systematic errors of the modeling/experiment comparison.
We further predict that TTET experiments probing contact formation between residue 1 and each of the other residues in HP35 should discover a finely varying spectrum of timescales (Fig. 5). The heterogeneous N terminus of the near-native state N′ places residues 1 and 15 in close contact with a rapid timescale of 20 ± 10 ns, despite the separation of these residues in the crystallographic conformation N. These nonnative contacts are quite specific. For example, residue 12 is also in this N-terminal region but is predicted to interact with residue 1 on a dramatically longer timescale (800 ± 200 ns) due to anchoring by the well-packed phenylalanine core. Further fine structure is apparent in contact-formation rates between residue 1 and residues in the remainder of the protein, including two sites (23 and 35) that have already been probed experimentally (12).
Fig. 5.

Timescales of contact formation involving residue 1. Experimental values for the 0–35 TTET process are plotted at 0, 1, and 2 M GdmCl. For the 0–23 TTET process, experimental data is shown as the interval [6 μs , 100 μs] to represent uncertainty due to the limited xanthone lifetime. No xanthone decay is assumed in the simulation values.
Discussion
Challenges of Direct Comparison of Simulation and Experiment.
Our investigation of HP35 is enabled by recent experimental methods that directly probe contact between specific molecular locations. Prior to these advances, comparisons of simulation and experiment were often limited by systematic error. Ideally, one would directly simulate the experimental observable, but it remains challenging to derive circular dichroism spectra or fluorescence changes (30) from structural models. Comparisons are further complicated by the presence of multiple structurally distinct states rather than two clearly separated folded and unfolded states. Kinetic measurements introduce additional difficulties, including the difficulty of performing measurements under native conditions and the extraction of relaxation rates rather than microscopic rates. In contrast, contact formation is easily compared to simulation and generally does not require assuming a small number of states.
These challenges appeared several times during our analysis. For example, the 2–4 kcal/mol difference in ΔGfold between the circular dichroism model and our own two-state approximation suggests an understabilized native state. However, the lack of a quantitative bridge between the spectroscopic measurement and simulation led to uncertainty in the two-state comparison. The TTET comparisons helped confirm the understabilization of simulated HP35, with the best agreement of traces and denaturant dependences to experiments under moderately denaturing conditions. Comparisons to structural models also highlighted the difficulty of interpreting experimental data. Simulations suggested heterogeneity at the terminal helices of HP35. Early experimental work made no mention of this, although crystallographic models exhibit high temperature factors in the N-terminal helix (Fig. S7) and NMR analysis produced few long-range restraints between this helix and the rest of the molecule (31). Contact-formation experiments identified a near-native state characterized by terminal fluctuations; its TTET signature permits quantitative comparison to simulation. Probes with nanosecond time resolution and residue-level structural resolution allow comparisons that may be unattainable using lower-resolution techniques.
Recent studies have pointed out other challenges in comparing simulation to experiment. Long simulations of HP35 in multiple force fields (32) have suggested that some mechanistic details, such as order of helix folding, may be sensitive to force field differences. This supports the approach used here, where independent datasets using two force fields (ff03 and ff99SB-ILDN) and two water models (TIP3P and GBSA) help evaluate the robustness of simulation-based predictions. Another recent study (33) points out that early simulation-based models of HP35 measured equilibrium constants that were too unfolded. The equilibrium estimates in the current work help ameliorate this deficiency by a more advanced scheme for MSM construction and improved datasets that contain more adequate sampling of the folded state. Even with advanced force field and analysis methods, some deviations between simulation and experiment are expected; for that reason, we stress that the current predictions appear most valid under mildly denaturing conditions.
Future Challenges.
In the future, faster simulations, well-calibrated denaturant models, and improved force fields should allow the simulation of HP35 across the wide range of solvent and temperature conditions accessible to experiment. Likewise, the present TTET model neglects the effect of fluorophores, except for the analysis presented in Fig. S2. Faster simulation methodologies, increasing computational power, and the facile incorporation of arbitrary chemical modifications will allow simulations to be performed with all choices of donor and acceptor locations. Finally, scaling to larger and more complex proteins may reveal near-native dynamics on a wider spectrum of timescales.
Conclusion.
Although direct, quantitative agreement between simulation and experiment remains challenging, we have demonstrated a straightforward method to simulate TTET in silico. Using this method, we have predicted 1,225 contact-formation timescales and shown them to be consistent with the four TTET-pairs experimentally probed so far. Simulations lead to a model consistent with existing TTET experiments but with structural differences compared to previous models. Beyond this comparison, atomic-detail simulations predict an ensemble of near-native states and a labyrinth of fine structure in HP35 contact-formation timescales, which will be testable with further TTET experiments. With an aggregate amount of information rivaling high-resolution structural experiments, contact formation provides a way to probe the kinetic, thermodynamic, and structural details of models at high temporal and spatial resolution. HP35 is not yet a “solved” problem, but the validation or refutation of our current predictions will be a powerful step toward that goal.
Methods
Theory: A Model For Simulating TTET.
TTET experiments involve stimulating (via laser) a triplet state in a donor (xanthone). Through the short-range Dexter mechanism, this triplet excitation is transferred to an acceptor (napthylalanine). The total population of the donor triplet state is recorded by monitoring the wavelength at which the donor triplet state decays. To simulate this experimental setup, we add an additional degree of freedom: a triplet state that starts on the donor and can transfer to the acceptor upon chromophore contact. Given an n-state MSM for the conformational dynamics of the protein, we model this effect by splitting each conformational state into two substates: one for each possible location of the triplet (Fig. 6). Because an experiment will typically monitor the wavelength of either the donor or acceptor, we call the two substates “light” and “dark.” To parameterize this 2n-state model requires 4n2 transition probabilities, which we can define with three assumptions. First, triplet transfer is irreversible. Second, excitation transfer only occurs within a given conformational state, because transitions between conformations modeled here occur on the 10-ns (or slower) timescale compared to the picosecond-timescale excitation transfer (12). Finally, the rate of triplet transfer within each conformational state is estimated using the distance between the donor and acceptor residues, quantified by transfer coefficients fi. The transition matrix for the joint conformational-triplet dynamics is thus given by the following equations, where P0is the conformational transition matrix and δi,j is the Kronecker delta:
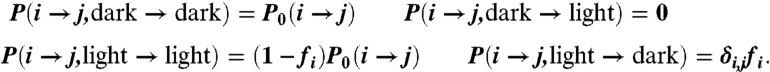 |
Fig. 6.

A schematic shows the splitting of each conformational state (Left) into two distinct electronic states (Right). The two electronic states represent two different possible locations for the excited triplet state (yellow). Experiments that monitor a single wavelength will detect only those molecules where the triplet is on the observable fluorophore. Thus, states with the excitation on the donor we call “light” (L) and the other states we call “dark” (D). Allowed transitions are marked by black arrows. Only conformational state 3 has the donor and acceptor in close contact; thus, the transition L → D is allowed only in state 3.
The transfer coefficients fi for each state were estimated using a 3-state model similar to the one found in ref. 12—see SI Methods. To simulate each raw TTET trace, the system is initially placed in its conformational equilibrium with all triplets on the donor (light). The total population with the triplet state on the donor was then monitored as a function of time, leading to the decaying traces in Fig. 2. In Fig. 2 and Figs. S1 and S4, plotted TTET amplitudes were reduced to account for the intrinsic xanthone decay (see SI Methods), which occurs on a 10-μs timescale. We note previous work using molecular dynamics to simulate contact formation (34).
Simulation Details.
The fast-folding double-norleucine mutant of HP35 (7) was simulated using Gromacs (35). Four independent datasets were used (ff03-TIP3P-300K, ff03-GBSA-300K, ff99SB-ILDN-TIP3P-300K, and ff99SB-ILDN-TIP3P-278K). Further details are provided in SI Methods.
MSM Details.
Simulation data were sampled at 1-ns intervals and clustered using the k-centers algorithm (21). Clustering rmsd calculations included all nonequivalent heavy atoms. The number of states was chosen by terminating k-centers when cluster radii reached 1.75 Å. Computations were carried out using version 2.0 of MSMBuilder (21, 36).
Supplementary Material
Acknowledgments.
We thank Greg Bowman for discussions on MSMs. Folding@Home donors provided computer resources. MSM construction was performed on the BioX2 cluster (National Science Foundation Award CNS-0619926). R.D. is funded by a Burroughs–Wellcome Career Award at the Scientific Interface. K.B. is funded by a Stanford Graduate Fellowship. We thank the National Science Foundation for support through Frontiers in Integrative Biological Research Grants EF-0623664, MCB-0954714, and DMS-0900700; the National Institutes of Health for support through Grants R01-GM062868 and Simbios U54-GM072970; and the National Science Foundation Award CNS-0619926 for computer resources. We thank Thomas Kiefhaber for discussing his results prior to publication.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: Contact formation time traces are provided at the National Institutes of Health Simbios Center for Biomedical Computing, https://simtk.org/home/hp35-datasets/.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1010880108/-/DCSupplemental.
References
- 1.McKnight JC, Doering DS, Matsudaira PT, Kim PS. A thermostable 35-residue subdomain within villin headpiece. J Mol Biol. 1996;260:126–134. doi: 10.1006/jmbi.1996.0387. [DOI] [PubMed] [Google Scholar]
- 2.Kubelka J, Eaton WA, Hofrichter J. Experimental tests of villin subdomain folding simulations. J Mol Biol. 2003;329:625–630. doi: 10.1016/s0022-2836(03)00519-9. [DOI] [PubMed] [Google Scholar]
- 3.Duan Y, Kollman PA. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 4.Zagrovic B, Snow CD, Shirts MR, Pande VS. Simulation of folding of a small alpha-helical protein in atomistic detail using worldwide-distributed computing. J Mol Biol. 2002;323:927–937. doi: 10.1016/s0022-2836(02)00997-x. [DOI] [PubMed] [Google Scholar]
- 5.Ensign DL, Kasson PM, Pande VS. Heterogeneity even at the speed limit of folding: Large-scale molecular dynamics study of a fast-folding variant of the villin headpiece. J Mol Biol. 2007;374:806–816. doi: 10.1016/j.jmb.2007.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freddolino PL, Schulten K. Common structural transitions in explicit-solvent simulations of villin headpiece folding. Biophys J. 2009;97:2338–2347. doi: 10.1016/j.bpj.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubelka J, Chiu TK, Davies DR, Eaton WA, Hofrichter J. Sub-microsecond protein folding. J Mol Biol. 2006;359:546–553. doi: 10.1016/j.jmb.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 8.Bowman GR, Pande VS. Protein folded states are kinetic hubs. Proc Natl Acad Sci USA. 2010;107:10890–10895. doi: 10.1073/pnas.1003962107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buscaglia M, Kubelka J, Eaton WA, Hofrichter J. Determination of ultrafast protein folding rates from loop formation dynamics. J Mol Biol. 2005;347:657–664. doi: 10.1016/j.jmb.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 10.Brewer SH, et al. Effect of modulating unfolded state structure on the folding kinetics of the villin headpiece subdomain. Proc Natl Acad Sci USA. 2005;102:16662–16667. doi: 10.1073/pnas.0505432102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meng W, Shan B, Tang Y, Raleigh DP. Native like structure in the unfolded state of the villin headpiece helical subdomain, an ultrafast folding protein. Protein Sci. 2009;18:1692–1701. doi: 10.1002/pro.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reiner A, Henklein P, Kiefhaber T. An unlocking/relocking barrier in conformational fluctuations of villin headpiece subdomain. Proc Natl Acad Sci USA. 2010;107:4955–4960. doi: 10.1073/pnas.0910001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lapidus LJ, Eaton WA, Hofrichter J. Measuring the rate of intramolecular contact formation in polypeptides. Proc Natl Acad Sci USA. 2000;97:7220–7225. doi: 10.1073/pnas.97.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindorff-Larsen K, et al. Improved side-chain torsion potentials for the amber ff99sb protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Best RB, Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J Phys Chem B. 2009;113:9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duan Y, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 17.Friedrichs MS, et al. Accelerating molecular dynamic simulation on graphics processing units. J Comput Chem. 2009;30:864–872. doi: 10.1002/jcc.21209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw DE, et al. Anton, a special-purpose machine for molecular dynamics simulation. ACM SIGARCH Computer Architecture News. 2007;35:1–12. [Google Scholar]
- 19.Chodera JD, Singhal N, Pande VS, Dill KA, Swope WC. Automatic discovery of metastable states for the construction of Markov models of macromolecular conformational dynamics. J Chem Phys. 2007;126:155101. doi: 10.1063/1.2714538. [DOI] [PubMed] [Google Scholar]
- 20.Noé F, Fischer S. Transition networks for modeling the kinetics of conformational change in macromolecules. Curr Opin Struct Biol. 2008;18:154–162. doi: 10.1016/j.sbi.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 21.Bowman GR, Beauchamp KA, Boxer G, Pande VS. Progress and challenges in the automated construction of Markov state models for full protein systems. J Chem Phys. 2009;131:124101. doi: 10.1063/1.3216567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noé F, Schütte C, Vanden-Eijnden E, Reich L, Weikl TR. Constructing the equilibrium ensemble of folding pathways from short off-equilibrium simulations. Proc Natl Acad Sci USA. 2009;106:19011–19016. doi: 10.1073/pnas.0905466106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snow CD, Nguyen H, Pande VS, Gruebele M. Absolute comparison of simulated and experimental protein-folding dynamics. Nature. 2002;420:102–106. doi: 10.1038/nature01160. [DOI] [PubMed] [Google Scholar]
- 24.Vugmeyster L, McKnight CJ. Slow motions in chicken villin headpiece subdomain probed by cross-correlated NMR relaxation of amide NH bonds in successive residues. Biophys J. 2008;95:5941–5950. doi: 10.1529/biophysj.108.134320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Camilloni C, et al. Urea and guanidinium chloride denature protein l in different ways in molecular dynamics simulations. Biophys J. 2008;94:4654–4661. doi: 10.1529/biophysj.107.125799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auton M, Holthauzen LMF, Bolen DW. Anatomy of energetic changes accompanying urea-induced protein denaturation. Proc Natl Acad Sci USA. 2007;104:15317–15322. doi: 10.1073/pnas.0706251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deuflhard P, Huisinga W, Fischer A, Schütte C. Identification of almost invariant aggregates in reversible nearly uncoupled Markov chains. Linear Algebra Appl. 2000;315:39–59. [Google Scholar]
- 29.Lei H, Wu C, Liu H, Duan Y. Folding free-energy landscape of villin headpiece subdomain from molecular dynamics simulations. Proc Natl Acad Sci USA. 2007;104:4925–4930. doi: 10.1073/pnas.0608432104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogers DM, Hirst JD. First-principles calculations of protein circular dichroism in the near ultraviolet. Biochemistry. 2004;43:11092–11102. doi: 10.1021/bi049031n. [DOI] [PubMed] [Google Scholar]
- 31.McKnight CJ, Matsudaira PT, Kim PS. NMR structure of the 35-residue villin headpiece subdomain. Nat Struct Biol. 1997;4:180–183. doi: 10.1038/nsb0397-180. [DOI] [PubMed] [Google Scholar]
- 32.Piana S, Lindorff-Larsen K, Shaw DE. How robust are protein folding simulations with respect to force field parameterization? Biophys J. 2011;100:L47–L49. doi: 10.1016/j.bpj.2011.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cellmer T, Buscaglia M, Henry ER, Hofrichter J, Eaton WA. Making connections between ultrafast protein folding kinetics and molecular dynamics simulations. Proc Natl Acad Sci USA. 2011;108:6103–6108. doi: 10.1073/pnas.1019552108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeh IC, Hummer G. Peptide loop-closure kinetics from microsecond molecular dynamics simulations in explicit solvent. J Am Chem Soc. 2002;124:6563–6568. doi: 10.1021/ja025789n. [DOI] [PubMed] [Google Scholar]
- 35.Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306–317. [Google Scholar]
- 36.Bowman GR, Huang X, Pande VS. Using generalized ensemble simulations and Markov state models to identify conformational states. Methods. 2009;49:197–201. doi: 10.1016/j.ymeth.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.