

Human MutSβ is a 232 kDa heterodimer (MSH2–MSH3) involved in the lesion-recognition step of mismatch repair. Here, the overexpression, purification, biochemical characterization and cocrystallization of MutSβ with a duplex DNA substrate are reported.
Keywords: MutSα, MutSβ, MSH2, MSH3, MSH6, mismatch repair, limited proteolysis
Abstract
MutSβ is a eukaryotic mismatch repair protein that preferentially targets extrahelical unpaired nucleotides and shares partial functional redundancy with MutSα (MSH2–MSH6). Although mismatch recognition by MutSα has been shown to involve a conserved Phe-X-Glu motif, little is known about the lesion-binding mechanism of MutSβ. Combined MSH3/MSH6 deficiency triggers a strong predisposition to cancer in mice and defects in msh2 and msh6 account for roughly half of hereditary nonpolyposis colorectal cancer mutations. These three MutS homologs are also believed to play a role in trinucleotide repeat instability, which is a hallmark of many neurodegenerative disorders. The baculovirus overexpression and purification of recombinant human MutSβ and three truncation mutants are presented here. Binding assays with heteroduplex DNA were carried out for biochemical characterization. Crystallization and preliminary X-ray diffraction analysis of the protein bound to a heteroduplex DNA substrate are also reported.
1. Introduction
The DNA mismatch repair (MMR) pathway is responsible for correcting base substitutions and extrahelical base lesions that arise from DNA-replication and proofreading errors (Hsieh & Yamane, 2008 ▶; Iyer et al., 2006 ▶). Additionally, it is involved in the suppression of homeologous recombination (Iyer et al., 2006 ▶; Jiricny, 2006 ▶; Li, 2008 ▶) and the activation of cell-cycle checkpoints and signal apoptosis as a response to DNA damage produced by chemical agents and carcinogens (Hsieh & Yamane, 2008 ▶; Iyer et al., 2006 ▶; Kunkel & Erie, 2005 ▶). MMR defects in mammals can lead to serious biological consequences, including an ∼1000-fold decrease in DNA-replication fidelity (Hsieh & Yamane, 2008 ▶; Iyer et al., 2006 ▶). In humans, defects in MMR genes are responsible for a predisposition to hereditary nonpolyposis colorectal cancer, development of a subset of sporadic tumors and resistance to certain chemotherapeutic agents (Iyer et al., 2006 ▶; Kunkel & Erie, 2005 ▶; Li, 2008 ▶).
Unlike the prokaryotic MutS homodimer, mismatch recognition in eukaryotes utilizes at least two MutS-homolog (MSH) heterodimers: MutSα (MSH2–MSH6) and MutSβ (MSH2–MSH3). The role of a third heterodimer, MSH4–MSH5, appears to be limited to meiotic recombination (Ross-Macdonald & Roeder, 1994 ▶; Snowden et al., 2004 ▶). While MutSα binds to base–base mispairs and insertion–deletion loops of 1–10 unpaired nucleotides, MutSβ preferentially targets two or more unpaired nucleotides (Acharya et al., 1996 ▶; Genschel et al., 1998 ▶; Palombo et al., 1996 ▶). Although the conserved Phe-X-Glu motif has been shown to play a major role in mismatch recognition in both prokaryotic MutS and eukaryotic MutSα (Lamers et al., 2000 ▶; Obmolova et al., 2000 ▶; Warren et al., 2007 ▶), the underlying molecular basis of the substrate specificity of MutSβ remains poorly understood. Despite the presence of a conserved Phe-X-Glu motif in the MSH3 mismatch binding domain, MutSβ is only weakly active towards base–base mispairs (Genschel et al., 1998 ▶; Harrington & Kolodner, 2007 ▶) and single unpaired nucleotides (Acharya et al., 1996 ▶; Genschel et al., 1998 ▶; Palombo et al., 1996 ▶). Additional studies in Saccharomyces cerevisiae indicated that the mismatch binding domain of MSH2 is involved in MutSβ-mediated MMR but not MutSα-mediated MMR (Lee et al., 2007 ▶) and the DNA-binding mode of MutSβ appears to vary depending on the size of the loop (Dowen et al., 2010 ▶). Taken together, these observations suggest a unique mechanism of mismatch binding in MutSβ.
Besides MMR, MutSβ plays an unclear role in trinucleotide repeat expansion, which is a critical step in the pathogenesis of diseases such as Huntington’s disease, fragile X syndrome and myotonic dystrophy (Mirkin, 2007 ▶; Pearson et al., 2005 ▶). Despite the requirement for msh2 and msh3 in the expansion of trinucleotide repeat sequences (Manley et al., 1999 ▶; Savouret et al., 2004 ▶; Foiry et al., 2006 ▶; van den Broek et al., 2002 ▶), which have the propensity to adopt unusual structures (Mitas et al., 1995 ▶; Pearson et al., 2002 ▶; Smith et al., 1995 ▶; Yu et al., 1995 ▶), MutSβ exhibits identical biochemical properties when interacting with CAG repeats and insertion–deletion loops (Tian, Hou et al., 2009 ▶). Microsatellite instability (changes in the length of short repetitive DNA sequences) has also been partially attributed to MSH3 deficiency (Risinger et al., 1996 ▶), and msh3 polymorphisms have been suggested to increase the risk of colorectal cancer (Berndt et al., 2007 ▶).
For MutS proteins, the central function of DNA binding is tightly regulated by their ATPase activity (Schofield & Hsieh, 2003 ▶). The nucleotide-binding and ATPase activities of MutSβ, as previously inferred from those of MutSα, appear to be distinct and lesion-dependent (Owen et al., 2009 ▶; Tian, Gu et al., 2009 ▶), implying an association between differential nucleotide-occupancy states and lesion specificity. This, together with a common binding mode of human MutSα for a variety of DNA lesions (Warren et al., 2007 ▶), necessitates a comparative structural analysis of both heterodimers. In the present study, recombinant human MutSβ (hMutSβ) and three deletion mutants have been overexpressed in a baculovirus system and purified to homogeneity. Here, we report the crystallization and preliminary X-ray diffraction analysis of the protein bound to a duplex DNA containing an insertion–deletion loop.
2. Materials and methods
2.1. Cloning, expression and purification of hMutSβ
Three hMutSβ deletion mutants were generated from a pFastBac Dual vector containing msh2 and msh3 by PCR mutagenesis. These constructs (MutSβΔ162, MutSβΔ175 and MutSβΔ223) lacked the first 162, 175 and 223 residues of MSH3, respectively. The truncation in MutSβΔ162 is based on sequence alignment with hMutSαΔ341 (Warren et al., 2007 ▶), whereas the MutSβΔ223 deletion eliminates predicted disordered regions before the putative MSH3 mismatch binding domain (Notredame et al., 2000 ▶). MutSβΔ175 is a proteolytic fragment identified by subtilisin digestion (see §3.1). These mutants were expressed in Sf9 insect cells and purified as described previously for the full-length protein (Genschel et al., 1998 ▶) using buffers supplemented with 10 µg ml−1 leupeptin, 10 µg ml−1 aprotinin, 10 µg ml−1 E-64, 1 µg ml−1 pepstatin A, 0.1%(v/v) saturated phenylmethylsulfonyl fluoride and 1 mM dithiothreitol. The cell lysate was centrifuged (18 500g) and loaded onto a Q-Sepharose column equilibrated with 250 mM KCl buffer (25 mM HEPES–KOH pH 7.5, 0.5 mM EDTA and 250 mM KCl) and directly connected to a single-stranded DNA-cellulose column. After the two columns had been disconnected, the single-stranded DNA-cellulose column was washed with 300 mM KCl buffer and subsequently with 300 mM KCl buffer containing 2.5 mM MgCl2. MutSβ was eluted with 300 mM KCl buffer containing 2.5 mM MgCl2 and 1 mM ATP. Pooled fractions were diluted to 100 mM KCl and loaded onto a second Q-Sepharose column equilibrated with 100 mM KCl buffer. Fractions eluted with 250 mM KCl buffer were diluted to 100 mM KCl and loaded onto an 8 ml Mono Q 10/100 GL column (GE Healthcare). The protein was eluted with an 80 ml linear gradient of 100–370 mM KCl buffer at 0.5 ml min−1 and stored at 193 K in 150 mM KCl buffer. The purity of the protein was confirmed on a 12% SDS–PAGE gel (Fig. 1 ▶).
Figure 1.

Purification of MutSβ and its deletion mutants. (a) Representative chromatogram (shown for MutSβΔ162) of the final purification step (Mono Q column) for MutSβ and its deletion mutants. Absorbance at 280 nm versus elution volume is shown in blue; percentage elution buffer versus elution volume is shown in red. (b) A 12% SDS–PAGE of purified human MutSβ and its deletion mutants. Lane 1 shows molecular-weight standards, with weights (in kDa) indicated on the left. Lanes 2, 3, 4 and 5 show full-length MutSβ (FL), MutSβΔ162, MutSβΔ175 and MutSβΔ223. The locations of both MSH2 and MSH3 subunits are indicated by labelled arrows.
2.2. Limited proteolysis of hMutSβ
Full-length hMutSβ (3.6 µM) was incubated with 5 mM MgCl2, a twofold molar excess of ADP and a 1.2-fold molar excess of a 14 bp duplex DNA containing a CA insert (5′-CCTAGCGCAGCGGTTC-3′ annealed with 5′-GAACCGCCGCTAGG-3′). The mixture was subjected to partial proteolysis by subtilisin (1:167 protease:protein molar ratio) at 310 K in a buffer consisting of 25 mM HEPES pH 7.5 and 150 mM NaCl. Samples were quenched with 5 mM phenylmethylsulfonyl fluoride at the indicated time points, run on a 12% SDS–PAGE gel and electroblotted onto PVDF membrane. After Coomassie Blue staining, the proteolytic fragment (Fig. 2 ▶) was excised and submitted to peptide sequencing (University of Texas Medical Branch Biomolecular Resource Facility).
Figure 2.

SDS–PAGE of limited proteolysis of human MutSβ by subtilisin. The leftmost lane shows molecular-weight standards, with weights (in kDa) indicated on the left. Digestion times (min) are shown at the top. An MSH3 fragment was detected starting 2 min after digestion. The locations of full-length (FL) MSH3, the MSH3 proteolysis fragment and MSH2 are indicated by labelled arrows.
2.3. DNA-binding studies of hMutSβ
The heteroduplex DNA substrates (41 bp) that were used for surface plasmon resonance spectroscopy (SPRS) experiments contained a centrally positioned CA dinucleotide insertion–deletion (I/D) loop and were prepared as described previously (Blackwell et al., 2001 ▶) by annealing the HPLC-purified synthetic oligonucleotides 5′-AGCCGAATTTTTAGACTCGATAGCTTGCTAGCAATTCGGCG-3′ (top strand) and 5′-CGCCGAATTGCTAGCAAGCTCAATCGAGTCTAAAAATTCGGCT-3′ (bottom strand; CA insertion shown in bold). Corresponding homoduplexes were prepared by annealing the top strand with the oligonucleotide 5′-AGCCGAATTTTTAGACTCGATAGCTTGCTAGCAATTCGGCG-3′. The top strand contained a 5′ biotin tag for immobilization on a streptavidin-coated surface.
MutSβ binding to DNA was analyzed by SPRS on a Biacore 2000 (GE Healthcare). The indicated concentrations of MutSβ or MutSβΔ162 (in 25 mM HEPES–KOH pH 7.5, 150 mM KCl, 5 mM MgCl2, 1 mM dithiothreitol, 0.02% surfactant P20) were allowed to flow at 20 µl min−1 over a streptavidin chip derivatized with 200 response units of a biotinylated 41 bp homoduplex or heteroduplex DNA. Protein flow was carried out for 5 min followed by a 1 min wash with running buffer. MutSβ dissociation from DNA upon ATP challenge was then monitored by the injection of 1 mM ATP in running buffer. Dissociation kinetic data were fitted by nonlinear regression to a single exponential decay function.
2.4. DNA substrates
Oligonucleotides (Midland Certified Reagent Co.) were annealed in 10 mM sodium cacodylate pH 7.4, 50 mM NaCl, 10 mM MgSO4 and 0.5 mM EDTA. For initial crystal screening, a 14 bp duplex DNA containing a CA insert (5′-CCTAGCGCAGCGGTTC-3′ annealed with 5′-GAACCGCCGCTAGG-3′) was derived from the G·T mispair heteroduplex used for the crystallization of hMutSα (Warren et al., 2007 ▶). DNA length (5′-CCTAGCGCAGCGGTC-3′ annealed with 5′-GACCGCCGCTAGG-3′, 5′-CTAGCGCAGCGGTTC-3′ annealed with 5′-GAACCGCCGCTAG-3′, 5′-CAGCGCAGCGGTTC-3′ annealed with 5′-GAACCGCCGCTG-3′, 5′-TCCCTAGCGCAGCGGTTCCGA-3′ annealed with 5′-CGGAACCGCCGCTAGGGAC-3′) and end base-pair sequences (5′-CCTAGCGCAGCGGTTT-3′ annealed with 5′-AAACCGCCGCTAGG-3′, 5′-GCTAGCGCAGCGGTTC-3′ annealed with 5′-GAACCGCCGCTAGC-3′, 5′-CCTAGCGCAGCGGTTG-3′ annealed with 5′-CAACCGCCGCTAGG-3′) were subsequently explored for screening and optimization.
2.5. Crystallization
Full-length hMutSβ, MutSβΔ162, MutSβΔ175 and MutSβΔ223 were used for crystallization trials. Protein samples at 29 µM were incubated with 2.5 mM MgCl2, 10 mM dithiothreitol, a twofold molar excess of ADP and a 1.2-fold molar excess of loop-containing duplex DNA of various lengths. The sitting-drop vapor-diffusion method and a set of sparse-matrix crystallization screens from Qiagen were used for initial crystallization trials at 290 K. Screening was performed using a Mosquito robot (TTP LabTech) in two-well MRC crystallization plates using a reservoir volume of 82 µl and drops consisting of 200 nl protein solution and 200 nl reservoir solution.
3. Results and discussion
3.1. Proteolytic fragment of human MSH3
A proteolytic fragment (Fig. 2 ▶) was identified by N-terminal sequencing to be an MSH3 fragment starting at residue 176. A similar proteolytic fragment of MSH6 was observed for human MutSα (Warren et al., 2007 ▶), suggesting similar domain structures for MSH3 and MSH6.
3.2. DNA-binding studies of hMutSβ
To determine whether the truncation of the N-terminal 162 amino acids of MSH3 alters the heteroduplex binding of MutSβ, we compared the affinity and specificity of MutSβ and MutSβΔ162 for a 41 bp I/D heteroduplex and an otherwise identical homoduplex DNA by SPRS (see §2.3). As shown in Fig. 3 ▶(a), the affinities of MutSβ and MutSβΔ162 for a CA dinucleotide I/D heteroduplex are similar (4 ± 1 and 5 ± 2 nM, respectively) and both proteins prefer heteroduplex over homoduplex DNA. Furthermore, both MutSβ (Fig. 3b ▶) and MutSβΔ162 (Fig. 3c ▶) dissociate with similar t 1/2 values (∼2.5–3 s) from heteroduplex DNA upon challenge with 1 mM ATP, indicating that that functional interaction of DNA-binding and nucleotide-binding sites is retained in MutSβΔ162. Similar results were obtained with MutSβΔ223 (data not shown).
Figure 3.

Binding of MutSβ and MutSβΔ162 to a 41 bp heteroduplex containing a CA insertion–deletion loop. (a) Binding of MutSβ (circles) and MutSβΔ162 (squares) to a 41 bp heteroduplex (closed symbols) or control homoduplex (open symbols) was assessed by SPRS (see §2.3). Mass response units at saturation (∼400 s) are plotted as a function of protein concentration. Data were fitted by nonlinear least-squares regression to a rectangular hyperbola, yielding K d values of 4 ± 1 nM for the wild-type protein and 5 ± 2 nM for MutSβΔ162 for heteroduplex binding. Binding to homoduplex DNA was not saturable up to 100 nM protein concentration. Dissociation of MutSβ (b) or MutSβΔ162 (c) from 41 bp heteroduplex (solid lines) and homoduplex (dotted lines) DNA substrates upon challenge (arrow) with 1 mM ATP was assessed by SPRS.
3.3. Crystallization
MutSβΔ162 was initially cocrystallized with ADP and a 14 bp duplex DNA containing a CA insert (5′-CCTAGCGCAGCGGTTC-3′ annealed with 5′-GAACCGCCGCTAGG-3′). By using 1 µl protein solution and 1 µl precipitant, crystals with a bipyramidal morphology (form 1; Fig. 4 ▶ a) grew out of drops containing a precipitating agent of 100 mM HEPES pH 7.5, 200 mM calcium acetate, 5 mM cadmium chloride and 9–12% PEG 8000 at 290 K. Further screening by varying the DNA-substrate length and end base-pair sequence led to a second crystal form using the MutSβΔ223 construct. Crystallization of MutSβΔ223 with a revised duplex (5′-TCCCTAGCGCAGCGGTTCCGA-3′ annealed with 5′CGGAACCGCCGCTAGGGAC-3′) yielded three-dimensional rectangular crystals (form 2; Fig. 4b ▶) from 100 mM trisodium citrate pH 5.6, 250 mM lithium sulfate and 12–15% PEG 4000 at 290 K. The crystals reached final dimensions of ∼200 × 80 × 40 µm in 5 d.
Figure 4.

Human MutSβ crystals. (a) Tetragonal form 1 crystal with approximate dimensions of 200 × 200 × 200 µm. (b) Monoclinic form 2 crystals of dimensions 200 × 80 × 40 µm.
Form 1 crystals were transferred into a stabilization solution (100 mM HEPES pH 7.5, 200 mM calcium acetate, 17% PEG 8000, 150 mM KCl and 0.5 mM EDTA) and serially to cryobuffer solutions supplemented with 7, 13.5, 20 and 27% ethylene glycol. Similar step-transfer cryocooling performed with glycerol, PEG 8000, calcium acetate and PEG 200 compromised the diffraction quality. Form 2 crystals were cryoprotected by stepwise transfer into mother-liquor solutions supplemented with ethylene glycol up to a final ethylene glycol concentration of 22% before flash-cooling in liquid nitrogen.
3.4. Data collection and processing
Diffraction data were collected at 100 K on beamline 22-ID (SER-CAT) at the Advanced Photon Source and beamline 12.3.1 (SIBYLS) at the Advanced Light Source. Crystal data are summarized in Table 1 ▶. Crystal form 1 diffracted to ∼10 Å resolution and belonged to a tetragonal space group, with unit-cell parameters a = b = 300, c = 260 Å. Crystal form 2 belonged to space group C2, with unit-cell parameters a = 185, b = 134, c = 102 Å. Form 2 crystals diffracted to a resolution of ∼3.1 Å (Fig. 5 ▶) and two data sets from these crystals were merged to produce a more complete data set. Data were reduced using XDS (Kabsch, 2010 ▶) and scaled to a resolution of 3.1 Å with an R merge of 7.5% and an overall completeness of 100.0%. Solvent-content analysis suggested a Matthews coefficient (Matthews, 1968 ▶) of 2.88 Å3 Da−1, corresponding to one molecule in the asymmetric unit with a solvent content of 57%. Analysis of reduced data was carried out using SCALA (Winn et al., 2011 ▶). Molecular replacement (MR) was carried out with the program Phaser (McCoy et al., 2007 ▶) using the structure of human MutSα (PDB entry 2o8b; Warren et al., 2007 ▶) as a search model. Although Phaser provided a clear MR solution (both rotational and translational Z scores > 20) with the expected one molecule per asymmetric unit, the resulting maps were not of sufficient quality to build the 976 residues necessary to complete the MSH3 subunit. Expression of selenomethionine-substituted protein to provide experimental phasing information is under way.
Table 1. Crystallographic data statistics for MutSβΔ223.
Values in parentheses are for the highest shell.
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 184.84, b = 134.14, c = 102.15, β = 90.30 |
| X-ray source | ALS SIBYLS 12.3.1 |
| Wavelength (Å) | 0.9795 |
| Resolution (Å) | 50–3.1 (3.27–3.1) |
| Rmerge† | 7.5 (70.8) |
| Rmeas‡ | 8.0 (76.0) |
| Rp.i.m.§ | 2.9 (27.4) |
| 〈I/σ(I)〉 | 20.3 (3.0) |
| Completeness (%) | 100.0 (100.0) |
| Total reflections | 343597 |
| Unique reflections | 45282 |
| Multiplicity | 7.6 (7.6) |
| Molecules per asymmetric unit | 1 |
| VM (Å3 Da−1) | 2.88 |
| Solvent content (%) | 57 |
| Wilson B factor (Å2) | 90.5 |
| Molecular weight (kDa) | 221 |
R
merge = 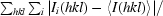
 .
.
R
meas = 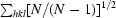
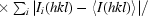 .
.
R
p.i.m. = 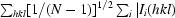 −
− 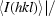 .
.
Figure 5.

A diffraction pattern of crystal form 2 collected on beamline 12.3.1 at the Advanced Light Source. The resolution ring corresponds to 3.05 Å.
4. Conclusions
In contrast to the repair of both base–base mismatches and insertion–deletion loops by a single homodimer in prokaryotes, eukaryotic MMR employs multiple heterodimeric MutS homologs. This partition of substrate recognition is puzzling given the relatively low cellular abundance of MutSβ (Drummond et al., 1997 ▶; Genschel et al., 1998 ▶) and its functional overlap with MutSα in repairing extrahelical nucleotides (Acharya et al., 1996 ▶; Genschel et al., 1998 ▶). Furthermore, despite the presence of a conserved Phe-X-Glu motif in both MSH3 and MSH6 mismatch binding domains, base–base mispairs are ideal substrates for MutSα but not MutSβ (Palombo et al., 1996 ▶). Structural characterization and comparison of both MutSβ and MutSα bound to heteroduplex DNA is likely to clarify this ambiguity, provide insight into the molecular mechanism of eukaryotic mismatch recognition and establish a basis for studying repeat instability and segregation fidelity.
Acknowledgments
We would like to thank Elisabeth Penland and Stephanie Armstrong for assistance in insect cell culture and Miaw-Sheue Tsai for the cloning of MutSβΔ175 and MutSβΔ223. This work was supported by R01 GM91487 to LSB and P01 CA92584 to LSB and PLM from the National Institutes of Health. PLM is an Investigator of the Howard Hughes Medical Institute. Data were collected on the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory and beamline 12.3.1 (SIBYLS) at the Advanced Light Source, Lawrence Berkeley National Laboratory. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CH11357. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231.
References
- Acharya, S., Wilson, T., Gradia, S., Kane, M. F., Guerrette, S., Marsischky, G. T., Kolodner, R. & Fishel, R. (1996). Proc. Natl Acad. Sci. USA, 93, 13629–13634. [DOI] [PMC free article] [PubMed]
- Berndt, S. I., Platz, E. A., Fallin, M. D., Thuita, L. W., Hoffman, S. C. & Helzlsouer, K. J. (2007). Int. J. Cancer, 120, 1548–1554. [DOI] [PubMed]
- Blackwell, L. J., Wang, S. & Modrich, P. (2001). J. Biol. Chem. 276, 33233–33240. [DOI] [PubMed]
- Broek, W. J. van den, Nelen, M. R., Wansink, D. G., Coerwinkel, M. M., te Riele, H., Groenen, P. J. & Wieringa, B. (2002). Hum. Mol. Genet. 11, 191–198. [DOI] [PubMed]
- Dowen, J. M., Putnam, C. D. & Kolodner, R. D. (2010). Mol. Cell. Biol. 30, 3321–3328. [DOI] [PMC free article] [PubMed]
- Drummond, J. T., Genschel, J., Wolf, E. & Modrich, P. (1997). Proc. Natl Acad. Sci. USA, 94, 10144–10149. [DOI] [PMC free article] [PubMed]
- Foiry, L., Dong, L., Savouret, C., Hubert, L., te Riele, H., Junien, C. & Gourdon, G. (2006). Hum. Genet. 119, 520–526. [DOI] [PubMed]
- Genschel, J., Littman, S. J., Drummond, J. T. & Modrich, P. (1998). J. Biol. Chem. 273, 19895–19901. [DOI] [PubMed]
- Harrington, J. M. & Kolodner, R. D. (2007). Mol. Cell. Biol. 27, 6546–6554. [DOI] [PMC free article] [PubMed]
- Hsieh, P. & Yamane, K. (2008). Mech. Ageing Dev. 129, 391–407. [DOI] [PMC free article] [PubMed]
- Iyer, R. R., Pluciennik, A., Burdett, V. & Modrich, P. L. (2006). Chem. Rev. 106, 302–323. [DOI] [PubMed]
- Jiricny, J. (2006). Nature Rev. Mol. Cell Biol. 7, 335–346. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kunkel, T. A. & Erie, D. A. (2005). Annu. Rev. Biochem. 74, 681–710. [DOI] [PubMed]
- Lamers, M. H., Perrakis, A., Enzlin, J. H., Winterwerp, H. H., de Wind, N. & Sixma, T. K. (2000). Nature (London), 407, 711–717. [DOI] [PubMed]
- Lee, S. D., Surtees, J. A. & Alani, E. (2007). J. Mol. Biol. 366, 53–66. [DOI] [PMC free article] [PubMed]
- Li, G.-M. (2008). Cell Res. 18, 85–98. [DOI] [PubMed]
- Manley, K., Shirley, T. L., Flaherty, L. & Messer, A. (1999). Nature Genet. 23, 471–473. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mirkin, S. M. (2007). Nature (London), 447, 932–940. [DOI] [PubMed]
- Mitas, M., Yu, A., Dill, J., Kamp, T. J., Chambers, E. J. & Haworth, I. S. (1995). Nucleic Acids Res. 23, 1050–1059. [DOI] [PMC free article] [PubMed]
- Notredame, C., Higgins, D. G. & Heringa, J. (2000). J. Mol. Biol. 302, 205–217. [DOI] [PubMed]
- Obmolova, G., Ban, C., Hsieh, P. & Yang, W. (2000). Nature (London), 407, 703–710. [DOI] [PubMed]
- Owen, B. A., Lang, W. H. & McMurray, C. T. (2009). Nature Struct. Mol. Biol. 16, 550–557. [DOI] [PMC free article] [PubMed]
- Palombo, F., Iaccarino, I., Nakajima, E., Ikejima, M., Shimada, T. & Jiricny, J. (1996). Curr. Biol. 6, 1181–1184. [DOI] [PubMed]
- Pearson, C. E., Nichol Edamura, K. & Cleary, J. D. (2005). Nature Rev. Genet. 6, 729–742. [DOI] [PubMed]
- Pearson, C. E., Tam, M., Wang, Y.-H., Montgomery, S. E., Dar, A. C., Cleary, J. D. & Nichol, K. (2002). Nucleic Acids Res. 30, 4534–4547. [DOI] [PMC free article] [PubMed]
- Risinger, J. I., Umar, A., Boyd, J., Berchuck, A., Kunkel, T. A. & Barrett, J. C. (1996). Nature Genet. 14, 102–105. [DOI] [PubMed]
- Ross-Macdonald, P. & Roeder, G. S. (1994). Cell, 79, 1069–1080. [DOI] [PubMed]
- Savouret, C., Garcia-Cordier, C., Megret, J., te Riele, H., Junien, C. & Gourdon, G. (2004). Mol. Cell. Biol. 24, 629–637. [DOI] [PMC free article] [PubMed]
- Schofield, M. J. & Hsieh, P. (2003). Annu. Rev. Microbiol. 57, 579–608. [DOI] [PubMed]
- Smith, G. K., Jie, J., Fox, G. E. & Gao, X. (1995). Nucleic Acids Res. 23, 4303–4311. [DOI] [PMC free article] [PubMed]
- Snowden, T., Acharya, S., Butz, C., Berardini, M. & Fishel, R. (2004). Mol. Cell, 15, 437–451. [DOI] [PubMed]
- Tian, L., Gu, L. & Li, G.-M. (2009). J. Biol. Chem. 284, 11557–11562. [DOI] [PMC free article] [PubMed]
- Tian, L., Hou, C., Tian, K., Holcomb, N. C., Gu, L. & Li, G.-M. (2009). J. Biol. Chem. 284, 20452–20456. [DOI] [PMC free article] [PubMed]
- Warren, J. J., Pohlhaus, T. J., Changela, A., Iyer, R. R., Modrich, P. L. & Beese, L. S. (2007). Mol. Cell, 26, 579–592. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yu, A., Dill, J. & Mitas, M. (1995). Nucleic Acids Res. 23, 4055–4057. [DOI] [PMC free article] [PubMed]