

Abstract
The all-ferric [Fe4S4]4+ cluster [Fe4S4{N(SiMe3)2}4] 1 and its one-electron reduced form [1]- serve as convenient precursors for the synthesis of 3∶1-site differentiated [Fe4S4] clusters and high-potential iron-sulfur protein (HiPIP) model clusters. The reaction of 1 with four equivalents (equiv) of the bulky thiol HSDmp (Dmp = 2,6-(mesityl)2C6H3, mesityl = 2,4,6-Me3C6H2) followed by treatment with tetrahydrofuran (THF) resulted in the isolation of [Fe4S4(SDmp)3(THF)3] 2. Cluster 2 contains an octahedral iron atom with three THF ligands, and its Fe(S)3(O)3 coordination environment is relevant to that in the active site of substrate-bound aconitase. An analogous reaction of [1]- with four equiv of HSDmp gave [Fe4S4(SDmp)4]- 3, which models the oxidized form of HiPIP. The THF ligands in 2 can be replaced by tetramethyl-imidazole (Me4Im) to give [Fe4S4(SDmp)3(Me4Im)] 4 modeling the [Fe4S4(Cys)3(His)] cluster in hydrogenases, and its one-electron reduced form [4]- was synthesized from the reaction of 3 with Me4Im. The reversible redox couple between 3 and [3]- was observed at E1/2 = -820 mV vs. Ag/Ag+, and the corresponding reversible couple for 4 and [4]- is positively shifted by +440 mV. The cyclic voltammogram of 3 also exhibited a reversible oxidation couple, which indicates generation of the all-ferric [Fe4S4]4+ cluster, [Fe4S4(SDmp)4].
Keywords: Fe4S4 cluster, thiolates
Cuboidal [Fe4S4] clusters are ubiquitous metal-centers in proteins, expediting electron transfer and enzymatic reactions. These [Fe4S4] cores are usually bound to four cysteinyl thiolates (Cys) as found in the high-potential iron-sulfur proteins (HiPIP) and widely distributed ferredoxins (Fd). Some [Fe4S4] clusters carrying an N- or O-donor ligand and three Cys ligands are also known, for example the [Fe4S4(Cys)3(His)] cluster (His = histidinyl imidazole) in [NiFe] and [FeFe] hydrogenases (Fig. 1) (1–6), and the [Fe4S4(Cys)3(O-donor)] cluster in aconitase (7–9) and protochlorophyllide reductase (10). All of these [Fe4S4] clusters are present in three oxidation states, [Fe4S4]3+ (HiPIPox), [Fe4S4]2+ (HiPIPred/Fdox), and [Fe4S4]+ (Fdred), while the [Fe4S4]0 state has been suggested for the cluster in the Fe-protein of nitrogenase (11, 12). To date, no [Fe4S4]4+ cluster has been found in proteins.
Fig. 1.

The iron-sulfur clusters in [NiFe] hydrogenases. The coordinates have been taken from the crystal data for the protein of D. v. Miyazaki F, where the PDB code is 1WUL (5).
The influence of the ligands around the [Fe4S4] core on the cluster properties is an important issue. For Fd and HiPIP the formation of hydrogen bonds with water has been suggested to account for most of the difference between the redox potentials of HiPIP and Fd (13), indicating the importance of hydrophobic shielding of the [Fe4S4] clusters in the more oxidized form. Synthetic analogues of HiPIPox with thiolates derived from bulky hydrocarbon groups are valuable for the investigation of the dependency of the redox potentials on hydrogen bonding. In the case of the hydrogenase [Fe4S4(Cys)3(His)] cluster, a possible role of the His ligand is to modulate the redox potential, as has been observed for the Rieske proteins featuring His-bound [Fe2S2] centers (14). However, there is only limited data on the effect on the redox potentials of [Fe4S4] clusters (4, 15). Thus synthetic analogues of [Fe4S4(Cys)3(His)] clusters, with a (thiolate)3(imidazole) ligand set, are required to elucidate the influence of histidine coordination. Although a number of synthetic [Fe4S4] clusters have been reported, most of these are of the type [Fe4S4(SR)4]2-, which has the [Fe4S4]2+ (HiPIPred/Fdox) oxidation state (16). However synthesis of other family members such as analogues of HiPIPox and [Fe4S4(Cys)3(His)] clusters remains difficult. There is only one isolated compound for the [Fe4S4]3+ cluster modeling HiPIPox, [Fe4S4(STip)4]- (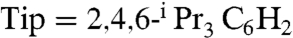 ) (17, 18), which was prepared by chemical oxidation of [Fe4S4(STip)4]2-. As for [Fe4S4(Cys)3(His)] analogues, generation of [Fe4S4(LS3)(imidazoles)]- [LS3 = 1,3,5-tris((4,6-dimethyl-3-mercaptophenyl)thio)-2,4,6-tris(p- tolylthio)benzenate)] was inferred from an 1H NMR spectrum of a mixture containing [Fe4S4(LS3)Cl]2-, excess imidazoles, and NaBF4 (19), however isolation and detailed characterization of the imidazole-bound [Fe4S4] cluster have not been accomplished.
) (17, 18), which was prepared by chemical oxidation of [Fe4S4(STip)4]2-. As for [Fe4S4(Cys)3(His)] analogues, generation of [Fe4S4(LS3)(imidazoles)]- [LS3 = 1,3,5-tris((4,6-dimethyl-3-mercaptophenyl)thio)-2,4,6-tris(p- tolylthio)benzenate)] was inferred from an 1H NMR spectrum of a mixture containing [Fe4S4(LS3)Cl]2-, excess imidazoles, and NaBF4 (19), however isolation and detailed characterization of the imidazole-bound [Fe4S4] cluster have not been accomplished.
We (20) and Lee and coworkers (21) have reported the synthesis of an all-ferric cluster in the [Fe4S4]4+ oxidation state carrying four amide ligands, [Fe4S4{N(SiMe3)2}4] 1. This [Fe4S4]4+ oxidation state is not yet known in proteins. This all-ferric cluster has two less electrons than the [Fe4S4]2+ state in the common [Fe4S4(SR)4]2- clusters, and its high oxidation state begs reduction. In fact, the cyclic voltammetry of 1 in THF shows that the first reduction process occurs at E1/2 = 0.10 V vs. Ag/Ag+, which opens a unique possibility for preparation of synthetic analogues of HiPIPox and [Fe4S4(Cys)3(His)] clusters, via reduction. Here we report the successful synthesis of an [Fe4S4]3+ cluster [Fe4S4(SDmp)4]- 3 modeling HiPIPox as well as 3∶1-site differentiated [Fe4S4] clusters having a tetramethyl-imidazole (Me4Im) ligand, [Fe4S4(SDmp)3(Me4Im)]n- (n = 0 4, n = 1 [4]-); Dmp = 2,6-(mesityl)2C6H3), starting from the all-ferric cluster 1 or from its one-electron reduced form [1]-. We have also prepared another 3∶1-site differentiated [Fe4S4] cluster [Fe4S4(SDmp)3(THF)3] 2 containing an octahedral iron atom with three THF ligands, which is structurally relevant to the active site of aconitase. The electrochemical properties of the unique [Fe4S4] clusters and the influence of the bulky -SDmp ligand on the redox potential are also discussed.
Results and Discussion
Synthesis of [Fe4S4{N(SiMe3)2}4]-[1]-.
Chemical reduction of 1 was attained by treatment of 1 with one equiv of sodium naphthalenide (NaC10H8) in THF at 0 °C (Scheme 1, top). Formation of the anionic cluster [1]- is manifested in the electro-spray ionization mass spectrum (ESI-MS), showing an anionic signal at m/z = 992.4. After removal of naphthalene by sublimation, Na(THF)2[Fe4S4{N(SiMe3)2}4] was isolated in 71% yield as black crystals. This anionic cluster has been also prepared recently using NaSH or Na2S as reductants (21). Two redox couples at E1/2 = 0.11 V and -0.98 V vs. Ag/Ag+ were observed in the cyclic voltammogram (CV) of [1]-, as in the case of 1.
Scheme 1.

The crystal structure of Na(THF)2[Fe4S4{N(SiMe3)2}4] was determined by X-ray analysis, using single crystals obtained from a hexane solution. As the structure of the same cluster has been reported (21), here we describe only its salient features. An amide nitrogen atom (N1) and a sulfur atom of [1]- interact weakly with a Na(THF)2 unit, and the Na-N1 and Na-S distances are 2.528(2) Å and 3.1992(11) Å, respectively. The Na-N1 interaction results in a slight pyramidalization at N1, displacing the N atom 0.3721(20) Å from the plane defined by the Fe and Si neighbors, while displacement of the other nitrogen atoms is less than 0.0948(22) Å. The planarity of the amide nitrogen suggests a strong N → Fe π-interaction, which shortens the Fe-N distances and efficiently stabilizes the oxidized [Fe4S4]3+ core of [1]-. Indeed the iron-amide (N1) distance of 1.9466(19) Å is notably longer than the other iron-amide distances (Fe-N2, N3, N4) [1.892(2)–1.897(2) Å]. Interestingly, the Fe-Fe distances of [1]-, ranging from 2.8044(4) Å to 2.9151(5) Å, are shorter than those of 1 [2.8667(7)–3.0014(5) Å], while they are longer than those of [Fe4S4(SR)4]2-(2.71–2.82 Å) (16).
Reactions of 1 and [1]- with HSDmp (Dmp = 2,6-(mesityl)2C6H3).
The amide nitrogen of N(SiMe3)2 bound to iron is a Brϕnstead base, and we have utilized this property to synthesize various iron-sulfide clusters (22–25). Here, we describe the reactions of clusters 1 and [1]- with the bulky thiol HSDmp (Dmp = 2,6-(mesityl)2C6H3) (26).
Reaction of 1.
Addition of four equiv of HSDmp to a toluene solution of 1 led to a color change from reddish black to purplish black, and the removal of volatile materials afforded a black solid. Although characterization of this black solid has been unsuccessful, treatment with THF resulted in the isolation of [Fe4S4(SDmp)3(THF)3] 2 in 67% yield as black crystals [Scheme 1, second-row (left)]. On the other hand, the reaction of 1 with HSDmp in THF did not afford cluster 2, and the product(s) of this reaction remains uncharacterized. Cluster 2 consists of one ferrous and three ferric iron atoms, and thus the all-ferric [Fe4S4]4+ core in 1 is reduced by one electron during the reaction with HSDmp. We presume that one of the SDmp- ligands introduced onto the [Fe4S4] core has been oxidized generating the disulfide (1/2 DmpS-SDmp), while the resulting vacant coordination site on iron is occupied by three THF molecules.
The generation of one unique iron site in a [Fe4S4]3+ cubane cluster by reduction of 1 is intriguing, and this reaction offers a new synthetic route to 3∶1-site differentiated [Fe4S4] clusters without invoking the use of tridentate thiolate auxiliaries (19, 27, 28). This route complements the recently reported oxidation reactions of the all-ferrous [Fe4S4]0 clusters [Fe4S4(Pi Pr 3)3]2 or [Fe4S4(Pi Pr 3)2]4 with disulfides, diselenides, or I2, yielding 3∶1-site differentiated [Fe4S4]+ clusters (29).
Cluster 2 was structurally identified by X-ray crystallographic analysis. The molecule displays threefold crystallographic symmetry, and there are two unique molecules in the asymmetric unit, and one of these is shown in Fig. 2, along with selected bond distances and angles. The threefold axis lies along the Fe1-S2 vector, and thus three of the four iron atoms (Fe2, Fe2*, Fe2′) are crystallographically equivalent. These iron atoms have a distorted tetrahedral geometry surrounded by four sulfur atoms, three from the [Fe4S4] core and fourth from a thiolate ligand. On the other hand, Fe1 is nearly octahedral, bound to three oxygen atoms of THF molecules and three sulfur atoms from the core, with the S1-Fe1-S1* and S1-Fe1-O1 angles being 95.53(4)° and 87.72(11)°, respectively. Due to the octahedral coordination geometry at Fe1, the Fe1-Fe2 distance [2.9600(9) Å] is significantly elongated compared with the Fe2-Fe2* distance [2.7328(11) Å], indicating no Fe1-Fe2 bonding interaction. The Fe1-S1 bond [2.3938(12) Å] is also longer than the Fe2-S1 and Fe2-S2 bonds [2.2412(10) and 2.2732(13) Å] by 0.1–0.15 Å. It is noteworthy that the octahedral geometry of Fe1 coordinated by three O-donors and three S-donors resembles that of the active site of aconitase (7–9), which catalyzes the reversible isomerization of citrate and isocitrate. In fact, the Fe1-S and Fe1-O distances of 2 [Fe-S; 2.3939(12) Å, Fe-O; 2.234(3) Å] are similar to those for the isocitrate complex of aconitase [Fe-S; 2.33–2.36 Å, Fe-O; 2.20 (water), 2.28 and 2.37 Å (isocitrate)] (7–9). This similarity opens the possibility that the unique iron atom of molecule 2 may have catalytic properties. The THF ligands bound to Fe1 may be weakly held, so that 2 may serve as a synthetic precursor to 3∶1-site differentiated [Fe4S4] clusters.
Fig. 2.

Molecular structure of [Fe4S4(SDmp)3(THF)3] 2 with thermal ellipsoids at the 50% probability level. The THF ligand on Fe1 is disordered over two positions, and one of these is shown for clarity. Selected distances (Å) and angles (°): Fe1-Fe2 2.9600(9), Fe2-Fe2* 2.7328(11), Fe1-S1 2.3938(12), Fe2-S1 2.2412(10), Fe2-S2 2.2732(13), Fe2-S3 2.2234(14), Fe1-O1 2.234(3), S1-Fe1-S1* 95.53(4), and S1-Fe1-O1 87.72(11).
Reaction of [1]-.
Treatment of [1]- with four equiv of HSDmp in toluene resulted in the isolation of [Fe4S4(SDmp)4]- 3 [Scheme 1, second-row (right)] in 69% yield as crystals. In this reaction, all four amide ligands of [1]- were all replaced by thiolates, without changing the [Fe4S4]3+ oxidation state. The ESI-MS spectrum of a THF solution of 3 gave an anionic signal at m/z = 1,732.4, and the isotope pattern matches that calculated. The oxidation state of 3, [Fe4S4]3+, is the same as that of HiPIPox, for which only one isolated model cluster, [Fe4S4(STip)4]-, has been reported (17, 18). The EPR spectrum of cluster 3 gave a rhombic signal at g = 2.076, 2.035, and 2.018 in frozen toluene at 8 K, whose g values are similar to those of [Fe4S4(STip)4]- with g = 2.10, 2.05, and 2.03 (18). The averaged g value of 3, gav = 2.043, is also close to those for HiPIPox with gav = 2.056 (Rubrivivax gelatinosa) (30), 2.065 (Thiobacillus ferrooxidans) (31), and 2.048 (Ectothiorhodospira halophila) (32), although these HiPIPox signals are axial.
The structure of 3 was determined by X-ray analysis, using single crystals obtained from a toluene-hexamethyldisiloxane (HMDSO) solution (Fig. 3). The mean values of the Fe-S(core), Fe-S(thiolate), and Fe-Fe distances of 3, are compared with those of [Fe4S4(STip)4]- (17, 18), [Fe4S4(SPh)4]2- (33), and [Fe4S4(SPh)4]3- (34), in Table 1. The Na cation of 3, which interacts with THF, one of the SDmp sulfur atoms, one of the core sulfur atoms, and one of the mesityl groups of SDmp, is disordered over two positions with 75∶25 occupancy factors, and only one of these is shown for clarity. The Na-C(mesityl) distances [2.885(4)–2.979(5) Å] are in the range of the Na-C(Tip) distances determined for NaS(2,6-Tip2C6H3) [2.839(5)–3.249(5) Å] (35). The higher oxidation state of 3 compared to the [Fe4S4(SPh)4]2-/3- clusters results in a smaller ionic radius for iron, and therefore the Fe-S(core) and Fe-S(thiolate) bond lengths of 3 are slightly but evidently shorter than those of the [Fe4S4(SPh)4]2-/3- clusters. The Fe-S(core) and Fe-S(thiolate) bond lengths of 3 and [Fe4S4(STip)4]- are similar, while the mean Fe-S(thiolate) length is longer for 3 by ca. 0.02 Å probably because of the Na-SDmp interaction. Whereas the charge on the [Fe4S4] core affects the Fe-S bond lengths, the Fe-Fe distances of 3 are similar to the [Fe4S4(SR)4]1-/2-/3- clusters.
Fig. 3.

Structure of [Na(THF)][Fe4S4(SDmp)4] 3 with thermal ellipsoids at the 50% probability level. The Na(THF) group is disordered over two positions, and one of these is shown for clarity. Selected distances (Å): Fe1-Fe2 2.7282(8), Fe1-Fe3 2.8127(10), Fe1-Fe4 2.7894(8), Fe2-Fe3 2.7561(6), Fe2-Fe4 2.7306(6), Fe3-Fe4 2.7889(7), Fe1-S1 2.2810(12), Fe1-S2 2.2885(11), Fe1-S3 2.2316(11), Fe2-S1 2.3020(12), Fe2-S2 2.2625(13), Fe2-S4 2.2716(13), Fe3-S1 2.2471(12), Fe3-S3 2.3057(13), Fe3-S4 2.2791(13), Fe4-S2 2.2177(11), Fe4-S3 2.2716(12), Fe4-S4 2.2674(14), Fe1-S5 2.2445(15), Fe2-S6 2.2615(9), Fe3-S7 2.2408(11), Fe4-S8 2.2128(10), and S6-Na1 2.894(3).
Table 1.
Selected distances (Å) for clusters 3, [Fe4S4(STip)4]-, [Fe4S4(SPh)4]2-, and [Fe4S4(SPh)4]3-
Synthesis and Structures of 3∶1-Site Differentiated [Fe4S4] Clusters with a Tetramethyl-Imidazole Ligand.
In toluene the THF ligands of 2 were replaced by one tetramethyl-imidazole (Me4Im), and [Fe4S4(SDmp)3(Me4Im)] 4 was isolated in 46% yield as crystals [Scheme 1, bottom (left)]. Alternatively, cluster 4 was synthesized directly from 1 in 54% yield, via successive treatment with four equiv of HSDmp and with one equiv of Me4Im. An analogous [Fe4S4]3+ cluster with histidine ligation has been generated as the oxidized form of the site-directed Cys → His mutant of the DNA repair enzyme MutY, and this cluster has been reported to degrade readily into an [Fe3S4]+ cluster by release of an iron atom (36).
The one-electron reduced form of 4, [Fe4S4(SDmp)3(Me4Im)]- [4]-, was obtained in 72% yield as black crystals from the reaction of [Fe4S4(SDmp)4]- 3 with one equiv of Me4Im in toluene [Scheme 1, bottom (right)], and the ESI-MS spectrum of a THF solution of [4]- exhibits an anionic signal at m/z = 1,511.6 (M-). Interestingly, liberation of a SDmp ligand in 3 induces one-electron reduction of the [Fe4S4]3+ core to [Fe4S4]2+ of [4]-, presumably via formation of DmpS-SDmp. Thus single-site ligand replacement via reduction is also applicable to the HiPIPox-type [Fe4S4]3+ cluster 3. In a similar manner to the direct synthesis of 4 from 1, the one-pot reaction of [1]- with four equiv of HSDmp and one equiv of Me4Im in toluene gave crystals of [4]- in 86% yield. As demonstrated here by the formation of the Me4Im-adducts 4 and [4]-, this ligand exchange reaction accompanied by one-electron reduction is a promising tool for the incorporation of one external biologically relevant ligand onto [Fe4S4(SR)4]n- clusters. We have also examined chemical oxidation/reduction of 4/[4]-. However, oxidation of [4]- with [Cp2Fe][PF6] and reduction of 4 with Na(C10H8) yielded uncharacterizable black solids.
The coordination of three thiolates and a tetramethyl-imidazole to the [Fe4S4] core of 4 and [4]- models the structure of the [Fe4S4(Cys)3(His)] cluster found in hydrogenases. The overall similarity among the core structures of 4, [4]-, and the [Fe4S4(Cys)3(His)] cluster from Desulfovibrio gigas can be seen in Fig. 4. One feature common to all three clusters is a slight bending of the imidazole coordinaton, as is manifested by the deviation of the S*-Fe1-N angles from 180°, namely, 167.70(15)° (4), 165.67(8)° ([4]-), and 160.9° (D. gigas). As a consequence, the tetrahedral geometry at Fe1 is distorted. The distances in D. gigas, Fe-Fe (2.70–2.75 Å), Fe-N(imidazole) (1.99 Å), Fe-S(thiolate) (2.25–2.42 Å), and Fe-S(core) (2.23–2.36 Å), are comparable to those listed in Table 2 for 4 and [4]-, taking the lower accuracy of distances in protein structures into consideration. The oxidation state of the [Fe4S4(Cys)3(His)] cluster in the crystal structure of D. gigas has not been clarified yet, although the [Fe4S4]2+/1+ states have been suggested to be involved in the enzymatic process. Successful isolation of 4 and [4]- suggests that the [Fe4S4]3+/2+ states might be another possibility for the [Fe4S4(Cys)3(His)] clusters. It should be also noted that 4 and [4]- are structurally analogous to the His-bound [Fe4S4(Cys)3(His)] clusters serving as one of the electron-transfer sites of the membrane-bound nitrate reductase from Escherichia coli (37), and as the active site of 4-hydroxybutyryl-CoA dehydratase (38).
Fig. 4.

The core structures of 4, [4]-, and the [Fe4S4(Cys)3(His)] cluster from D. gigas (PDB code 2FRV) (2). For clarity, only the central aromatic rings of -SDmp ligands are shown for 4 and [4]-.
Table 2.
Selected distances (Å) and angle (°) for clusters 4 and [4]-
| 4 | [4]- | |
| Oxidation state | [Fe4S4]3+ | [Fe4S4]2+ |
| av. Fe-Fe | 2.7450(11) | 2.7116(8) |
| Fe1-Fe | 2.7272(11) | 2.7102(7) |
| other Fe-Fe | 2.7628(10) | 2.7130(8) |
| av. Fe-S(core) | 2.2681(17) | 2.2846(12) |
| Fe1-S(core) | 2.2782(17) | 2.2796(12) |
| other Fe-S(core) | 2.2647(16) | 2.2862(12) |
| av. Fe-S(thiolate) | 2.2179(17) | 2.2627(10) |
| Fe-N(imidazole) | 1.992(5) | 2.017(2) |
| S4-Fe1-N | 167.70(15) | 165.67(8) |
While the structures of 4 and [4]- shown in Fig. 5 are very much alike, there are some differences in distances due to the different oxidation states, as listed in Table 2. The higher oxidation state for 4 leads to slightly shorter Fe-N(imidazole), Fe-S(core), and Fe-S(thiolate) distances than those for [4]-. The most notable difference among these can be found in the Fe-S(thiolate) distances, 2.2090(17)–2.2351(15) Å [average (av.) 2.218 Å] for 4 and 2.2361(10)–2.2866(10) Å (av. 2.263 Å) for [4]-, indicating more flexibility for the thiolate ligands than the core sulfur atoms in response to the change of oxidation state. Similarly, the Fe-S(thiolate) distances in the [Fe4S4]3+ cluster 3, 2.2128(10)–2.2615(9) Å (av. 2.227 Å), are shorter than those for [4]-, although the Na-S(thiolate) interaction elongates one of the Fe-S(thiolate) bonds of 3 to 2.2615(9) Å. In contrast to the Fe-N(imidazole), Fe-S(core), and Fe-S(thiolate) distances, the Fe-Fe distances for 4, 2.6946(10)–2.7978(10) Å (av. 2.745 Å), and 3, 2.7282(8)–2.8127(10) Å (av. 2.768 Å), are slightly longer than those for [4]-, 2.6834(6)–2.7335(7) Å (av. 2.712 Å).
Fig. 5.

Structures of [Fe4S4(SDmp)3(Me4Im)] 4 and [Fe4S4(SDmp)3(Me4Im)]- [4]- with thermal ellipsoids at the 50% probability level.
Electronic Properties of 3, 4, and [4]- as Models of HiPIP and [Fe4S4(Cys)3(His)].
The CV for the HiPIP model 3 and the [Fe4S4(Cys)3(His)] models 4 and [4]- have been measured and compared to assess the influence of the imidazole ligand on the redox potentials. Fig. 6 shows the CV data for 3 and [4]- in the region of 0 ∼ -1.2 V, where the potentials are referenced to Ag/Ag+.
Fig. 6.

Redox couples of 3/[3]- and 4/[4]- measured by CV in THF at room temperature.
The [Fe4S4]3+/2+ redox couple between 4 and [4]- was found to be reversible at E1/2 = -380 mV, and the corresponding reversible redox couple for 3 and [3]- appeared at E1/2 = -820 mV. Thus the redox potential is shifted by +440 mV, when one of the -SDmp ligands in 3 is replaced by Me4Im. The positive shift is reasonable, as the electron-donating abilities of anionic thiolate ligands should be greater than those of neutral imidazoles. Similarly, the in situ generated [Fe4S4] cluster with a 4-methyl-imidazole (4-MeIm) ligand, [Fe4S4(LS3)(4-MeIm)]-, shows a redox couple more positive by 320 mV than the cluster having an ethane-thiolate instead of 4-MeIm, [Fe4S4(LS3)(SEt)]2- (19).
The significantly higher redox potential for 4 relative to 3 implies that facile electron transfer could occur from the [Fe4S4(Cys)4] cluster, which is proximal to the Ni-Fe active site of [NiFe] hydrogenases, to the distal-[Fe4S4(Cys)3(His)] cluster. This direction is the electron flow that promotes the oxidation of H2 at the active site. On the other hand, hydrogenases also promote the reduction of H+ that requires the reverse electron transfer from the distal- to proximal-[Fe4S4] clusters. Therefore, there must be a mechanism, which promotes the reverse electron transfer process, by modulating the redox potentials of the [Fe4S4] clusters. The unique positioning of the distal-[Fe4S4(Cys)3(His)] cluster in the protein may give a clue. The distal cluster is located at the protein surface with the imidazole N-H group oriented toward outside of the protein (Fig. 1). We hypothesize here that the imidazole N-H group is exposed to water, and that coordination of the imidazole to iron enhances the N-H acidity facilitating its deprotonation. This deprotonation at imidazole would cause a substantial negative shift of the potential of the [Fe4S4(Cys)3(His)] cluster, due to the addition of an extra negative charge with retention of the oxidation state of the iron atoms. Therefore, protonation/deprotonation at the imidazole may act as a changeover switch reversing the electron transfer between the distal- and proximal-[Fe4S4] clusters.
Interestingly, upon scanning a CV of 3 toward positive potential, a reversible redox couple at E1/2 = +80 mV was observed (Fig. 7), which indicates the generation of an all-ferric [Fe4S4]4+ cluster, [Fe4S4(SDmp)4]0. Cluster 1 is the sole example of an all-ferric [Fe4S4]4+ cluster, and a thiolate-bound [Fe4S4]4+ cluster is unprecedented. Stabilization of the all-ferric state of the [Fe4S4]4+ core requires a strongly basic, electron donating set of ligands to counter the large positive charge of the iron atoms. Even the removal of a small amount of negative charge due to the formation of hydrogen bonds appears to destabilize the all-ferric oxidation state. As for the HiPIPox cluster, the [Fe4S4]3+ core is buried in a hydrophobic pocket of the protein, while the [Fe4S4]2+/1+ core of Fd is exposed to hydrogen bonding by water (13). In this context, the unique ability of higher oxidation states of 3 may arise from the environment provided by the bulky hydrocarbon groups of the -SDmp ligands, which mimics a hydrophobic pocket of the protein.
Fig. 7.

CV of 3 in the region of 0.4 ∼ -0.2 V vs. Ag/Ag+.
Materials and Methods
All reactions and the manipulations were carried out using standard Schlenk techniques and a glove box with a nitrogen atmosphere. Toluene, THF, hexane, and HMDSO were purified by columns of activated alumina and a supported copper catalyst supplied by Hansen and Co. Ltd.
All experimental conditions and procedures, spectroscopic data, and details of characterization of complexes are given in SI Text, which is published on the PNAS web site.
Supplementary Material
Acknowledgments.
We thank Roger E. Cramer for his help in X-ray crystallographic analysis and for careful reading of the manuscript. This research was financially supported by Grant-in-Aids for Scientific Research (Nos. 18GS0207 and 18064009) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited with the Cambridge Crystallographic Data Centre, Cambridge CB2 1EK, United Kingdom (CSD reference nos.: CCDC823086–823090).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1106472108/-/DCSupplemental.
References
- 1.Volbeda A, et al. Crystal structure of the [NiFe] hydrogenase from Desulfovibrio gigas. Nature. 1995;373:580–587. doi: 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]
- 2.Volbeda A, et al. Structure of the [NiFe] hydrogenase active site: evidence for biologically uncommon Fe ligands. J Am Chem Soc. 1996;118:12989–12996. [Google Scholar]
- 3.Higuchi Y, Yagi T, Yasuoka N. Unusual ligand structure in Ni-Fe active center and an additional Mg site in hydrogenase revealed by high resolution X-ray structure analysis. Structure. 1997;5:1671–1680. doi: 10.1016/s0969-2126(97)00313-4. [DOI] [PubMed] [Google Scholar]
- 4.Rousset M, et al. [3Fe-4S] to [4Fe-4S] cluster conversion in Desulfovibrio fructosovorans [NiFe] hydrogenase by site-directed mutagenesis. Proc Natl Acad Sci USA. 1998;95:11625–11630. doi: 10.1073/pnas.95.20.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogata H, et al. Activation process of [NiFe] hydrogenase elucidated by high-resolution X-ray analyses: conversion of the ready to the unready state. Structure. 2005;13:1635–1642. doi: 10.1016/j.str.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 6.Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC. X-ray crystal structure of the Fe-only hydrogenase(CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science. 1998;282:1853–1858. doi: 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]
- 7.Lauble H, Kennedy MC, Beinert H, Stout CD. Crystal structures of aconitase with isocitrate and nitroisocitrate bound. Biochemistry. 1992;31:2735–2748. doi: 10.1021/bi00125a014. [DOI] [PubMed] [Google Scholar]
- 8.Lloyd SJ, Lauble H, Prasad GS, Stout CD. The mechanism of aconitase: 1.8 Å resolution crystal structure of the S642A:citrate complex. Protein Sci. 1999;8:2655–2662. doi: 10.1110/ps.8.12.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dupuy J, et al. Crystal structure of human iron regulatory protein 1 as cytosolic aconitase. Structure. 2006;14:129–139. doi: 10.1016/j.str.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 10.Muraki N, et al. X-ray crystal structure of the light-independent protochlorophyllide reductase. Nature. 2010;465:110–114. doi: 10.1038/nature08950. [DOI] [PubMed] [Google Scholar]
- 11.Watt GD, Reddy KRN. Formation of an all ferrous Fe4S4 cluster in the iron protein component of Azotobacter vinelandii nitrogenase. J Inorg Biochem. 1994;53:281–294. [Google Scholar]
- 12.Angove HC, Yoo SJ, Münck E, Burgess BK. An all-ferrous state of the Fe protein of nitrogenase. J Biol Chem. 1998;273:26330–26337. doi: 10.1074/jbc.273.41.26330. [DOI] [PubMed] [Google Scholar]
- 13.Dey A, et al. Solvent tuning of electrochemical potentials in the active sites of HiPIP versus ferredoxin. Science. 2007;318:1464–1468. doi: 10.1126/science.1147753. [DOI] [PubMed] [Google Scholar]
- 14.Stephens PJ, Jollie DR, Warshel A. Protein control of redox potentials of iron-sulfur proteins. Chem Rev. 1996;96:2491–2514. doi: 10.1021/cr950045w. [DOI] [PubMed] [Google Scholar]
- 15.Teixeira M, et al. Redox intermediates of Desulfovibrio gigas [NiFe] hydrogenase generated under hydrogen. J Biol Chem. 1989;264:16435–16450. [PubMed] [Google Scholar]
- 16.Rao PV, Holm RH. Synthetic analogues of the active sites of iron-sulfur proteins. Chem Rev. 2004;104:527–559. doi: 10.1021/cr020615+. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 17.O’Sullivan T, Millar MM. Synthesis and study of an analogue for the [Fe4S4]3+ center of oxidized high-potential iron-sulfur proteins. J Am Chem Soc. 1985;107:4096–4097. [Google Scholar]
- 18.Papaefthymiou V, Millar MM, Münck E. Mössbauer and EPR studies of a synthetic analogue for the Fe4S4 core of oxidized and reduced high-potential proteins. Inorg Chem. 1986;25:3010–3014. [Google Scholar]
- 19.Zhou C, Holm RH. Comparative isotropic shifts, redox potentials, and ligand binding propensities of [1∶3] site-differentiated cubane-type [Fe4Q4]2+ clusters (Q = S, Se) Inorg Chem. 1997;36:4066–4077. [Google Scholar]
- 20.Ohki Y, Sunada Y, Tatsumi K. Synthesis of [2Fe-2S] and [4Fe-4S] clusters having terminal amide ligands from an iron(II) amide complex. Chem Lett. 2005;34:172–173. [Google Scholar]
- 21.Sharp CR, Duncan JS, Lee SC. [Fe4S4]q cubane clusters (q = 4+, 3+, 2+) with terminal amide ligands. Inorg Chem. 2010;49:6697–6705. doi: 10.1021/ic100742c. [DOI] [PubMed] [Google Scholar]
- 22.Ohki Y, Sunada Y, Honda M, Katada M, Tatsumi K. Synthesis of the P-cluster inorganic core of nitrogenases. J Am Chem Soc. 2003;125:4052–4053. doi: 10.1021/ja029383m. [DOI] [PubMed] [Google Scholar]
- 23.Ohki Y, Ikagawa Y, Tatsumi K. Synthesis of new [8Fe-7S] clusters: a topological link between the core structures of P-cluster, FeMo-co, and FeFe-co of nitrogenases. J Am Chem Soc. 2007;129:10457–10465. doi: 10.1021/ja072256b. [DOI] [PubMed] [Google Scholar]
- 24.Ohki Y, Murata A, Imada M, Tatsumi K. C-H bond activation of decamethylcobaltocene mediated by a nitrogenase Fe8S7 P-cluster model. Inorg Chem. 2009;48:2358–2360. doi: 10.1021/ic900284f. [DOI] [PubMed] [Google Scholar]
- 25.Ohki Y, et al. Synthesis, structures, and electronic properties of [8Fe-7S] cluster complexes modeling the nitrogenase P-cluster. J Am Chem Soc. 2009;131:13168–13178. doi: 10.1021/ja9055036. [DOI] [PubMed] [Google Scholar]
- 26.Ellison JJ, Ruhlandt-Senge K, Power PP. Synthesis and characterization of thiolato complexes with two-coordinate iron(II) Angew Chem Int Ed Engl. 1994;33:1178–1180. [Google Scholar]
- 27.Stack TD, Holm RH. Subsite-differentiated analogues of biological [4Fe-4S]2+ clusters: synthesis, solution, and solid-state structures, and subsite-specific reactions. J Am Chem Soc. 1988;110:2484–2494. [Google Scholar]
- 28.Walsdorff C, Saak W, Pohl S. A new preorganized tridentate ligand bearing three indolethiolate groups. Preparation of 3∶1 subsite-differentiated Fe4S4 clusters. J Chem Soc Dalton. 1997:1857–1861. [Google Scholar]
- 29.Deng L, Majumdar A, Lo W, Holm RH. Stabilization of 3∶1 site-differentiated cubane-type clusters in the [Fe4S4]1+ core oxidation state by tertiary phosphine ligation: synthesis, core structural diversity, and S = 1/2 ground states. Inorg Chem. 2010;49:11118–11126. doi: 10.1021/ic101702b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schoepp B, et al. In vivo participation of a high potential iron-sulfur protein as electron donor to the photochemical reaction center of Rubrivivax gelatinosus. Biochemistry. 1995;34:11736–11742. doi: 10.1021/bi00037a010. [DOI] [PubMed] [Google Scholar]
- 31.Cavazza C, Guigliarelli B, Bertrand P, Bruschi M. Biochemical and EPR characterization of a high potential iron-sulfur protein in Thiobacillus ferrooxidans. FEMS Microbiol Lett. 1995;130:193–200. [Google Scholar]
- 32.Beinert H, Thomson AJ. Three-iron clusters in iron-sulfur proteins. Arch Biochem Biophys. 1983;222:333–361. doi: 10.1016/0003-9861(83)90531-3. [DOI] [PubMed] [Google Scholar]
- 33.Que L, Bobrik MA, Ibers JA, Holm RH. Synthetic analogs of the active sites of iron-sulfur proteins. VII. Ligand substitution reactions of the tetranuclear clusters [Fe4S4(SR)4]2- and the structure of [(CH3)4N]2[Fe4S4(SC6H5)4] J Am Chem Soc. 1974;96:4168–4178. doi: 10.1021/ja00820a018. [DOI] [PubMed] [Google Scholar]
- 34.Laskowski EJ, et al. Synthetic analogues of the 4-Fe active sites of reduced ferredoxins. Electric properties of the tetranuclear trianions [Fe4S4(SR)4]3- and the structure of [(C2H5)3(CH3)N]3[Fe4S4(SC6H5)4] J Am Chem Soc. 1978;100:5322–5337. doi: 10.1021/ja00449a061. [DOI] [PubMed] [Google Scholar]
- 35.Niemeyer M, Power PP. Donor-free alkali metal thiolates: synthesis and structure of dimeric, trimeric, and tetrameric complexes with sterically encumbered terphenyl substituents. Inorg Chem. 1996;35:7264–7272. doi: 10.1021/ic960570t. [DOI] [PubMed] [Google Scholar]
- 36.Messick TE, et al. Noncysteinyl coordination to the [4Fe-4S]2+ cluster of the DNA repair adenine glycosylase MutY introduced via site-directed mutagenesis. Structural characterization of an unusual histidinyl-coordinated cluster. Biochemistry. 2002;41:3931–3942. doi: 10.1021/bi012035x. [DOI] [PubMed] [Google Scholar]
- 37.Bertero MG, et al. Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat Struct Biol. 2003;10:681–687. doi: 10.1038/nsb969. [DOI] [PubMed] [Google Scholar]
- 38.Martins BM, Dobbek H, Cinkaya I, Buckel W, Messerschmidt A. Crystal structure of 4-hydroxybutyryl-CoA dehydratase: radical catalysis involving a [4Fe-4S] cluster and flavin. Proc Natl Acad Sci USA. 2004;101:15645–15649. doi: 10.1073/pnas.0403952101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.