

Summary
Asthma and allergy are common conditions with complex etiologies involving both genetic and environmental contributions. Recent genome-wide association studies (GWAS) and meta-analyses of GWAS have begun to shed light on both common and distinct pathways that contribute to asthma and allergic diseases. Associations with variation in genes encoding the epithelial cell-derived cytokines, interleukin-33 (IL-33) and thymic stromal lymphopoietin (TSLP), and the IL1RL1 gene encoding the IL-33 receptor, ST2, highlight the central roles for innate immune response pathways that promote the activation and differentiation of T-helper 2 (Th2) cells in the pathogenesis of both asthma and allergic diseases. In contrast, variation at the 17q21 asthma locus, encoding the ORMDL3 and GSDML genes, is specifically associated with risk for childhood onset asthma. These and other genetic findings are providing a list of well-validated asthma and allergy susceptibility genes that are expanding our understanding of the common and unique biological pathways that are dysregulated in these related conditions. Ongoing studies will continue to broaden our understanding of asthma and allergy and unravel the mechanisms for the development of these complex traits.
Keywords: association studies, linkage studies, GWAS, asthma, allergic disease, atopy
Introduction
Asthma and allergy are common conditions with complex and heterogeneous etiologies. Asthma and allergic disease often co-occur in the same individual or in different individuals within the same families. However, whether this co-occurrence reflects distinct conditions with shared pathogenic pathways or different clinical manifestations of the same disease is currently unknown.
Both asthma and allergic diseases (defined below) have significant genetic contributions, with heritability estimates varying between 35% and 95% for asthma (1-15), 33% and 91% for allergic rhinitis (hayfever) (2, 11-16), 71% and 84% for atopic dermatitis (eczema) (12-14), 35% and 84% for total serum immunoglobulin E (IgE) levels (9, 17-36), 34% and 68% for specific serum IgE levels (33-38), 30% and 66% for bronchial hyperresponsiveness (BHR) (32, 35, 36), and 24% to 41% for blood eosinophil counts (26, 35-37, 39). Overall, therefore, significant proportions of the inter-individual risks for these conditions are due to genetic differences. Nonetheless, identifying specific genes and variation within those genes that account for this risk has been one of the major challenges in asthma/allergy research over the past 40 years. Characterizing the genetic architecture of these conditions is the first step in beginning to understand whether they share a common etiology (i.e. different manifestations of the same disease) or overlapping pathogenic pathways (i.e. different diseases due to perturbations of a shared pathway). Recent genetic studies are beginning to shed light on this important question.
In this review, we summarize the genetic studies of asthma and allergic diseases that have been conducted over the past 40 years, with emphasis on studies completed in the past 5 years and, in particular, on results of genome-wide association studies (GWAS). Ultimately, we will discuss these results in light of the relationship between asthma and allergic diseases.
Asthma and allergic diseases
Allergy refers to a detrimental immune-mediated inflammatory response to normally harmless environmental substances known as allergens, resulting in one or more allergic diseases such as asthma, allergic rhinitis, atopic dermatitis, and food allergy. In contrast, atopy is defined by evidence of allergic sensitization to known allergens, reflected in either a positive skin prick test (SPT) or the presence of specific IgE to one or more known allergens in the serum. Elevated total serum IgE is sometimes also used as a proxy for atopy. The relationship between allergic diseases and atopy is complicated by two facts: not everyone with atopy develops clinical manifestations of allergy and not everyone with clinical symptoms of allergic disease is atopic when tested for specific IgE to a wide range of environmental allergens (40). In this section, we focus on the epidemiology and burden of asthma and allergy, as well as the heterogeneous nature of the asthma phenotypes, and highlight the link between asthma and allergic diseases.
Epidemiology and burden of asthma and allergy
The increase in the prevalence of asthma and allergic diseases in most countries around the world in recent decades poses a substantial global health burden to people of all ages and all ethnic backgrounds. The global prevalence of asthma ranges from 1% to 18% of the population in different countries (41-45). It is estimated that asthma affects as many as 300 million people worldwide; this is expected to increase to 400 million by 2025 (41, 42). Asthma is estimated to account for 1 in every 250 deaths worldwide, resulting in 250,000 deaths each year worldwide (41, 42). Allergic rhinitis, clinically defined as a symptomatic disorder of the nose induced after allergen exposure by an IgE-mediated inflammation, affects 10% to 20% of the population, with an estimated over 500 million affected people worldwide (46, 47). Atopic dermatitis, a chronic inflammatory pruritic skin disease, affects 10% to 20% of children and 1% to 3% of adults worldwide (48-50). Food allergy, defined as an adverse immune response to food proteins, affects up to 6% of young children and 3% to 4% of adults (51). The prevalence of allergic sensitization (based on positive SPT to allergens or the presence of allergen-specific serum IgE) ranges from 16% to 57% in the populations of the United States, Europe, Australia, New Zealand, and Taiwan (52-56). Allergic diseases cause widespread morbidity, leading to impaired work productivity and school performance, reduced quality of life, and substantial medical and non-medical costs.
Asthma: a complex, multifactorial disease
Asthma is a complex syndrome characterized by the presence of chronic inflammation in the lower airways, resulting in variable airflow obstruction and BHR and causing recurrent episodes of coughing, wheezing, breathlessness, and chest tightness (41, 57). Because the pathogenesis of asthma is unknown and there are no tests or biomarkers that definitively diagnose asthma or distinguish it from other diseases, diagnosis is based on clinical features, demonstration of reversible expiratory airflow obstruction (or BHR when lung function is normal), and exclusion of alternative diagnoses that mimic asthma (41, 57). It is important to note that although tests for BHR are sensitive for a diagnosis of asthma, they have limited specificity (58). Therefore, a negative test may help exclude a diagnosis of current asthma in a subject who is not taking asthma medications. However, a positive test does not indicate that a subject has asthma (59), and BHR may be caused by conditions other than asthma, such as allergic rhinitis (60), chronic obstructive pulmonary disease (COPD) (61), and cystic fibrosis (62). Allergic sensitization commonly occurs in 80% of asthmatic children and approximately 60% of asthmatic adults (63). Tests for allergic sensitization may identify allergic triggers that cause asthma symptoms in individual patients, but individuals with allergic sensitization may develop other allergic diseases, and some sensitized individuals may never experience allergic symptoms (64, 65). Thus, skin prick tests or measurements of specific IgE are also not specific for a diagnosis of asthma or even for allergic diseases.
Clinical heterogeneity and intermediate phenotypes
Asthma is not a single disease but rather an umbrella for multiple diseases with similar clinical features, and likely with different genetic and environmental contributors. On the one hand, more precise phenotypic definitions would allow identification of more homogeneous subgroups of patients with asthma and facilitate genetic studies. On other hand, the discovery of asthma and allergy susceptibility loci could provide insights into phenotypic subgroups and ultimately inform the selection of targeted/effective treatment. Because of this clinical heterogeneity and challenge in defining asthma subgroups, intermediate phenotypes that can be measured more objectively are often the focus of genetic studies (66-73). These phenotypes include BHR, lung function, serum IgE, allergen skin test response, and blood eosinophil count, as described in Table 1. Genetic studies of intermediate phenotypes may ultimately facilitate the discovery of susceptibility genes for asthma or allergic diseases (66-68), or they may only identify genes that influence normal variation in the trait itself.
Table 1.
Allergic diseases and associated phenotypes that have been considered in genetic studies
Allergic diseases
|
Clinical or physiological phenotypes
|
Atopy-related phenotypes
|
Trigger-related phenotypes
|
Inflammatory phenotypes
|
Link between asthma and allergic diseases
The clinical relationship between asthma, allergic rhinitis, and atopic dermatitis, the so-called ‘allergic triad’, is well known (74). These diseases often have their roots during childhood. Several longitudinal studies provide evidence for a characteristic sequential development of atopic manifestations during childhood: atopic dermatitis and food allergy typically develop in infancy followed by asthma and/or allergic rhinitis in childhood. This is referred to as the ‘atopic march’ (75-80). About 30% of children with atopic dermatitis develop asthma, and nearly 66% develop allergic sensitization and symptoms of allergic rhinitis (81). The vast majority (~80%) of patients with asthma have allergic rhinitis, whereas 19% to 38% of patients with allergic rhinitis have coexisting asthma (82-84). Of particular note is that asthma and allergic rhinitis are linked by common epidemiologic, clinical, and pathophysiologic mechanisms, as well as common comorbidities and therapeutic approaches, leading to the concept of ‘one airway one disease’ (47, 85-88), although also differences have also been noted (89). Yet, an open question remains as to whether asthma and allergic diseases are part of the same disease continuum, the ‘atopic syndrome’, occurring in different organs in the body, or they are distinct disease entities that have their own specific causes.
Environment modifiers of asthma and allergy
Asthma and allergy are caused by the complex interplay between genetic factors and environmental exposures that occur at critical times in development. Epidemiologic studies have highlighted many associations between environmental exposures and subsequent risk for asthma and allergy. Modifiers of risk that have been reported in the literature include genetics (discussed below), race/ethnicity (90), sex (91), passive (92-96) and active (97-99) tobacco smoke exposure, farm animals and related products (100-104), domestic cats and dogs (105-108), family size and birth order (109-112), daycare attendance in early childhood (113-115), respiratory viral infections (116-119), microbial exposures (120), vaccination (121-124), antibiotics and antipyretics (125-129), mode of delivery (130-133), breastfeeding (134-137), intake of diet and nutrition (138-143), air pollution (144-146), obesity (147-149), allergens (150-152), and occupational exposures (153-155). Further research is needed to clarify the importance of each and to elucidate the mechanisms through which each of these factors modifies risk for asthma and allergy.
Approaches for discovering asthma and allergy genes
The approaches used for asthma and allergy gene discovery have evolved over time as the various genotyping technologies have become both more high throughput and less costly. As we begin to adopt 21st century technologies, such as Next Generation sequencing (156) of whole genomes, gene discovery will no longer be limited by genotyping technologies but rather by the enormous computational and bioinformatic demands, and the interpretation and synthesis of the vast amounts of data generated by these approaches. In this section, we take a historical perspective and review the approaches that have been used to discover or better characterize asthma and allergy susceptibility loci. A summary of these approaches is shown in Table 2. We finish this section with a discussion of the implications of using each approach for gene discovery.
Table 2.
Approaches for gene discovery
| Approach | Advantages | Disadvantages |
|---|---|---|
| Candidate Gene Association Studies (1970’s-present) |
|
|
| Genome-Wide Linkage Studies (1980’s-1990’s) |
|
|
| Genome-Wide Association Studies (2007-present) |
|
|
| Re-Sequencing Studies in Genes, Exomes, or Whole Genomes (ongoing) |
|
|
Candidate gene association studies
Candidate gene association studies are conceptually straightforward. The most common approach is a case-control study in which one searches for an enrichment of a marker (e.g., single nucleotide polymorphisms (SNPs) or insertion deletion (indel) polymorphisms) allele or haplotype (combination of alleles) in cases (individuals with a specific disease) compared to controls (either individuals without the disease, referred to as ‘super controls’, or subjects from the general population for whom disease status is unknown, referred to as ‘population controls’). Alternative approaches are cohort and cross-sectional study designs in which individuals are not selected on the basis of disease status. In these studies, one searches for an enrichment of the disease in individuals with a certain genotype at the marker locus or haplotype. The effect sizes of associated alleles, genotypes, or haplotypes are typically reported as either odds ratios (ORs) for case-control studies or relative risks (RRs) for cohort and cross-sectional studies.
As its name implies, genes are selected for these studies based on their known function (functional candidates), or their chromosomal position in a linkage peak (see ‘Genome-wide linkage studies’ below) or near a previous association signal (positional candidate). The specific variants used in candidate gene studies can be selected based on function (for example, an amino acid substitution that is known to influence protein function or a promoter SNP that is known to influence transcription), previous associations with the same or related disease, or to comprehensively survey variation in and around the gene of interest (which may also include the first two categories of variants). The latter strategy requires knowledge about the haplotype structure of the gene and the ability to select SNPs that ‘tag’ all common haplotypes. Such ‘tag SNPs’ would not be in strong linkage disequilibrium (LD) with each other but would be selected to be in LD with other SNPs that are not genotyped in the sample (see references (157-159) for further description of this strategy). Thus, a relatively small number of SNPs (10 to several hundred depending on the size of the gene, the haplotype structure, and the stringency used for determining LD) are selected for each gene. Because genes are selected based on prior hypotheses (related to function or position or both), associations discovered by this method are easy to interpret. The latter is one of the advantages of candidate gene association studies.
The vast majority of genetic studies of asthma and allergic diseases have been candidate gene association studies in which genes were selected based on their known function and likely involvement in pathogenesis, as previously reviewed (160-162). As a result, these studies are heavily biased towards studies of immune-related genes. In fact, the most significant limitation of this approach is that it is confined to what we know (or what we think we know) about disease pathogenesis and gene functions. The candidate gene approach cannot, by itself, discovery novel pathways or genes.
Genome-wide approaches
In contrast to candidate gene association studies, the other gene discovery approaches that we will discuss are genome-wide approaches, which consider all regions of the genome without any prior hypotheses about the location of the most significant genetic contributors to risk. As a result, genome-wide approaches are referred to as ‘hypothesis-free’ or ‘hypothesis-generating’. The greatest advantage of genome-wide approaches is that they can discover novel genes and pathways involved in disease pathogenesis. The greatest limitation of these approaches is the statistical burden that results from the large number of tests performed and the resulting requirement for very large sample sizes to achieve statistical significance. In addition, the relationship between the discovered variants or loci and disease pathogenesis is not always obvious.
Genome-wide linkage studies
The first genome-wide approach that was technically feasible to apply in large samples was linkage studies. A requirement of linkage studies is the availability of families with at least two affected relatives (most commonly affected sibling pairs). The basic premise of linkage studies is that the ‘disease locus’ will co-segregate with the disease in families. As a result, regions of the genome that harbor susceptibility loci will be shared among affected relatives more often than expected by chance. For example, using a sibling pair design, one looks for alleles at marker loci that are shared by both siblings more than 50% of the time (the expected amount of allele sharing between siblings). Depending on whether both parents are also genotyped, the precision of the estimate that the genotypes are truly identical by descent (each shared allele inherited from the same parent) or identical by state (same allele but not necessarily inherited from the same parent) varies. The evidence for linkage is reported as a likelihood ratio, or a LOD score (log of the odds in favor of linkage). Linkage studies are performed in many families, and LOD scores are summed across families. Typically LOD scores >>3 (representing greater than 1,000 to 1 odds in favor of linkage), preferably >3.4, are required to declare that an observed linkage to a complex disease is significant.
The advantage of linkage studies is that they require relatively few genetic markers, at least by 21st century standards. Genome-wide linkage studies in the 1990s typically used 400-800 highly polymorphic microsatellite markers (also referred to as short tandem repeat polymorphisms, or STRPs), but today it is easier to use ~8,000-10,000 SNPs, which are available on genotyping arrays and provide equivalent or better coverage and information as 1,000 STRPs (163). Moreover, linkage studies will reveal regions or genes that harbor multiple rare variants that confer risk for disease, even if the specific variant differs among families. This is an often-overlooked advantage of linkage studies, although discovering multiple rare variants that are contributing to a linkage peak is a daunting task. In addition to the requirement for studying families, the most significant limitations of linkage studies are that it identifies very broad regions that can contain hundreds of genes (i.e., has poor resolution) and has low power to detect risk variants with modest effect sizes on disease risk.
Genome-wide association studies (GWAS)
In contrast to genome-wide linkage studies, the main advantages of GWAS are its excellent resolution, its power to detect risk variants with modest effect sizes, and that it does not require studying families (164). The GWAS extends the candidate gene approach described above to include markers that ‘tag’ (at least in theory) all common variation in the genome. GWAS became feasible in the past five years as high throughput genotyping platforms, such as those made by Affymetrix or Illumina, with 300,000 to over 1 million SNPs became available and affordable. Moreover, using the data available through the HapMap project (165-168), >3 million genotypes can currently be ‘imputed’ for subjects from most racial/ethnic groups using the genotypes generated by the Affymetrix or Illumina SNP arrays and the known haplotype structure of the HapMap samples (168-170). Therefore, current GWAS typically test for associations with a million or more SNPs. The statistical burden resulting from performing this many tests requires very stringent thresholds of significance (typically with p<10−7) and, therefore, very large sample sizes to achieve genome-wide levels of significance. The latter requirement is one of limitations of this approach and has encouraged many national and international collaborations and meta-analyses, in which results of many smaller GWAS are combined to increase power. Two recent meta-analyses of asthma GWAS in Europe (171) and the U.S. (172) demonstrate the power of this approach (discussed below).
The greatest limitation of the GWAS is that it will primarily detect common risk variants, both because the genotyping platforms include mostly common variants and because of the reduced power to detect associations with SNPs with low (<1%) minor allele frequencies (MAFs). In fact, the GWAS experiences to date, across many diseases including asthma, suggest that the risk alleles identified by this approach account for a very small proportion of the genetic risk. There are many possible explanations for this observation, (see ‘Missing Heritability’ section below). However, one currently popular explanation for poor predictive power of the risk variants identified by GWAS is that rare variants, which will not be detected by GWAS, have overall larger effects than common variants on disease risk, challenging the ‘common disease-common variant hypothesis’ (173-175), one of the basic rationales for performing GWAS (164, 176).
Re-sequencing studies
Whereas GWAS have dominated common disease genetic studies in the past five years, sequencing studies will dominate the next five. The current explosion of sequencing studies, most of which are ongoing at the time of this writing, has been facilitated by the development of high throughput, massively parallel sequencing, referred to as Next Generation (NextGen) sequencing (156, 177). The rationale for these studies is the belief that rare variants, with larger effect sizes on disease risk than common variants, explain a significant portion of the genetic risk for common diseases (reviewed in 178, 179). Interestingly, theoretical modeling performed a decade ago suggested that multiple, low frequency alleles at susceptibility loci are more likely than one or a few common alleles to contribute to risk for common diseases (180, 181). Such allelic heterogeneity will not be detected by the classical statistical approaches to association studies, including GWAS. In addition, as mentioned above, the most commonly used genotyping platforms nearly exclusively interrogate common variation. In contrast, the detection of rare variants requires the re-sequencing of genes, exomes, or even whole genomes in large samples of individuals to discover rare variation that is not present or tagged by variation on the genotyping platforms. There are currently many ongoing studies to re-sequence the exomes of hundreds or thousands of individuals to discover rare variants that are enriched in cases compared to controls, but the jury is still out as to whether rare variants will account for a significant proportion of the genetic risk for complex diseases, including asthma and allergic diseases. However, it is already clear that the cost of sequencing genomes or exomes is dropping so rapidly that this technology may even replace SNP genotyping in the near future. In contrast, the bioinformatic and computational requirements for storage, analysis, and interpreting sequence data are enormous, and developments in this area are barely keeping apace with the generation of these data.
The requirement for replication
Because of the large number of tests performed in genetic studies there is always the possibility that some variants will show very significant associations by chance alone. This is true for candidate gene association studies as well, although to a lesser extent. As a result, the gold standard for validating an association is to replicate the association in at least one independent sample. The replication sample(s) should be of sufficient size to minimize the likelihood of a type 2 error (false negative result), and the replicated association should ideally be with the same allele and in the same direction as in the original study. However, because of the complexity of these diseases, and the likely important roles for early life environmental exposures (see ‘Environmental modifiers’ section above), negative replication studies of very strong association signals can be difficult to interpret and the evidence for lack of replication should be as carefully evaluated as those supporting the original association (182).
Summary: choice of approach for gene discovery and the role of allele frequency spectrum and effect sizes
The approaches to gene discovery reviewed above have fallen in and out of favor, as the limitations of each approach become more apparent and developing technologies allow us to adopt newer strategies that were previously technically implausible or financially prohibitive. However, we believe that all of these approaches should have a place in the gene mapper’s toolbox, because it is unlikely that any one approach will be sufficient to identify all genetic risk factors. The best choice of method, of course, depends on the true underlying genetic architecture of asthma and allergic disease, which is currently unknown (179, 183) (Fig. 1). If, for example, the most important genetic risk factors for asthma and allergy are common variants at many loci with modest effect sizes, then GWAS, and meta-analyses of GWAS, should be a powerful approach for finding these variants. Under that scenario, the variants identified by GWAS should collectively explain most of the genetic risk, or heritability, of the trait. If, however, most of the genetic risk for these diseases is due to multiple rare or low frequency variants with relatively larger effect sizes, then re-sequencing or even linkage studies might be the best or only way to detect these risk variants. In fact, it is likely that the genetic architecture of asthma and allergy encompasses both common and rare alleles, which will be revealed by a combination of approaches. As sequence data become available over the next few years, it will be particularly interesting to see whether genes with common risk alleles (such as those discovered by GWAS) also harbor rare variants that confer risk or whether the genes with common vs. rare risk variants are mutually exclusive. If the latter is the case, it will be additionally interesting to determine whether the genes that harbor rare variation with larger effects on disease risk share any features in common with each other, such as a molecular signatures of positive or negative selection.
Fig. 1. Genetic architecture of asthma and allergic diseases.
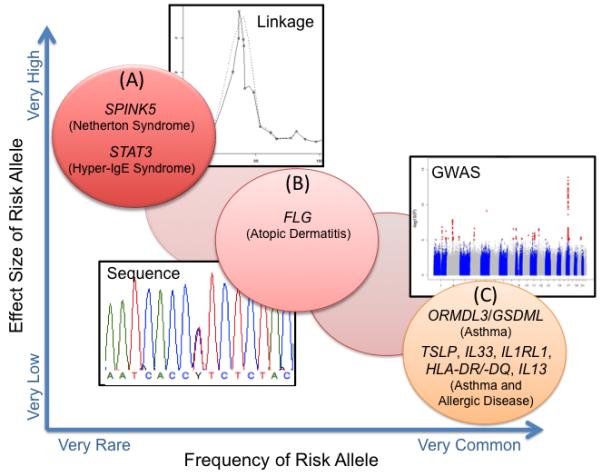
Examples are shown for (A) Monogenic diseases with allergic phenotypes that are caused by highly penetrant rare (<1%) mutations. These disease genes can be discovered by linkage studies in families segregating the disease. (B) Complex diseases or phenotypes with low frequency (1-5%) risk alleles with intermediate effect sizes. The relative paucity of genes in his category reflects the limited ability for re-sequencing studies in the past. (C) Complex diseases and phenotypes with common disease risk alleles (>5%) with very low effect sizes and penetrances can be discovered by GWAS. Modified from references (179, 183).
An overview of asthma and allergy gene discoveries
Candidate gene association studies
The vast majority of the >100 loci harboring variation that has been associated with risk for asthma and allergic phenotypes were first identified by candidate gene association studies (160-162), starting with the early studies of HLA and allergies in the Blumenthal and Marsh laboratories in the 1970s (184, 185). As of 2007, variation in more than 30 genes had been associated with asthma or allergic phenotypes in at least five independent (candidate gene) association studies (161, 162). Because these studies have been extensively reviewed elsewhere (160-162), an overview of all genes associated with asthma or allergy are not further discussed here.
Linkage followed by positional candidate studies
Identifying genes via linkage studies proved to be quite challenging. Among the >25 linkage studies reported between 1996 and 2009 (reviewed in 186, 187), only nine genes were successfully identified by positional candidate studies in linked regions (188-196) (Table 3). All but one of these genes (IRAK3) were novel at the time, i.e. they had not been considered as asthma ‘candidate’ genes prior to their identification through linkage and positional cloning studies.
Table 3.
Asthma/atopy genes discovered through linkage and positional cloning studies
| Gene | Chromosome Location |
Associated Phenotypes |
Reference |
|---|---|---|---|
|
ADAM33 (a disintegrin and metalloproteinase 33) |
20p13 | Asthma, BHR |
Van Eerdewegh et al. 2002 (192) |
|
DPP10 (dipeptidyl peptidase 10) |
2q14 | Asthma | Allen et al. 2003 (188) |
|
PHF11 (PHD finger protein 11) |
13q14 | Asthma, IgE | Zhang et al. 2003 (193) |
|
NPSR1 (neuropeptide S receptor 1) (also called GPRA, G protein-coupled protein receptor for asthma susceptibility) |
7p14 | Asthma, IgE | Laitinen et al. 2004 (189) |
|
HLA-G (human leukocyte antigen G) |
6p21 | Asthma, BHR, atopy |
Nicolae et al. 2005 (190) |
|
CYFIP2 (cytoplasmic FXMR interacting protein 2) |
5q33 | Atopic asthma |
Noguchi et al. 2005 (191) |
|
IRAK3 (interleukin-1 receptor-associated kinase 3) (also called IRAK-M) |
12q14 | Early onset, persistent asthma |
Balaci et al. 2007 (194) |
|
COL6A5 (collagen, type VI, alpha 6) (also called COL29A1) |
3q21 | Atopic dermatitis |
Söderhäll et al. 2007 (195) |
|
OPN3/CHML (opsin 3/choroideremia-like) |
1qter | Atopic asthma |
White et al. 2008 (196) |
Two recent meta-analyses of asthma and atopy linkage studies combined results for nine (186) and 20 (187) independent populations, providing data for 5,832 individuals of European ancestry in 1,267 pedigrees with asthma diagnoses and 5,425 asthmatic individuals in 2,053 racially and ethnically diverse families, respectively. Genome-wide significant linkages for asthma were reported for regions on 2p21-p14 (187), for asthma or BHR on an overlapping region on 6p22.3-21.1 (186, 187), and for BHR on 2q22.1-q23.3, 7q12.11-q31.1, and 5q23.2-q34 (186). Genome-wide significant linkages for atopy phenotypes were reported on 2q32-q34 for eosinophil count (187), for overlapping regions on 3p25.3-q24 and on 17p12-q25 for positive SPT (186, 187), on 5q23-q33 for a quantitative measure of SPT (187), and on 5q11.2-q14.3 and 6pter-6p22.3 for IgE (186). The region linked to asthma and BHR on 6p includes the HLA region, the location of one of the positionally cloned asthma genes, HLA-G (190), and for one of the most significant associations in a European meta-analysis of a GWAS for asthma (HLA-DQ) and for IgE (HLA-DRB1) (discussed below in ‘Meta-analysis of GWAS’) (171). The atopy locus on 5q harbors the ADRB2 gene and another positionally cloned asthma gene, CYPIP2 (191). The atopy loci on 2q and 3p include many outstanding candidates, including ICOS, CD28, and CTLA4 on 2q and TLR9, CCR3, and CCR5 on 3p. Curiously, both meta-analyses identified a broad region on chromosome 17 linked to atopy (positive SPT) that includes the 17q21 asthma susceptibility locus (ORMDL3/GSDML) (197), which does not show associations with atopy (discussed below in ‘Genome-wide association studies’). This suggests that at least one additional locus in this region contributes to atopic phenotypes and that the asthma and atopy susceptibility loci in this region are distinct.
In general, linkage studies of asthma and allergy were plagued by poor replication of linkage signals from study to study, possibly due to small sample sizes. Whether the recent meta-analyses of linkage studies provides some clarity on the location of the most important asthma or atopy loci remains to be seen. However, as revealed by the meta-analyses, many of the more modest linkage regions are consistent across studies. Nonetheless, the variants that explained the linkage signals have not yet been identified for most of these regions in families showing linkage. A possible explanation for why the latter step has been so difficult could be the presence of multiple risk variants or even multiple genes within the same linkage peak, with different variants or genes contributing to risk in different families. Because the follow-up studies to linkage are basically association studies of variation under linkage peaks, linkage signals that result from allelic or locus heterogeneity would have been (and still are) difficult to unravel. However, it is noteworthy that variation in many genes located within replicated linked regions, such as those on 6p, 5q, and 12q have been associated with asthma and allergic phenotypes. Although never demonstrated empirically, it is possible that different families are segregating variation in different genes (or different combinations of variation at each gene) in these regions. This would generate linkage signals that would not be explained by common variation at a single locus. Only direct sequencing of the linked regions in families showing evidence for linkage would allow direct testing of this hypothesis.
Genome-wide association studies
The first GWAS of asthma, published in 2007, identified ORMDL3 as a novel asthma susceptibility locus on chromosome 17p21 (197). Common variation in this locus was associated with asthma in 1,000 cases and 1,000 controls, and then replicated in >5,000 subjects. Moreover, one of the associated variants was an expression quantitative trait locus (eQTL) for ORMDL3 in lymphoblastoid cell lines (LCLs) (197), indicating that at least one of the associated SNPs (among many that are in very strong LD) regulates the expression of ORMDL3 in a genotype specific manner. Subsequent studies showed that the eQTL also regulated expression of a neighboring gene, GSDML (also called GSDMB) (197-200). In fact, due to the extensive LD between SNPs and the co-regulation of the expression of genes in this region, it has not been possible to delineate between the effects of these two genes and, as a result, this is now referred to as the 17q21 asthma locus. Many studies have since replicated associations with SNPs on 17q21 and asthma, further showing that the association is primarily with childhood onset asthma (199, 201-203) and is more pronounced in children with exacerbations, respiratory viral infections, severe asthma, and exposure to environment tobacco smoke in infancy (199, 201, 204-206). However, variation at this locus is not associated with atopy (171, 199, 201, 203), providing evidence for an asthma susceptibility locus that acts through non-atopic (i.e. non-IgE-mediated) pathways. The functions of ORMDL3 and GSDML are only beginning to be understood (207-209), but how one or both of these genes influence risk for childhood asthma is currently unknown.
This first asthma GWAS was followed by many others of asthma (210-214), atopic dermatitis (215), eosinophilic esophagitis (EoE) (216), atopy (217) and the asthma/allergy intermediate quantitative traits, YKL-40 levels (66), total serum IgE (67), eosinophil count (68), and lung function (FEV1, FEV1/FVC) (218). Despite the relatively large sample sizes of the published GWAS [compared to those in the earlier candidate studies (161)], no associations replicated across all studies and few reached genome-wide levels of significance (Table 4A). This is expected if the variants associated with these phenotypes have very small effects on disease risk, thereby requiring even larger samples to detect significant associations. In addition, heterogeneity within each sample could dilute the effects of risk variants if a particular variant confers risk only in some individuals but not in others. In particular, early life environmental exposures may interact with genotype to determine risk so that associations will only be apparent in exposed individuals. Such gene-environment interactions are likely widespread in asthma and allergic diseases, as recently reviewed (219), and variants involved in interactions will be very difficult, if not impossible, to detect in a GWAS.
Table 4.
Published GWAS and meta-analyses of GWAS for asthma, allergic diseases, or intermediate phenotypes
| Phenotype | Study | Gene(s) Discovered |
Approx. Size of Discovery Sample |
Ethnicity of Discovery Sample |
Ethnicity of Replication Sample(s) |
Replications |
|---|---|---|---|---|---|---|
| A. GWAS for asthma, allergic diseases, and intermediate quantitative phenotypes. | ||||||
| Asthma | Moffatt et al. 2007 (197) |
ORMDL3* (17q21) |
1,000 cases/1,000 controls |
British | German | Replicated association of ORMDL3 with asthma |
| YKL-40 levels | Ober et al. 2008 (66) |
CHI3L1* | 753 Hutterites | European American |
European American, German |
Replicated association of CHI3L1 with YKL-40 levels and asthma |
| Total serum IgE | Weidinger et al. 2008 (67) |
FCER1A, RAD50, STAT6 |
1,530 subjects | German | Replicated associations of FCER1A, RAD50, STAT6 with IgE; RAD50 variants associated with AD and asthma |
|
| Asthma | Himes et al. 2009 (211) |
PDE4D | 422 cases/1,533 controls from ‘Illumina iControlDB’ |
European American |
African American, European American, U.S. Hispanic, British |
Replicated association of PDE4D with asthma in European Americans |
| Eosinophil count | Gudbjartsson et al. 2009 (68) |
IL1RL1*, IKZF2*, GATA2*, IL5*, SH2B3*, MYB, WDR36, IL33, GRFA2 |
9,393 subjects | Icelander | European, European American, Asian |
Replicated associations of IL1RL1, IL33, MYB, WDR36 with asthma in all replication samples |
| Atopic dermatitis (AD) |
Esparza-Gordilla et al. 2009 (215) |
FLG, C11orf30 | 939 cases/975 controls; 270 nuclear families |
German | German | Replicated association of C11orf30 with AD |
| Lung function (FEV1/FVC ratio) |
Wilk et al. 2009 (218) |
4q31* (near HHIP) |
7,691 subjects | European Americans |
European Americans |
Replicated association of SNP near HHIP with FEV1/FVC |
| Asthma | Hancock et al. 2010 (210) |
TLE4 | 492 asthma case- parent trios |
Mexican | Mexican | Replicated association of TLE4 with asthma |
| Eosinophilic esophagitis (EoE) |
Rothenberg et al. 2010 (216) |
TSLP/WDR36, 13q31.1* |
181 cases/1,974 controls |
European American |
European American |
Replicated association of TSLP/WDR36 with EoE |
| Asthma | Mathias et al. 2010 (213) |
ADRA1B, PRNP, DPP10, GNA13 |
498 cases/500 controls; 1,028 subjects in 163 families |
African American, African Caribbean |
African American, European |
Replicated association of DPP10 with asthma |
| Asthma | Sleiman et al. 2010 (214) |
DENND1B*, ORMDL3* |
793 cases/1,988 controls |
European American |
African American. European American, European |
Replicated association of DENND1B with asthma in European Americans and with opposite allele in African Americans |
| Asthma | Li et al. 2010 (212) |
RAD50, IL13, HLA-DR-DQ, LRPB1, SNX10, CA10, KCNJ2 |
473 cases/1,892 controls from ’Illumina iControlDB’ |
European | No replication sample included |
|
| Atopy | Wan et al. 2011 (217) |
FNDC31* | 1,083 cases/2,770 controls |
European | European | Association with FNDC31 not replicated |
| B. Meta-analyses of asthma and lung function GWAS | ||||||
|---|---|---|---|---|---|---|
| Lung function (FEV1, FEV1/FVC) |
SpiroMeta Consortium Repapi et al. 2010 (69) |
GSTCD*, TNS1*, 4q31* (near HHIP), HTR4*, AGER*, THSD4* |
20,288 subjects | European | European | Replicated associations of GSTCD, TNS1, and HTR4 with FEV1 and AGER and THSD4 with FEV1/FVC |
| Lung function (FEV1, FEV1/FVC) |
CHARGE Consortium Hancock et al. 2010 (70) |
HHIP*, GRP126*, ADAM19*, AGER/PPT2*, FAM13A*, PID1*, HTR4*, INTS12/ GSTCD/NPNT* |
20,890 subjects | European, European American |
in silico replication in SpiroMeta Consortium |
Replicated associations of HHIP, GPR126, ADAM19, AGER-PPT2, and HTR4 with FEV1 and INTS12-GSTCD-NPNT with FEV1/FVC |
| Asthma, total serum IgE |
GABRIEL Consortium Moffatt et al. 2010 (171) |
IL18R1*, HLA- DRB1*, HLA- DQ*, IL33*, ORMDL3/ GSDML*, SMAD3*, IL2RB*, SLCA22A5, IL13, RORA |
10,365 cases/ 16,110 controls |
European | No replication sample included |
|
| Asthma |
EVE Consortium Torgerson et al. 2011 (172) |
ORMDL3/ GSDML*, IL1RL1*, TSLP*, RTP2*, IL33, PYHIN1 |
3,601 cases/3,853 controls/1,702 case-parent trios |
European American, African American/ African Caribbean, Latino |
European American, African American, Latino |
Replicated associations of ORMDL3/GSDML, IL1RL1, TSLP, IL33 in all race/ethnic groups; replicated association of PYHIN1 in African Americans |
Asterisk denotes genes with associations that met criteria for genome-wide significance in each study.
Meta-analyses of GWAS
To maximize the power to detect risk alleles with modest effect sizes, the results of individual GWAS can be combined in a meta-analysis. Two meta-analyses of asthma GWAS have recently been completed, one by the GABRIEL Consortium (171) and one by the EVE Consortium (172) of European and U.S. investigators, respectively. While the GABRIEL meta-analysis included only subjects of European ancestry, the EVE meta-analysis included racially and ethnically diverse subjects from the U.S. and Mexico. The combined results of these two large meta-analyses yielded a set of highly replicated associations, some of which are evident in ethnically diverse subjects and some that may be ethnic- or race-specific (Table 5). These two meta-analyses are discussed in detail below. Two meta-analyses for lung function (FEV1 and FEV1/FVC ratio) have been reported by the SpiroMeta (69) and CHARGE (70) Consortiums. The results of those studies are summarized in Table 4B but are not discussed in more detail here because, to date, none of the SNPs or genes associated with lung function are also associated with asthma or allergic disease, although some have also been associated with COPD (220).
Table 5.
Combined results of EVE (172) and GABRIEL (171) meta-analyses of asthma GWAS in European American, African American/African Caribbean, and Latino samples (EVE) and in European samples (GABRIEL).
| Gene/Region | EVE | GABRIEL | Race/Ethnic Groups |
|---|---|---|---|
| 17q21 (ORMDL3/GSDML) | 1.2×10−14 | 6.4×10−23* | All |
| IL1RL1/IL18R1 (chr. 2) | 1.4×10−8 | 3.4×10−9 | All |
| TSLP (chr. 5) | 7.3×10−10 | 7.52−8 | All |
| IL33 (chr. 9) | 2.5×10−7 | 9.2×10−10 | All |
| SMAD3 (chr. 15) | 1.9×10−4 | 3.9×10−9 | Europeans, European Americans |
| RORA (chr. 15) | 2.1×10−3 | 1.1×10−7 | Europeans, European Americans |
| HLA-DQ (chr. 6) | 0.014 | 7.0×10−14 | All |
| PYHIN1 (chr. 1) | 3.6×10−7 | -- | African Americans/African Caribbeans |
| IL2RB (chr. 22) | ns | 1.1×10−8 | Europeans |
| SLC22A5 (chr. 5) | ns | 2.2×10−7 | Europeans |
| IL13 (chr. 5) | ns | 1.4×10−7 | Europeans |
The smallest p-values are shown for SNPs in the most significant genes in either study.
p-value for association with childhood onset asthma
The GABRIEL Consortium meta-analysis of European asthma GWAS
The GABRIEL Consortium (A Multidisciplinary Study to Identify the Genetic and Environmental Causes of Asthma in the European Community) reported the first meta-analysis of asthma GWAS in 10,365 cases and 16,110 controls recruited from 23 studies, and genotyped for 582,892 SNPs (171). This sample included the subjects in the first GWAS that discovered ORMDL3/GSDML as a novel asthma susceptibility locus (197). In the meta-analysis, SNPs in or near the following six genes reached genome-wide levels of significance (p ≤ 7.2 × 10−8): HLA-DQ, ORMDL3/GSDML, IL33, IL18R1, IL2RB, and SMAD3; SNPs in or near three additional genes had p-values ≤ 5.0 × 10−7: SLCA22A5, IL13, and RORA (Table 5). Associations were generally more significant in childhood onset asthma. This was particularly true for the associations with SNPs at the 17q21 ORMDL3/GSDML locus, for which there was significant heterogeneity in the ORs between childhood onset and adult onset asthma (smallest p-value for childhood onset asthma = 6.0 × 10−23, OR 0.76; smallest p-value for adult onset asthma = 0.49). In contrast, the HLA-DQ association was more significant in cases with adult onset asthma, although the heterogeneity in the ORs between adult cases (p = 4.0 × 10−8, OR 1.26) and child onset cases (p = 2 × 10−5, OR 1.14) was not significant. There were no significant associations observed for cases with severe asthma or occupational asthma; or for interactions between any of the associated SNPs.
A second GWAS of total serum IgE was conducted in 7,087 cases and 7,667 controls. Significant associations were reported with SNPs in the class II region of the MHC (p = 8.0 × 10−15), which was independent of the asthma association with HLA-DQ SNPs. Previously reported associations were replicated with SNPs in or near the FCER1A, IL13, STAT6, and IL4R/IL21R, although none reached genome-wide levels of significance in the meta-analysis. Surprisingly, there was no overlap between the most significant SNPs associated with asthma and the most significant SNPs associated with total serum IgE, suggesting that asthma and IgE-mediated pathways may be genetically distinct.
The investigators used the seven SNPs associated with childhood asthma to classify individuals with a predicted probability of disease ≥0.5. In this analysis, the sensitivity for classifying subjects as having or not having asthma was only 35%, with specificity of 75%. Thus, although these variants show very significant associations with asthma they are overall poor predictors of disease status, reflecting both their small effect size and common allele frequencies in the population.
The EVE Consortium meta-analysis of asthma GWAS in ethnically diverse populations
The EVE Consortium is a collaboration between U.S. investigators to identify asthma genes in ethnically diverse populations. Nine groups of investigators contributed asthma GWAS results for European Americans (1,486 cases, 1,539 controls, 620 case-parent trios), African American/African Caribbeans (1,154 cases, 1,054 controls, 413 case-parent trios), and Latinos (606 cases, 792 controls, 1,082 case-parent trios), yielding a combined sample of 3,246 cases, 3,385 controls, and 2,115 case-parent trios (172). Meta-analyses were performed within each racial/ethnic group and in the combined sample of racially and ethnically diverse subjects. SNPs with the most significant associations within each group or in the combined sample were genotyped in a replication sample of 7,202 cases, 6,426 controls, and 518 case-parent trios, comprised of European Americans (3,019 cases, 2,511 controls, 69 case-parent trios), African Americans (2,639 cases, 2,292 controls, 11 case-parent trios), and Latinos (1,544 cases, 1,623 controls, 438 case-parent trios). Each sample was genotyped with either an Illumina or Affymetrix array so a common set of approximately 2 million HapMap SNPs were either directly genotyped or imputed in each sample. In these analyses, the threshold for genome-wide significance was p < 2 × 10−8.
The EVE meta-analysis revealed SNPs in or near four loci that met genome-wide criteria for significance: ORMDL3/GSDML on 17q21, IL1RL1 on 2q12, and TSLP on 5q22.1 in the combined sample, and RTP2 on 3q27.3 in the Latino sample. SNPs in 11 other regions had p-values <10−6, including two potentially ethnic-specific associations in African Americans/African Caribbeans, one in European Americans, and one in Latinos. The most significant SNP in each of these 15 regions was genotyped in the replication samples; two SNPs failed genotyping, one with a potentially European American-specific association and one with potential association in the combined sample. Among the remaining 13 SNPs, those in or near four genes replicated associations with asthma: IL1RL1 (combined meta+replication p = 1.5 × 10−15), TSLP (combined meta+replication p = 1.0 × 10−14), IL33 (combined meta+replication p = 1.7 × 10−12), and ORMDL3/GSDML (combined meta+replication p = 2.2 × 10−16). One additional African American/African Caribbean-specific association replicated in the African American samples: PYHIN1 on 1q23.1 (combined meta+replication p = 3.9 × 10−9).
Interestingly, among the four pan-ethnic associations, none reached genome-wide levels of significance within any one group. Although SNPs in the ORMDL3/GSDML region achieved p-values <10−6 in the European American and Latino samples (smallest p-value in the African American/African Caribbean sample at this locus was 0.0012), none of the other three replicated regions had SNPs with p < 10−5 in any one racial/ethnic group despite sample sizes of 2-3,000 subjects per group. This reflects, at least in part, the modest effects of these common variants on disease risk. In fact, the odds ratios associated with the risk alleles at the four replicated pan-ethnic asthma susceptibility loci ranged from 1.10 (IL1RL1 in the African Americans/African Caribbeans) to 1.31 (ORMDL3/GSDML in the African Americans/African Caribbeans).
The association with SNPs in PHYHIN1 in subjects of African ancestry is quite interesting. The associated SNPs (rs1101999 and rs1102000) are in perfect LD with each other in populations of African descent, in whom minor allele frequencies are approximately 0.35. However, the associated (minor) alleles at these SNPs are absent in European populations. It is possible, therefore, that these associations are truly African-specific due to a common variant that is present primarily in populations of African descent or as a result of admixture (the alleles are present in Latino populations at about 0.02 frequency). Alternatively, there may be other variation in this gene in Europeans that is associated with asthma risk, but not well interrogated on the current genotyping platforms. Future re-sequencing studies of this gene in ethnically diverse cohorts will allow us to address this question and to determine whether PYHIN1 is a race-specific asthma susceptibility locus.
Summary of meta-analyses of asthma GWAS
Two large meta-analyses of asthma GWAS in European and diverse U.S. populations produced remarkably similar results (Table 5). SNPs in or near seven loci were associated with asthma in both studies, and SNPs in or near four of these loci had p-values at or near genome-wide levels of significance in both studies, with contributions from ethnically diverse samples: the 17q21 locus, the IL1RL1/IL18R1 locus, TSLP, and IL33. Thus, these four loci can be considered robustly associated asthma susceptibility genes. Moreover, three of these associations highlight the importance of epithelial cell-derived cytokines (TSLP and IL-33) that promote differentiation and activation of T helper (Th) 2 cells and their receptors (IL1RL1 encodes ST2, the receptor for IL-33 on mast cells, Th2 cells, T regulatory cells, and macrophages) (221-225). Notably, associations between allergic diseases and variants in or near these three genes had been previously reported. In a GWAS of eosinophil count, SNPs in IL1RL1 and IL33 were associated first with eosinophil count and then with asthma (68) and SNPs near TSLP were among the most significant in a GWAS of eosinophilic esophagitis (a food allergy-related disorder) (216). Candidate gene studies of TSLP (226-229) and IL1RL1 (202, 230-232) have further implicated these loci in asthma and atopy susceptibility. The combined data from candidate gene studies and GWAS suggest that variation in these three genes influence asthma risk and atopy phenotypes through shared pathways. As discussed above, the SNPs at the 17q21 asthma locus do not show associations with total serum IgE, positive SPT, or allergic diseases. Although the function of 17q21 genes and their role in asthma pathogenesis is currently unknown, one or more of these genes influence asthma risk through non-atopic, and perhaps novel, pathways.
Notably, the asthma-associated SNPs detected in the GABRIEL and EVE meta-analysis have modest effects on risk. The seven most associated SNPs in the GABRIEL meta-analysis (Table 5) could accurately classify individuals with asthma with only 35% sensitivity (with a predictive probability of 0.5, i.e., assuming the model is correct at least 50% of the time) (171). This is also reflected in the small ORs reported in the EVE meta-analysis for the most associated SNPs (172). It is possible that additional asthma-associated risk alleles with main effects will be revealed in future, even larger meta-meta-analyses, but it is unlikely that those as yet undiscovered asthma risk variants will significantly increase the sensitivity of predicting disease status based on genotypes or explain significantly more of the genetic risk for asthma. Importantly, however, these studies have and will continue to highlight the key pathways in asthma disease onset and progression and to identify potential therapeutic targets, which could lead to more effective treatment of asthma. Nonetheless, the GWAS (and meta-analyses of GWAS) experience to date suggests that common variants with significant main effects on asthma risk have only modest effect sizes and account for only a very small proportion of the genetic risk, or heritability, of this common disease. Thus, it is timely to ask questions about the nature of the remaining genetic contributions to asthma risk.
‘Missing heritability’ in asthma and allergic diseases
Despite the successes of GWAS for discovering common risk alleles for many complex diseases and quantitative phenotypes, only a small proportion of the heritability are accounted for by these variants, as is now apparent for asthma. This has been termed the ‘dark matter’ or ‘missing heritability’, which has been discussed extensively in the literature (178, 179, 233-244). Because the genetic architecture of asthma seems similar to other common diseases in this regard, we will review here the potential explanations for these observations, which we will consider in three categories. First, the genotyping platforms typically used for GWAS do not interrogate all variation. Missing from the typical GWAS are rare variants and structural variants, such as copy number variation (CNVs) and indels, which are not tagged or covered by SNPs on the SNP genotyping arrays. The former will only be detected by direct sequencing, and a number of ongoing studies of asthma as well as many other common diseases should shed light on the importance of rare variants in the genetic architecture of these diseases, as discussed above. Structural variation is quite common in the human genome and because these polymorphisms can result in complete deletions or duplications of genes they are a potentially important source of variation to consider in genetic studies of common diseases (245-248). Currently, however, studies of asthma (249, 250) have been limited by the fact that structural variants are poorly represented on the SNP arrays (251), so it is still unknown as to whether structural variants contribute significantly to the genetic architecture of asthma and allergic diseases. Alternative approaches for comprehensively surveying CNVs and indels are becoming more available and affordable so we should also know the answer to this question in the near future (252-254).
The second source of the ‘missing heritability’ is genetic risk due to interaction effects. Current GWAS approaches test for associations between each SNP and the disease or phenotype under investigation. As a result, GWAS will be most powerful for detecting SNPs with main effects on disease risk or phenotypic distributions. Testing for gene-gene interactions (i.e. epistasis) in a GWAS is severely hampered by the enormous multiple comparison burden incurred when testing all possible pairs of genes (i.e. for a GWAS using 500,000 SNPs, there are potentially 500,000 × 500,000 or 250 billion interactions to test). Although there are considerable efforts ongoing to develop methods for studying gene-gene interactions using systems biology and network approaches, this area of investigation is still in its infancy and has not yet shed light on whether a significant proportion of the heritability of asthma is due to gene-gene interactions. In contrast, we already know that gene-environment interactions are important determinants of risk for asthma and allergic disease, as recently reviewed (219). Although genome-wide gene-environment interaction studies (GWIS) do not carry the same statistical burden as genome-wide gene-gene interaction studies, they still require very large samples (255). Moreover, very large datasets used for GWAS do not typically have uniformly measured environmental exposure data. Nonetheless, based on the many well-replicated gene-environment interaction effects on risk for asthma and allergic disease (219, 256-258), it is likely that a significant proportion of the missing heritability for these phenotypes will be accounted for by gene-environment interactions.
It is possible that estimates of heritability for many complex phenotypes are inflated. This is particularly true for estimates based on twin studies because monozygotic twins not only share more of their genes [100%, including the sharing of de novo mutations (235)] compared to dizygotic twins (~50%), but they also share a more ‘identical’ environment than do dizygotic twins. In addition, the heritability of any particular trait may vary significantly in different environments (243) and for different phenotypic subgroups (242), all of which are pooled in the large samples required for GWAS studies. As a result, the heritability for the trait under investigation may in fact be much smaller in the samples included in GWAS due to environmental and phenotypic heterogeneity.
Other considerations
In this review, we chose to focus on the current status of genetic linkage and association studies and the light that those studies have shed on the genetic architecture of asthma and allergic phenotypes. We believe that the field is at a critical juncture with respect to moving beyond GWAS to unravel the molecular, cellular, and physiological effects of variation at associated SNPs and characterizing the roles of associated genes in pathogenesis, as proposed by a recent workshop sponsored by the National Heart Lung and Blood Institute (NHLBI) of the National Institutes of Health (NIH) (259). We think that such studies are still in their infancy and will, therefore, be the subjects of future reviews on asthma and allergy genetics. We include under this umbrella characterizing the importance of epigenetic modifications on gene expression during development, as a genome modifier in response to environmental exposures, and as a potential mechanism for gene-environment interactions (260-265). From a genetic perspective, it will be particularly important to determine whether any of the risk-association variation discovered through genetic association (or re-sequencing) studies alters epigenetic modifications as its mechanism of affecting disease risk.
We have also neglected the important field of pharmacogenetics (266), although a number of pharmacogenetic examples in asthma can be cited, including the seminal work on genotype at the ALOX5 locus and response to leukotriene modifiers (267-271) and at the ADRB2 locus and response to inhaled beta-agonists (272-275). We expect many more examples of pharmacogenetic interactions being reported as GWAS for drug response are completed during the next year. Such studies may open the door for personalized medicine in the care of asthma patients, and will certainly be the focus of future reviews.
Asthma and allergic disease: same disease or shared pathways?
In the introduction of this review, we posed this question. Here we consider whether genetic studies have shed light on the answer. As mentioned above, because the asthma genetics field was so dominated by candidate gene studies in which ‘candidates’ were selected on their known function, most genes selected for study were based on their roles in the adaptive or innate immune responses in general, or in Th2-mediated responses in particular. Thus, the paradigm of asthma as an ‘allergic’ disease predominated in genetic circles. Early linkage studies in ‘candidate’ regions furthered this notion, by reporting linkages to the 11q region harboring the gene encoding the high affinity Fcβ receptor, FCERB1 (276), and to the 5q region harboring genes encoding Th2 cytokines, including IL4 and IL13 (71, 277).
The first key piece of evidence to suggest that genes in non-allergic, non-immune pathways may have important roles in asthma pathogenesis was the report of ADAM33 as the first positionally cloned asthma/BHR gene (192) (Table 3); associations with variation in this gene have since replicated in many independent studies (reviewed in 161). This gene is expressed in airway smooth muscle cells and is likely involved in smooth muscle and vascular modeling and remodeling (278, 279), although associations with allergic phenotypes have also been reported (161). More recently a large multi-center study of SNPs in 14 candidate genes for wheeze and allergy revealed that very few candidates showed evidence of an association with both asthma symptoms and elevated IgE levels (280), further suggesting that asthma and atopy are not different manifestations of the same disease. The results of the meta-analyses of asthma GWAS further highlighted the importance of non-immune/non-allergy genes as evidenced by the strong and consistent association of SNPs at the 17q21 locus with childhood onset asthma but not with atopy. Recent functional studies of ORMDL3 have suggested that it might play a role in epithelial cell remodeling (207, 208). Because typically approximately 80% of children with asthma are also atopic or have allergic disease, very large samples are required to delineate associations that are specific to asthma or to atopy. Thus, the demonstration that associations with variation at the 17q21 locus is specific to asthma and not to IgE levels in the GABRIEL Consortium meta-analysis is compelling evidence that this important locus is a primary asthma-susceptibility locus that acts independently of atopic, or IgE-mediated, pathways.
In contrast, the remaining asthma genes that emerged from the meta-analyses of asthma GWAS have also been associated with atopic phenotypes and allergic diseases. As discussed above, variations in the TSLP, IL33, and IL1RL1 genes have shown associations with many atopic phenotypes, including eosinophil count, total serum IgE, allergic disease (eosinophilic esophagitis), and atopy. Thus, these data indicate that asthma and allergic phenotypes may result from genetic alterations in a shared pathway of immunoregulation and promotion of Th2 cytokine production by epithelial cell cytokines. It is tempting to speculate that dysregulation of these pathways predisposes individuals toward the subsequent development of a potentially broad spectrum of diseases. Aberrations in other pathways may confer disease specificity. Thus, common alleles at the 17q21 locus and/or ADAM33 may be specific for the development of asthma, whereas relatively rare variants in the filaggrin gene (FLG) may be specific for atopic dermatitis (281). We predict that additional genes with specificity for upper airway diseases and food allergies, for example, are yet to be identified.
Concluding remarks and future directions
This is an exciting time in the field of asthma and allergy genetics, as a list of well validated susceptibility genes are emerging and defining important biological pathways. As additional GWAS and meta-analyses for asthma and allergic phenotypes are completed, this gene list will expand and our understanding of the common and unique pathways that are dysregulated in individuals with disease will broaden. The great challenges that we now face are to elucidate the effects of associated variation on gene regulation or function and of associated genes on asthma pathogenesis.
Acknowledgements
This work was supported by NIH grants HL101651, HL101543, HL70831, HL85197 to C.O.; T.C.Y. is supported by the National Science Council of Taiwan grant NSC 98-2314-B-182A-008-MY3 and a Chang Gung Memorial Hospital grant CMRPG260293.
Footnotes
The authors have no disclosures or conflicts of interest.
References
- 1.Edfors-Lubs M-L. Allergy in 7000 twin pairs. Acta Allergol. 1971;26:249–285. doi: 10.1111/j.1398-9995.1971.tb01300.x. [DOI] [PubMed] [Google Scholar]
- 2.Duffy DL, Martin NG, Battistutta D. Genetics of asthma and hay fever in Australian twins. Am Rev Respir Dis. 1990;142:1351–1358. doi: 10.1164/ajrccm/142.6_Pt_1.1351. [DOI] [PubMed] [Google Scholar]
- 3.Nieminen MM, Kaprio J, Koskenvuo M. A population-based study of bronchial asthma in adult twin pairs. Chest. 1991;100:70–75. doi: 10.1378/chest.100.1.70. [DOI] [PubMed] [Google Scholar]
- 4.Harris JR, Magnus P, Samuelsen SO, Tambs K. No evidence for effects of family environment on asthma. A retrospective study of Norwegian twins. Am J Respir Crit Care Med. 1997;156:43–49. doi: 10.1164/ajrccm.156.1.9609094. [DOI] [PubMed] [Google Scholar]
- 5.Laitinen T, Rasanen M, Kaprio J, Koskenvuo M, Laitinen LA. Importance of genetic factors in adolescent asthma: a population-based twin-family study. Am J Respir Crit Care Med. 1998;157:1073–1078. doi: 10.1164/ajrccm.157.4.9704041. [DOI] [PubMed] [Google Scholar]
- 6.Skadhauge LR, Christensen K, Kyvik KO, Sigsgaard T. Genetic and environmental influence on asthma: a population-based study of 11,688 Danish twin pairs. Eur Respir J. 1999;13:8–14. doi: 10.1183/09031936.99.13100899. [DOI] [PubMed] [Google Scholar]
- 7.Koeppen-Schomerus G, Stevenson J, Plomin R. Genes and environment in asthma: a study of 4 year old twins. Arch Dis Child. 2001;85:398–400. doi: 10.1136/adc.85.5.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hallstrand TS, Fischer ME, Wurfel MM, Afari N, Buchwald D, Goldberg J. Genetic pleiotropy between asthma and obesity in a community-based sample of twins. J Allergy Clin Immunol. 2005;116:1235–1241. doi: 10.1016/j.jaci.2005.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilkinson J, Grimley S, Collins A, Thomas NS, Holgate ST, Morton N. Linkage of asthma to markers on chromosome 12 in a sample of 240 families using quantitative phenotype scores. Genomics. 1998;53:251–259. doi: 10.1006/geno.1998.5485. [DOI] [PubMed] [Google Scholar]
- 10.Thomsen SF, van der Sluis S, Kyvik KO, Skytthe A, Backer V. Estimates of asthma heritability in a large twin sample. Clin Exp Allergy. 2010;40:1054–1061. doi: 10.1111/j.1365-2222.2010.03525.x. [DOI] [PubMed] [Google Scholar]
- 11.Willemsen G, van Beijsterveldt TC, van Baal CG, Postma D, Boomsma DI. Heritability of self-reported asthma and allergy: a study in adult Dutch twins, siblings and parents. Twin Res Hum Genet. 2008;11:132–142. doi: 10.1375/twin.11.2.132. [DOI] [PubMed] [Google Scholar]
- 12.Lichtenstein P, Svartengren M. Genes, environments, and sex: factors of importance in atopic diseases in 7-9-year-old Swedish twins. Allergy. 1997;52:1079–1086. doi: 10.1111/j.1398-9995.1997.tb00179.x. [DOI] [PubMed] [Google Scholar]
- 13.Nystad W, Roysamb E, Magnus P, Tambs K, Harris JR. A comparison of genetic and environmental variance structures for asthma, hay fever and eczema with symptoms of the same diseases: a study of Norwegian twins. Int J Epidemiol. 2005;34:1302–1309. doi: 10.1093/ije/dyi061. [DOI] [PubMed] [Google Scholar]
- 14.van Beijsterveldt CE, Boomsma DI. Genetics of parentally reported asthma, eczema and rhinitis in 5-yr-old twins. Eur Respir J. 2007;29:516–521. doi: 10.1183/09031936.00065706. [DOI] [PubMed] [Google Scholar]
- 15.Fagnani C, et al. Heritability and shared genetic effects of asthma and hay fever: an Italian study of young twins. Twin Res Hum Genet. 2008;11:121–131. doi: 10.1375/twin.11.2.121. [DOI] [PubMed] [Google Scholar]
- 16.Rasanen M, Laitinen T, Kaprio J, Koskenvuo M, Laitinen LA. Hay fever--a Finnish nationwide study of adolescent twins and their parents. Allergy. 1998;53:885–890. doi: 10.1111/j.1398-9995.1998.tb03996.x. [DOI] [PubMed] [Google Scholar]
- 17.Bazaral M, Orgel HA, Hamburger RN. IgE levels in normal infants and mothers and an inheritance hypothesis. J Immunol. 1971;107:794–801. [PubMed] [Google Scholar]
- 18.Bazaral M, Orgel HA, Hamburger RN. Genetics of IgE and allergy: Serum IgE levels in twins. Journal of Allergy and Clinical Immunology. 1974;54:288–304. [Google Scholar]
- 19.Grundbacher FJ. Causes of variation in serum IgE levels in normal populations. J Allergy Clin Immunol. 1975;56:104–111. doi: 10.1016/0091-6749(75)90114-1. [DOI] [PubMed] [Google Scholar]
- 20.Hasstedt SJ, Meyers DA, Marsh DG. Inheritance of immunoglobulin E: genetic model fitting. Am J Med Genet. 1983;14:61–66. doi: 10.1002/ajmg.1320140110. [DOI] [PubMed] [Google Scholar]
- 21.Meyers DA, Beaty TH, Freidhoff LR, Marsh DG. Inheritance of total serum IgE (basal levels) in man. Am J Hum Genet. 1987;41:51–62. [PMC free article] [PubMed] [Google Scholar]
- 22.Hanson B, McGue M, Roitman-Johnson B, Segal NL, Bouchard TJ, Blumenthal MN. Atopic disease and immunoglobulin e in twins reared apart and together. Am J Hum Genet. 1991;48:873–879. [PMC free article] [PubMed] [Google Scholar]
- 23.Blumenthal MN, Bonini S. Immunogenetics of specific immune responses to allergens in twins and families. In: Marsh DG, Blumenthal MN, editors. Genetic and environmental factors in clinical allergy. University of Minnesota; Minneapolis: 1990. pp. 132–142. [Google Scholar]
- 24.Blumenthal MN. Genetics of asthma and allergy. Allergy Asthma Proc. 2000;21:55–59. doi: 10.2500/108854100778249042. [DOI] [PubMed] [Google Scholar]
- 25.Abney M, McPeek MS, Ober C. Heritabilities of quantitative traits in a founder population. Am J Hum Genet. 2001;68:1302–1307. doi: 10.1086/320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ober C, Abney M, McPeek MS. The genetic dissection of complex traits in a founder population. Am J Hum Genet. 2001;69:1068–1079. doi: 10.1086/324025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strachan DP, Wong HJ, Spector TD. Concordance and interrelationship of atopic diseases and markers of allergic sensitization among adult female twins. J Allergy Clin Immunol. 2001;108:901–907. doi: 10.1067/mai.2001.119408. [DOI] [PubMed] [Google Scholar]
- 28.Jacobsen HP, Herskind AM, Nielsen BW, Husby S. IgE in unselected like-sexed monozygotic and dizygotic twins at birth and at 6 to 9 years of age: high but dissimilar genetic influence on IgE levels. J Allergy Clin Immunol. 2001;107:659–663. doi: 10.1067/mai.2001.113565. [DOI] [PubMed] [Google Scholar]
- 29.Mathias RA, et al. A genome-wide linkage analyses of total serum IgE using variance components analysis in asthmatic families. Genet Epidemiol. 2001;20:340–355. doi: 10.1002/gepi.5. [DOI] [PubMed] [Google Scholar]
- 30.Liebeler CL, Basu S, Jackola DR. Allergen-specific IgG1 provides parsimonious heritability estimates for atopy-associated immune responses to allergens. Hum Immunol. 2007;68:113–121. doi: 10.1016/j.humimm.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grant AV, et al. High heritability but uncertain mode of inheritance for total serum IgE level and Schistosoma mansoni infection intensity in a schistosomiasis-endemic Brazilian population. J Infect Dis. 2008;198:1227–1236. doi: 10.1086/591946. [DOI] [PubMed] [Google Scholar]
- 32.Hopp RJ, Bewtra AK, Watt GD, Nair NM, Townley RG. Genetic analysis of allergic disease in twins. J Allergy Clin Immunol. 1984;73:265–270. doi: 10.1016/s0091-6749(84)80018-4. [DOI] [PubMed] [Google Scholar]
- 33.Manolio TA, Barnes KC, Beaty TH, Levett PN, Naidu RP, Wilson AF. Sex differences in heritability of sensitization to Blomia tropicalis in asthma using regression of offspring on midparent (ROMP) methods. Hum Genet. 2003;113:437–446. doi: 10.1007/s00439-003-1005-6. [DOI] [PubMed] [Google Scholar]
- 34.Tsai HJ, et al. Familial aggregation of food allergy and sensitization to food allergens: a family-based study. Clin Exp Allergy. 2009;39:101–109. doi: 10.1111/j.1365-2222.2008.03111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palmer LJ, Burton PR, Faux JA, James AL, Musk AW, Cookson WO. Independent inheritance of serum immunoglobulin E concentrations and airway responsiveness. Am J Respir Crit Care Med. 2000;161:1836–1843. doi: 10.1164/ajrccm.161.6.9805104. [DOI] [PubMed] [Google Scholar]
- 36.Palmer LJ, Burton PR, James AL, Musk AW, Cookson WO. Familial aggregation and heritability of asthma-associated quantitative traits in a population-based sample of nuclear families. Eur J Hum Genet. 2000;8:853–860. doi: 10.1038/sj.ejhg.5200551. [DOI] [PubMed] [Google Scholar]
- 37.Koppelman GH, et al. Genome-wide search for atopy susceptibility genes in Dutch families with asthma. J Allergy Clin Immunol. 2002;109:498–506. doi: 10.1067/mai.2002.122235. [DOI] [PubMed] [Google Scholar]
- 38.Liu X, et al. Genetic and environmental contributions to allergen sensitization in a Chinese twin study. Clin Exp Allergy. 2009;39:991–998. doi: 10.1111/j.1365-2222.2009.03228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dal Colletto GM, Fulker DW, Barretto OC, Kolya M. Genetic and environmental effects on blood cells. Acta Genet Med Gemellol (Roma) 1993;42:245–252. doi: 10.1017/s000156600000324x. [DOI] [PubMed] [Google Scholar]
- 40.Jarvis D, Burney P. ABC of allergies. The epidemiology of allergic disease. BMJ. 1998;316:607–610. doi: 10.1136/bmj.316.7131.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Global Initiative for Asthma (GINA) [(updated 2009). [accessed 2011 March 1]];Global stratetgy for asthma management and prevention. Available from: www.ginasthma.org.
- 42.Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59:469–478. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- 43.Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee. Lancet. 1998;351:1225–1232. [PubMed] [Google Scholar]
- 44.Lai CK, Beasley R, Crane J, Foliaki S, Shah J, Weiland S. Global variation in the prevalence and severity of asthma symptoms: phase three of the International Study of Asthma and Allergies in Childhood (ISAAC) Thorax. 2009;64:476–483. doi: 10.1136/thx.2008.106609. [DOI] [PubMed] [Google Scholar]
- 45.Variations in the prevalence of respiratory symptoms, self-reported asthma attacks, and use of asthma medication in the European Community Respiratory Health Survey (ECRHS) Eur Respir J. 1996;9:687–695. doi: 10.1183/09031936.96.09040687. [DOI] [PubMed] [Google Scholar]
- 46.Brozek JL, et al. Allergic Rhinitis and its Impact on Asthma (ARIA) guidelines: 2010 revision. J Allergy Clin Immunol. 2010;126:466–476. doi: 10.1016/j.jaci.2010.06.047. [DOI] [PubMed] [Google Scholar]
- 47.Bousquet J, et al. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008 update (in collaboration with the World Health Organization, GA(2)LEN and AllerGen) Allergy. 2008;63(Suppl 86):8–160. doi: 10.1111/j.1398-9995.2007.01620.x. [DOI] [PubMed] [Google Scholar]
- 48.Larsen FS, Hanifin JM. Epidemiology of atopic dermatitis. Immunology and Allergy Clinics of North America. 2002;22:1–24. [Google Scholar]
- 49.Leung DY, et al. Disease management of atopic dermatitis: an updated practice parameter. Joint Task Force on Practice Parameters. Ann Allergy Asthma Immunol. 2004;93:S1–21. doi: 10.1016/s1081-1206(10)61385-3. [DOI] [PubMed] [Google Scholar]
- 50.Williams H, et al. Worldwide variations in the prevalence of symptoms of atopic eczema in the International Study of Asthma and Allergies in Childhood. J Allergy Clin Immunol. 1999;103:125–138. doi: 10.1016/s0091-6749(99)70536-1. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Sampson HA. Food allergy. J Clin Invest. 2011;121:827–835. doi: 10.1172/JCI45434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arbes SJ, Jr., Gergen PJ, Elliott L, Zeldin DC. Prevalences of positive skin test responses to 10 common allergens in the US population: results from the third National Health and Nutrition Examination Survey. J Allergy Clin Immunol. 2005;116:377–383. doi: 10.1016/j.jaci.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 53.Burney P, Malmberg E, Chinn S, Jarvis D, Luczynska C, Lai E. The distribution of total and specific serum IgE in the European Community Respiratory Health Survey. J Allergy Clin Immunol. 1997;99:314–322. doi: 10.1016/s0091-6749(97)70048-4. [DOI] [PubMed] [Google Scholar]
- 54.Sunyer J, et al. Geographic variations in the effect of atopy on asthma in the European Community Respiratory Health Study. J Allergy Clin Immunol. 2004;114:1033–1039. doi: 10.1016/j.jaci.2004.05.072. [DOI] [PubMed] [Google Scholar]
- 55.Sears MR, Herbison GP, Holdaway MD, Hewitt CJ, Flannery EM, Silva PA. The relative risks of sensitivity to grass pollen, house dust mite and cat dander in the development of childhood asthma. Clin Exp Allergy. 1989;19:419–424. doi: 10.1111/j.1365-2222.1989.tb02408.x. [DOI] [PubMed] [Google Scholar]
- 56.Yao TC, Ou LS, Yeh KW, Lee WI, Chen LC, Huang JL. Associations of Age, Gender and BMI with Prevalence of Allergic Diseases in Children: PATCH Study. J Asthma. 2011 doi: 10.3109/02770903.2011.576743. in press. [DOI] [PubMed] [Google Scholar]
- 57. [accessed 2011 March 1];National Heart, Lung, and Blood Institute and the National Asthma Education ad Prevention Program. Expert Panel Report 3 (EPR3): Guidelines for the Diagnosis and Management of Asthma. Available from: http://www.nhlbi.nih.gov/guidelines/asthma/asthgdln.htm. [PubMed]
- 58.Cockcroft DW, Murdock KY, Berscheid BA, Gore BP. Sensitivity and specificity of histamine PC20 determination in a random selection of young college students. J Allergy Clin Immunol. 1992;89:23–30. doi: 10.1016/s0091-6749(05)80037-5. [DOI] [PubMed] [Google Scholar]
- 59.Boulet LP. Asymptomatic airway hyperresponsiveness: a curiosity or an opportunity to prevent asthma? Am J Respir Crit Care Med. 2003;167:371–378. doi: 10.1164/rccm.200111-084PP. [DOI] [PubMed] [Google Scholar]
- 60.Ramsdale EH, Morris MM, Roberts RS, Hargreave FE. Asymptomatic bronchial hyperresponsiveness in rhinitis. J Allergy Clin Immunol. 1985;75:573–577. doi: 10.1016/0091-6749(85)90032-6. [DOI] [PubMed] [Google Scholar]
- 61.Ramsdale EH, Morris MM, Roberts RS, Hargreave FE. Bronchial responsiveness to methacholine in chronic bronchitis: relationship to airflow obstruction and cold air responsiveness. Thorax. 1984;39:912–918. doi: 10.1136/thx.39.12.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Haren EH, Lammers JW, Festen J, Heijerman HG, Groot CA, van Herwaarden CL. The effects of the inhaled corticosteroid budesonide on lung function and bronchial hyperresponsiveness in adult patients with cystic fibrosis. Respir Med. 1995;89:209–214. doi: 10.1016/0954-6111(95)90249-x. [DOI] [PubMed] [Google Scholar]
- 63.Johansson SG, Lundahl J. Asthma, atopy, and IgE: what is the link? Curr Allergy Asthma Rep. 2001;1:89–90. doi: 10.1007/s11882-001-0071-x. [DOI] [PubMed] [Google Scholar]
- 64.Bousquet J, et al. Factors responsible for differences between asymptomatic subjects and patients presenting an IgE sensitization to allergens. A GA2LEN project. Allergy. 2006;61:671–680. doi: 10.1111/j.1398-9995.2006.01048.x. [DOI] [PubMed] [Google Scholar]
- 65.Gold MS, Kemp AS. Atopic disease in childhood. Med J Aust. 2005;182:298–304. doi: 10.5694/j.1326-5377.2005.tb06707.x. [DOI] [PubMed] [Google Scholar]
- 66.Ober C, et al. Effect of variation in CHI3L1 on serum YKL-40 level, asthma risk, and lung function. NEJM. 2008;358:1682–1691. doi: 10.1056/NEJMoa0708801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weidinger S, et al. Genome-wide scan on total serum IgE levels identifies FCER1A as novel susceptibility locus. PLoS Genet. 2008;4:e1000166. doi: 10.1371/journal.pgen.1000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gudbjartsson DF, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet. 2009;41:342–347. doi: 10.1038/ng.323. [DOI] [PubMed] [Google Scholar]
- 69.Repapi E, et al. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2010;42:36–44. doi: 10.1038/ng.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hancock DB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42:45–52. doi: 10.1038/ng.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marsh DG, et al. Linkage analysis of IL4 and other chromosome 5q31.1 markers and total serum immunoglobulin E concentrations. Science. 1994;264:1152–1156. doi: 10.1126/science.8178175. [DOI] [PubMed] [Google Scholar]
- 72.Postma DS, et al. Genetic susceptibility to asthma--bronchial hyperresponsiveness coinherited with a major gene for atopy. N Engl J Med. 1995;333:894–900. doi: 10.1056/NEJM199510053331402. [DOI] [PubMed] [Google Scholar]
- 73.Daniels SE, et al. A genome-wide search for quantitative trait loci underlying asthma. Nature. 1996;383:247–250. doi: 10.1038/383247a0. [DOI] [PubMed] [Google Scholar]
- 74.Spergel JM. From atopic dermatitis to asthma: the atopic march. Ann Allergy Asthma Immunol. 2010;105:99–106. doi: 10.1016/j.anai.2009.10.002. quiz 107-109, 117. [DOI] [PubMed] [Google Scholar]
- 75.Rhodes HL, Sporik R, Thomas P, Holgate ST, Cogswell JJ. Early life risk factors for adult asthma: a birth cohort study of subjects at risk. J Allergy Clin Immunol. 2001;108:720–725. doi: 10.1067/mai.2001.119151. [DOI] [PubMed] [Google Scholar]
- 76.Rhodes HL, Thomas P, Sporik R, Holgate ST, Cogswell JJ. A birth cohort study of subjects at risk of atopy: twenty-two-year follow-up of wheeze and atopic status. Am J Respir Crit Care Med. 2002;165:176–180. doi: 10.1164/ajrccm.165.2.2104032. [DOI] [PubMed] [Google Scholar]
- 77.Gustafsson D, Sjoberg O, Foucard T. Development of allergies and asthma in infants and young children with atopic dermatitis--a prospective follow-up to 7 years of age. Allergy. 2000;55:240–245. doi: 10.1034/j.1398-9995.2000.00391.x. [DOI] [PubMed] [Google Scholar]
- 78.Ohshima Y, et al. Early sensitization to house dust mite is a major risk factor for subsequent development of bronchial asthma in Japanese infants with atopic dermatitis: results of a 4-year followup study. Ann Allergy Asthma Immunol. 2002;89:265–270. doi: 10.1016/S1081-1206(10)61953-9. [DOI] [PubMed] [Google Scholar]
- 79.Kulig M, Bergmann R, Klettke U, Wahn V, Tacke U, Wahn U. Natural course of sensitization to food and inhalant allergens during the first 6 years of life. J Allergy Clin Immunol. 1999;103:1173–1179. doi: 10.1016/s0091-6749(99)70195-8. [DOI] [PubMed] [Google Scholar]
- 80.Lau S, et al. The development of childhood asthma: lessons from the German Multicentre Allergy Study (MAS) Paediatr Respir Rev. 2002;3:265–272. doi: 10.1016/s1526-0542(02)00189-6. [DOI] [PubMed] [Google Scholar]
- 81.Illi S, et al. The natural course of atopic dermatitis from birth to age 7 years and the association with asthma. J Allergy Clin Immunol. 2004;113:925–931. doi: 10.1016/j.jaci.2004.01.778. [DOI] [PubMed] [Google Scholar]
- 82.Corren J. Allergic rhinitis and asthma: how important is the link? J Allergy Clin Immunol. 1997;99:S781–786. doi: 10.1016/s0091-6749(97)70127-1. [DOI] [PubMed] [Google Scholar]
- 83.Pawankar R. Allergic rhinitis and asthma: are they manifestations of one syndrome? Clin Exp Allergy. 2006;36:1–4. doi: 10.1111/j.1365-2222.2006.02420.x. [DOI] [PubMed] [Google Scholar]
- 84.Settipane RJ, Hagy GW, Settipane GA. Long-term risk factors for developing asthma and allergic rhinitis: a 23-year follow-up study of college students. Allergy Proc. 1994;15:21–25. doi: 10.2500/108854194778816634. [DOI] [PubMed] [Google Scholar]
- 85.Wallace DV, et al. The diagnosis and management of rhinitis: an updated practice parameter. J Allergy Clin Immunol. 2008;122:S1–84. doi: 10.1016/j.jaci.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 86.Spector SL. Overview of comorbid associations of allergic rhinitis. J Allergy Clin Immunol. 1997;99:S773–780. doi: 10.1016/s0091-6749(97)70126-x. [DOI] [PubMed] [Google Scholar]
- 87.Rowe-Jones JM. The link between the nose and lung, perennial rhinitis and asthma--is it the same disease? Allergy. 1997;52:20–28. doi: 10.1111/j.1398-9995.1997.tb04818.x. [DOI] [PubMed] [Google Scholar]
- 88.Vignola AM, Chanez P, Godard P, Bousquet J. Relationships between rhinitis and asthma. Allergy. 1998;53:833–839. doi: 10.1111/j.1398-9995.1998.tb03988.x. [DOI] [PubMed] [Google Scholar]
- 89.Bachert C, Vignola AM, Gevaert P, Leynaert B, Van Cauwenberge P, Bousquet J. Allergic rhinitis, rhinosinusitis, and asthma: one airway disease. Immunol Allergy Clin North Am. 2004;24:19–43. doi: 10.1016/S0889-8561(03)00104-8. [DOI] [PubMed] [Google Scholar]
- 90.Barnes KC, Grant AV, Hansel NN, Gao P, Dunston GM. African Americans with asthma: genetic insights. Proc Am Thorac Soc. 2007;4:58–68. doi: 10.1513/pats.200607-146JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ober C, Loisel DA, Gilad Y. Sex-specific genetic architecture of human disease. Nat Rev Genet. 2008;9:911–922. doi: 10.1038/nrg2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gergen PJ, Fowler JA, Maurer KR, Davis WW, Overpeck MD. The burden of environmental tobacco smoke exposure on the respiratory health of children 2 months through 5 years of age in the United States: Third National Health and Nutrition Examination Survey, 1988 to 1994. Pediatrics. 1998;101:E8. doi: 10.1542/peds.101.2.e8. [DOI] [PubMed] [Google Scholar]
- 93.Gilliland FD, Li YF, Peters JM. Effects of maternal smoking during pregnancy and environmental tobacco smoke on asthma and wheezing in children. Am J Respir Crit Care Med. 2001;163:429–436. doi: 10.1164/ajrccm.163.2.2006009. [DOI] [PubMed] [Google Scholar]
- 94.Stein RT, et al. Influence of parental smoking on respiratory symptoms during the first decade of life: the Tucson Children’s Respiratory Study. Am J Epidemiol. 1999;149:1030–1037. doi: 10.1093/oxfordjournals.aje.a009748. [DOI] [PubMed] [Google Scholar]
- 95.Lewis S, Richards D, Bynner J, Butler N, Britton J. Prospective study of risk factors for early and persistent wheezing in childhood. Eur Respir J. 1995;8:349–356. doi: 10.1183/09031936.95.08030349. [DOI] [PubMed] [Google Scholar]
- 96.Tariq SM, Hakim EA, Matthews SM, Arshad SH. Influence of smoking on asthmatic symptoms and allergen sensitisation in early childhood. Postgrad Med J. 2000;76:694–699. doi: 10.1136/pmj.76.901.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Strachan DP, Butland BK, Anderson HR. Incidence and prognosis of asthma and wheezing illness from early childhood to age 33 in a national British cohort. BMJ. 1996;312:1195–1199. doi: 10.1136/bmj.312.7040.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gilliland FD, et al. Regular smoking and asthma incidence in adolescents. Am J Respir Crit Care Med. 2006;174:1094–1100. doi: 10.1164/rccm.200605-722OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Polosa R, et al. Cigarette smoking is associated with a greater risk of incident asthma in allergic rhinitis. J Allergy Clin Immunol. 2008;121:1428–1434. doi: 10.1016/j.jaci.2008.02.041. [DOI] [PubMed] [Google Scholar]
- 100.Douwes J, et al. Farm exposure in utero may protect against asthma, hay fever and eczema. Eur Respir J. 2008;32:603–611. doi: 10.1183/09031936.00033707. [DOI] [PubMed] [Google Scholar]
- 101.Von Ehrenstein OS, Von Mutius E, Illi S, Baumann L, Bohm O, von Kries R. Reduced risk of hay fever and asthma among children of farmers. Clin Exp Allergy. 2000;30:187–193. doi: 10.1046/j.1365-2222.2000.00801.x. [DOI] [PubMed] [Google Scholar]
- 102.Riedler J, et al. Exposure to farming in early life and development of asthma and allergy: a cross-sectional survey. Lancet. 2001;358:1129–1133. doi: 10.1016/S0140-6736(01)06252-3. [DOI] [PubMed] [Google Scholar]
- 103.Perkin MR, Strachan DP. Which aspects of the farming lifestyle explain the inverse association with childhood allergy? J Allergy Clin Immunol. 2006;117:1374–1381. doi: 10.1016/j.jaci.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 104.von Mutius E, Vercelli D. Farm living: effects on childhood asthma and allergy. Nat Rev Immunol. 2010;10:861–868. doi: 10.1038/nri2871. [DOI] [PubMed] [Google Scholar]
- 105.Almqvist C, et al. Direct and indirect exposure to pets - risk of sensitization and asthma at 4 years in a birth cohort. Clin Exp Allergy. 2003;33:1190–1197. doi: 10.1046/j.1365-2222.2003.01764.x. [DOI] [PubMed] [Google Scholar]
- 106.Ownby DR, Johnson CC, Peterson EL. Exposure to dogs and cats in the first year of life and risk of allergic sensitization at 6 to 7 years of age. JAMA. 2002;288:963–972. doi: 10.1001/jama.288.8.963. [DOI] [PubMed] [Google Scholar]
- 107.Bufford JD, et al. Effects of dog ownership in early childhood on immune development and atopic diseases. Clin Exp Allergy. 2008;38:1635–1643. doi: 10.1111/j.1365-2222.2008.03018.x. [DOI] [PubMed] [Google Scholar]
- 108.Simpson A, Custovic A. Pets and the development of allergic sensitization. Curr Allergy Asthma Rep. 2005;5:212–220. doi: 10.1007/s11882-005-0040-x. [DOI] [PubMed] [Google Scholar]
- 109.Matricardi PM, et al. Sibship size, birth order, and atopy in 11,371 Italian young men. J Allergy Clin Immunol. 1998;101:439–444. doi: 10.1016/s0091-6749(98)70350-1. [DOI] [PubMed] [Google Scholar]
- 110.Bernsen RM, de Jongste JC, van der Wouden JC. Birth order and sibship size as independent risk factors for asthma, allergy, and eczema. Pediatr Allergy Immunol. 2003;14:464–469. doi: 10.1046/j.0905-6157.2003.00108.x. [DOI] [PubMed] [Google Scholar]
- 111.Kinra S, Smith G Davey, Jeffreys M, Gunnell D, Galobardes B, McCarron P. Association between sibship size and allergic diseases in the Glasgow Alumni Study. Thorax. 2006;61:48–53. doi: 10.1136/thx.2004.034595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Goldberg S, et al. Asthma prevalence, family size, and birth order. Chest. 2007;131:1747–1752. doi: 10.1378/chest.06-2818. [DOI] [PubMed] [Google Scholar]
- 113.Caudri D, et al. Early daycare is associated with an increase in airway symptoms in early childhood but is no protection against asthma or atopy at 8 years. Am J Respir Crit Care Med. 2009;180:491–498. doi: 10.1164/rccm.200903-0327OC. [DOI] [PubMed] [Google Scholar]
- 114.Haby MM, Marks GB, Peat JK, Leeder SR. Daycare attendance before the age of two protects against atopy in preschool age children. Pediatr Pulmonol. 2000;30:377–384. doi: 10.1002/1099-0496(200011)30:5<377::aid-ppul3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 115.Nystad W. Daycare attendance, asthma and atopy. Ann Med. 2000;32:390–396. doi: 10.3109/07853890008995945. [DOI] [PubMed] [Google Scholar]
- 116.Stein RT, et al. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet. 1999;354:541–545. doi: 10.1016/S0140-6736(98)10321-5. [DOI] [PubMed] [Google Scholar]
- 117.Sigurs N, et al. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med. 2005;171:137–141. doi: 10.1164/rccm.200406-730OC. [DOI] [PubMed] [Google Scholar]
- 118.Lemanske RF, Jr., et al. Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J Allergy Clin Immunol. 2005;116:571–577. doi: 10.1016/j.jaci.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 119.Lemanske RF, Jr., Busse WW. Asthma: Factors underlying inception, exacerbation, and disease progression. J Allergy Clin Immunol. 2006;117:S456–461. doi: 10.1016/j.jaci.2005.07.006. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bisgaard H, et al. Childhood asthma after bacterial colonization of the airway in neonates. N Engl J Med. 2007;357:1487–1495. doi: 10.1056/NEJMoa052632. [DOI] [PubMed] [Google Scholar]
- 121.Hviid A, Melbye M. Measles-mumps-rubella vaccination and asthma-like disease in early childhood. Am J Epidemiol. 2008;168:1277–1283. doi: 10.1093/aje/kwn253. [DOI] [PubMed] [Google Scholar]
- 122.McDonald KL, Huq SI, Lix LM, Becker AB, Kozyrskyj AL. Delay in diphtheria, pertussis, tetanus vaccination is associated with a reduced risk of childhood asthma. J Allergy Clin Immunol. 2008;121:626–631. doi: 10.1016/j.jaci.2007.11.034. [DOI] [PubMed] [Google Scholar]
- 123.El-Zein M, Parent ME, Benedetti A, Rousseau MC. Does BCG vaccination protect against the development of childhood asthma? A systematic review and meta-analysis of epidemiological studies. Int J Epidemiol. 2010;39:469–486. doi: 10.1093/ije/dyp307. [DOI] [PubMed] [Google Scholar]
- 124.Balicer RD, Grotto I, Mimouni M, Mimouni D. Is childhood vaccination associated with asthma? A meta-analysis of observational studies. Pediatrics. 2007;120:e1269–1277. doi: 10.1542/peds.2006-3569. [DOI] [PubMed] [Google Scholar]
- 125.McKeever TM, Lewis SA, Smith C, Hubbard R. The importance of prenatal exposures on the development of allergic disease: a birth cohort study using the West Midlands General Practice Database. Am J Respir Crit Care Med. 2002;166:827–832. doi: 10.1164/rccm.200202-158OC. [DOI] [PubMed] [Google Scholar]
- 126.Kozyrskyj AL, Ernst P, Becker AB. Increased risk of childhood asthma from antibiotic use in early life. Chest. 2007;131:1753–1759. doi: 10.1378/chest.06-3008. [DOI] [PubMed] [Google Scholar]
- 127.Kummeling I, et al. Early life exposure to antibiotics and the subsequent development of eczema, wheeze, and allergic sensitization in the first 2 years of life: the KOALA Birth Cohort Study. Pediatrics. 2007;119:e225–231. doi: 10.1542/peds.2006-0896. [DOI] [PubMed] [Google Scholar]
- 128.Risnes KR, Belanger K, Murk W, Bracken MB. Antibiotic Exposure by 6 Months and Asthma and Allergy at 6 Years: Findings in a Cohort of 1,401 US Children. Am J Epidemiol. 2011;173:310–318. doi: 10.1093/aje/kwq400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Marra F, et al. Does antibiotic exposure during infancy lead to development of asthma?: a systematic review and metaanalysis. Chest. 2006;129:610–618. doi: 10.1378/chest.129.3.610. [DOI] [PubMed] [Google Scholar]
- 130.Xu B, Pekkanen J, Hartikainen AL, Jarvelin MR. Caesarean section and risk of asthma and allergy in adulthood. J Allergy Clin Immunol. 2001;107:732–733. doi: 10.1067/mai.2001.113048. [DOI] [PubMed] [Google Scholar]
- 131.Kero J, et al. Mode of delivery and asthma -- is there a connection? Pediatr Res. 2002;52:6–11. doi: 10.1203/00006450-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 132.Tollanes MC, Moster D, Daltveit AK, Irgens LM. Cesarean section and risk of severe childhood asthma: a population-based cohort study. J Pediatr. 2008;153:112–116. doi: 10.1016/j.jpeds.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 133.Roduit C, et al. Asthma at 8 years of age in children born by caesarean section. Thorax. 2009;64:107–113. doi: 10.1136/thx.2008.100875. [DOI] [PubMed] [Google Scholar]
- 134.Kull I, Almqvist C, Lilja G, Pershagen G, Wickman M. Breast-feeding reduces the risk of asthma during the first 4 years of life. J Allergy Clin Immunol. 2004;114:755–760. doi: 10.1016/j.jaci.2004.07.036. [DOI] [PubMed] [Google Scholar]
- 135.Oddy WH. Breastfeeding and asthma in children: findings from a West Australian study. Breastfeed Rev. 2000;8:5–11. [PubMed] [Google Scholar]
- 136.Dell S, To T. Breastfeeding and asthma in young children: findings from a population-based study. Arch Pediatr Adolesc Med. 2001;155:1261–1265. doi: 10.1001/archpedi.155.11.1261. [DOI] [PubMed] [Google Scholar]
- 137.Gdalevich M, Mimouni D, Mimouni M. Breast-feeding and the risk of bronchial asthma in childhood: a systematic review with meta-analysis of prospective studies. J Pediatr. 2001;139:261–266. doi: 10.1067/mpd.2001.117006. [DOI] [PubMed] [Google Scholar]
- 138.Devereux G, Seaton A. Diet as a risk factor for atopy and asthma. J Allergy Clin Immunol. 2005;115:1109–1117. doi: 10.1016/j.jaci.2004.12.1139. quiz 1118. [DOI] [PubMed] [Google Scholar]
- 139.Willers SM, et al. Maternal food consumption during pregnancy and asthma, respiratory and atopic symptoms in 5-year-old children. Thorax. 2007;62:773–779. doi: 10.1136/thx.2006.074187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Romieu I, et al. Maternal fish intake during pregnancy and atopy and asthma in infancy. Clin Exp Allergy. 2007;37:518–525. doi: 10.1111/j.1365-2222.2007.02685.x. [DOI] [PubMed] [Google Scholar]
- 141.Devereux G, et al. Low maternal vitamin E intake during pregnancy is associated with asthma in 5-year-old children. Am J Respir Crit Care Med. 2006;174:499–507. doi: 10.1164/rccm.200512-1946OC. [DOI] [PubMed] [Google Scholar]
- 142.Devereux G, et al. Maternal vitamin D intake during pregnancy and early childhood wheezing. Am J Clin Nutr. 2007;85:853–859. doi: 10.1093/ajcn/85.3.853. [DOI] [PubMed] [Google Scholar]
- 143.Whitrow MJ, Moore VM, Rumbold AR, Davies MJ. Effect of supplemental folic acid in pregnancy on childhood asthma: a prospective birth cohort study. Am J Epidemiol. 2009;170:1486–1493. doi: 10.1093/aje/kwp315. [DOI] [PubMed] [Google Scholar]
- 144.Islam T, et al. Relationship between air pollution, lung function and asthma in adolescents. Thorax. 2007;62:957–963. doi: 10.1136/thx.2007.078964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schwartz J. Air pollution and children’s health. Pediatrics. 2004;113:1037–1043. [PubMed] [Google Scholar]
- 146.Trasande L, Thurston GD. The role of air pollution in asthma and other pediatric morbidities. J Allergy Clin Immunol. 2005;115:689–699. doi: 10.1016/j.jaci.2005.01.056. [DOI] [PubMed] [Google Scholar]
- 147.Chen Y, Dales R, Tang M, Krewski D. Obesity may increase the incidence of asthma in women but not in men: longitudinal observations from the Canadian National Population Health Surveys. Am J Epidemiol. 2002;155:191–197. doi: 10.1093/aje/155.3.191. [DOI] [PubMed] [Google Scholar]
- 148.Gilliland FD, et al. Obesity and the risk of newly diagnosed asthma in school-age children. Am J Epidemiol. 2003;158:406–415. doi: 10.1093/aje/kwg175. [DOI] [PubMed] [Google Scholar]
- 149.Schaub B, von Mutius E. Obesity and asthma, what are the links? Curr Opin Allergy Clin Immunol. 2005;5:185–193. doi: 10.1097/01.all.0000162313.64308.b5. [DOI] [PubMed] [Google Scholar]
- 150.Brussee JE, et al. Allergen exposure in infancy and the development of sensitization, wheeze, and asthma at 4 years. J Allergy Clin Immunol. 2005;115:946–952. doi: 10.1016/j.jaci.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 151.Torrent M, et al. Early-life allergen exposure and atopy, asthma, and wheeze up to 6 years of age. Am J Respir Crit Care Med. 2007;176:446–453. doi: 10.1164/rccm.200607-916OC. [DOI] [PubMed] [Google Scholar]
- 152.Tovey ER, Almqvist C, Li Q, Crisafulli D, Marks GB. Nonlinear relationship of mite allergen exposure to mite sensitization and asthma in a birth cohort. J Allergy Clin Immunol. 2008;122:114–118. e111–115. doi: 10.1016/j.jaci.2008.05.010. 118. [DOI] [PubMed] [Google Scholar]
- 153.Kogevinas M, et al. Exposure to substances in the workplace and new-onset asthma: an international prospective population-based study (ECRHS-II) Lancet. 2007;370:336–341. doi: 10.1016/S0140-6736(07)61164-7. [DOI] [PubMed] [Google Scholar]
- 154.Bakerly ND, Moore VC, Vellore AD, Jaakkola MS, Robertson AS, Burge PS. Fifteen-year trends in occupational asthma: data from the Shield surveillance scheme. Occup Med (Lond) 2008;58:169–174. doi: 10.1093/occmed/kqn007. [DOI] [PubMed] [Google Scholar]
- 155.Dykewicz MS. Occupational asthma: current concepts in pathogenesis, diagnosis, and management. J Allergy Clin Immunol. 2009;123:519–528. doi: 10.1016/j.jaci.2009.01.061. quiz 529-530. [DOI] [PubMed] [Google Scholar]
- 156.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 157.Johnson GC, et al. Haplotype tagging for the identification of common disease genes. Nat Genet. 2001;29:233–237. doi: 10.1038/ng1001-233. [DOI] [PubMed] [Google Scholar]
- 158.Montpetit A, et al. An evaluation of the performance of tag SNPs derived from HapMap in a Caucasian population. PLoS Genet. 2006;2:e27. doi: 10.1371/journal.pgen.0020027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chi PB, et al. Comparison of SNP tagging methods using empirical data: association study of 713 SNPs on chromosome 12q14.3-12q24.21 for asthma and total serum IgE in an African Caribbean population. Genet Epidemiol. 2006;30:609–619. doi: 10.1002/gepi.20172. [DOI] [PubMed] [Google Scholar]
- 160.Bosse Y, Hudson TJ. Toward a comprehensive set of asthma susceptibility genes. Annu Rev Med. 2007;58:171–184. doi: 10.1146/annurev.med.58.071105.111738. [DOI] [PubMed] [Google Scholar]
- 161.Ober C, Hoffjan S. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun. 2006;7:95–100. doi: 10.1038/sj.gene.6364284. [DOI] [PubMed] [Google Scholar]
- 162.Vercelli D. Discovering susceptibility genes for asthma and allergy. Nat Rev Immunol. 2008;8:169–182. doi: 10.1038/nri2257. [DOI] [PubMed] [Google Scholar]
- 163.Evans DM, Cardon LR. Guidelines for genotyping in genomewide linkage studies: single-nucleotide-polymorphism maps versus microsatellite maps. Am J Hum Genet. 2004;75:687–692. doi: 10.1086/424696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 165.The International HapMap Project Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 166.Manolio TA, Collins FS. The HapMap and genome-wide association studies in diagnosis and therapy. Annu Rev Med. 2009;60:443–456. doi: 10.1146/annurev.med.60.061907.093117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118:1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Moffatt MF, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Torgerson DG, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. 2011. in review. [DOI] [PMC free article] [PubMed]
- 173.Lander ES. The new genomics: global views of biology. Science. 1996;274:536–539. doi: 10.1126/science.274.5287.536. [DOI] [PubMed] [Google Scholar]
- 174.Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 175.Wang WY, Barratt BJ, Clayton DG, Todd JA. Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet. 2005;6:109–118. doi: 10.1038/nrg1522. [DOI] [PubMed] [Google Scholar]
- 176.Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 1994;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 177.Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet. 2010;19:R145–151. doi: 10.1093/hmg/ddq333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Eichler EE, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pritchard JK, Cox NJ. The allelic architecture of human disease genes: common disease-common variant…or not? Hum Mol Genet. 2002;11:2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- 182.Nicolae DL, Ober C. (Too) great expectations: the challenges in replicating asthma disease genes. Am J Respir Crit Care Med. 2009;179:1078–1079. doi: 10.1164/rccm.200903-0456ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.McCarthy MI, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 184.Blumenthal MN, Amos DB, Noreen H, Mendell NR, Yunis EJ. Genetic mapping of Ir locus in man: linkage to second locus of HLA. Science. 1974;184:1301–1303. doi: 10.1126/science.184.4143.1301. [DOI] [PubMed] [Google Scholar]
- 185.Marsh DG, Bias WB. HL-A linked antigen E immune response genes: an unproven hypothesis. Science. 1975;188:375–377. doi: 10.1126/science.1118733. [DOI] [PubMed] [Google Scholar]
- 186.Denham S, et al. Meta-analysis of genome-wide linkage studies of asthma and related traits. Respir Res. 2008;9:38. doi: 10.1186/1465-9921-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bouzigon E, et al. Meta-analysis of 20 genome-wide linkage studies evidenced new regions linked to asthma and atopy. Eur J Hum Genet. 2010;18:700–706. doi: 10.1038/ejhg.2009.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Allen M, et al. Positional cloning of a novel gene influencing asthma from chromosome 2q14. Nat Genet. 2003;35:258–263. doi: 10.1038/ng1256. [DOI] [PubMed] [Google Scholar]
- 189.Laitinen T, et al. Characterization of a common susceptibility locus for asthma-related traits. Science. 2004;304:300–304. doi: 10.1126/science.1090010. [DOI] [PubMed] [Google Scholar]
- 190.Nicolae D, et al. Fine mapping and positional candidate studies identify HLA-G as an asthma susceptibility gene on chromosome 6p21. Am J Hum Genet. 2005;76:349–357. doi: 10.1086/427763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Noguchi E, et al. Positional identification of an asthma susceptibility gene on human chromosome 5q33. Am J Respir Crit Care Med. 2005;172:183–188. doi: 10.1164/rccm.200409-1223OC. [DOI] [PubMed] [Google Scholar]
- 192.Van Eerdewegh P, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. 2002;418:426–430. doi: 10.1038/nature00878. [DOI] [PubMed] [Google Scholar]
- 193.Zhang Y, et al. Positional cloning of a quantitative trait locus on chromosome 13q14 that influences immunoglobulin E levels and asthma. Nat Genet. 2003;34:181–186. doi: 10.1038/ng1166. [DOI] [PubMed] [Google Scholar]
- 194.Balaci L, et al. IRAK-M is involved in the pathogenesis of early-onset persistent asthma. Am J Hum Genet. 2007;80:1103–1114. doi: 10.1086/518259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Soderhall C, et al. Variants in a novel epidermal collagen gene (COL29A1) are associated with atopic dermatitis. PLoS Biol. 2007;5:e242. doi: 10.1371/journal.pbio.0050242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.White JH, et al. Identification of a novel asthma susceptibility gene on chromosome 1qter and its functional evaluation. Hum Mol Genet. 2008;17:1890–1903. doi: 10.1093/hmg/ddn087. [DOI] [PubMed] [Google Scholar]
- 197.Moffatt MF, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 198.Dixon AL, et al. A genome-wide association study of global gene expression. Nat Genet. 2007;39:1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 199.Halapi E, et al. A sequence variant on 17q21 is associated with age at onset and severity of asthma. Eur J Hum Genet. 2010;18:902–908. doi: 10.1038/ejhg.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Verlaan DJ, et al. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am J Hum Genet. 2009;85:377–393. doi: 10.1016/j.ajhg.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bouzigon E, et al. Effect of 17q21 variants and smoking exposure in early-onset asthma. N Engl J Med. 2008;359:1985–1994. doi: 10.1056/NEJMoa0806604. [DOI] [PubMed] [Google Scholar]
- 202.Ferreira MA, et al. Association between ORMDL3, IL1RL1 and a deletion on chromosome 17q21 with asthma risk in Australia. Eur J Hum Genet. 2011;19:458–464. doi: 10.1038/ejhg.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sleiman PM, et al. ORMDL3 variants associated with asthma susceptibility in North Americans of European ancestry. J Allergy Clin Immunol. 2008;122:1225–1227. doi: 10.1016/j.jaci.2008.06.041. [DOI] [PubMed] [Google Scholar]
- 204.Bisgaard H, et al. Chromosome 17q21 gene variants are associated with asthma and exacerbations but not atopy in early childhood. Am J Respir Crit Care Med. 2009;179:179–185. doi: 10.1164/rccm.200809-1436OC. [DOI] [PubMed] [Google Scholar]
- 205.Smit LA, et al. 17q21 variants modify the association between early respiratory infections and asthma. Eur Respir J. 2010;36:57–64. doi: 10.1183/09031936.00154509. [DOI] [PubMed] [Google Scholar]
- 206.Tavendale R, Macgregor DF, Mukhopadhyay S, Palmer CN. A polymorphism controlling ORMDL3 expression is associated with asthma that is poorly controlled by current medications. J Allergy Clin Immunol. 2008;121:860–863. doi: 10.1016/j.jaci.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 207.Breslow DK, et al. Orm family proteins mediate sphingolipid homeostasis. Nature. 2010;463:1048–1053. doi: 10.1038/nature08787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Cantero-Recasens G, Fandos C, Rubio-Moscardo F, Valverde MA, Vicente R. The asthma-associated ORMDL3 gene product regulates endoplasmic reticulum-mediated calcium signaling and cellular stress. Hum Mol Genet. 2010;19:111–121. doi: 10.1093/hmg/ddp471. [DOI] [PubMed] [Google Scholar]
- 209.Carl-McGrath S, Schneider-Stock R, Ebert M, Rocken C. Differential expression and localisation of gasdermin-like (GSDML), a novel member of the cancer-associated GSDMDC protein family, in neoplastic and non-neoplastic gastric, hepatic, and colon tissues. Pathology. 2008;40:13–24. doi: 10.1080/00313020701716250. [DOI] [PubMed] [Google Scholar]
- 210.Hancock DB, et al. Genome-wide association study implicates chromosome 9q21.31 as a susceptibility locus for asthma in mexican children. PLoS Genet. 2009;5:e1000623. doi: 10.1371/journal.pgen.1000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Himes BE, et al. Genome-wide association analysis identifies PDE4D as an asthma-susceptibility gene. Am J Hum Genet. 2009;84:581–593. doi: 10.1016/j.ajhg.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Li X, et al. Genome-wide association study of asthma identifies RAD50-IL13 and HLA-DR/DQ regions. J Allergy Clin Immunol. 2010;125:328–335. e311. doi: 10.1016/j.jaci.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mathias RA, et al. A genome-wide association study on African-ancestry populations for asthma. J Allergy Clin Immunol. 2010;125:336–346. e334. doi: 10.1016/j.jaci.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Sleiman PM, et al. Variants of DENND1B associated with asthma in children. N Engl J Med. 2010;362:36–44. doi: 10.1056/NEJMoa0901867. [DOI] [PubMed] [Google Scholar]
- 215.Esparza-Gordillo J, et al. A common variant on chromosome 11q13 is associated with atopic dermatitis. Nat Genet. 2009;41:596–601. doi: 10.1038/ng.347. [DOI] [PubMed] [Google Scholar]
- 216.Rothenberg ME, et al. Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nat Genet. 2010;42:289–291. doi: 10.1038/ng.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wan YI, et al. A genome-wide association study to identify genetic determinants of atopy in subjects from the United Kingdom. J Allergy Clin Immunol. 2011;127:223–231. e221–223. doi: 10.1016/j.jaci.2010.10.006. 231. [DOI] [PubMed] [Google Scholar]
- 218.Wilk JB, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5:e1000429. doi: 10.1371/journal.pgen.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ober C, Vercelli D. Gene-environment interactions in human disease: nuisance or opportunity? Trends Genet. 2011;27:107–115. doi: 10.1016/j.tig.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hancock DB, London SJ. Determinants of lung function, COPD, and asthma. N Engl J Med. 2011;364:86–87. doi: 10.1056/NEJMc1011921. [DOI] [PubMed] [Google Scholar]
- 221.Borish L, Steinke JW. Interleukin-33 in asthma: how big of a role does it play? Curr Allergy Asthma Rep. 2011;11:7–11. doi: 10.1007/s11882-010-0153-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lloyd CM. IL-33 family members and asthma - bridging innate and adaptive immune responses. Curr Opin Immunol. 2010;22:800–806. doi: 10.1016/j.coi.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172–190. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Smith DE. IL-33: a tissue derived cytokine pathway involved in allergic inflammation and asthma. Clin Exp Allergy. 2010;40:200–208. doi: 10.1111/j.1365-2222.2009.03384.x. [DOI] [PubMed] [Google Scholar]
- 225.Ziegler SF. The role of thymic stromal lymphopoietin (TSLP) in allergic disorders. Curr Opin Immunol. 2010;22:795–799. doi: 10.1016/j.coi.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Bunyavanich S, et al. Thymic stromal lymphopoietin (TSLP) is associated with allergic rhinitis in children with asthma. Clin Mol Allergy. 2011;9:1. doi: 10.1186/1476-7961-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hunninghake GM, et al. Sex-stratified linkage analysis identifies a female-specific locus for IgE to cockroach in Costa Ricans. Am J Respir Crit Care Med. 2008;177:830–836. doi: 10.1164/rccm.200711-1697OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hunninghake GM, et al. TSLP polymorphisms are associated with asthma in a sex-specific fashion. Allergy. 2010 doi: 10.1111/j.1398-9995.2010.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Harada M, et al. TSLP Promoter Polymorphisms are Associated with Susceptibility to Bronchial Asthma. Am J Respir Cell Mol Biol. 2010 doi: 10.1165/rcmb.2009-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Reijmerink NE, et al. Association of IL1RL1, IL18R1, and IL18RAP gene cluster polymorphisms with asthma and atopy. J Allergy Clin Immunol. 2008;122:651–654. e658. doi: 10.1016/j.jaci.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 231.Savenije OE, et al. Interleukin-1 receptor-like 1 polymorphisms are associated with serum IL1RL1-a, eosinophils, and asthma in childhood. J Allergy Clin Immunol. 2011;127:750–756. e755. doi: 10.1016/j.jaci.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 232.Wu H, et al. Evaluation of candidate genes in a genome-wide association study of childhood asthma in Mexicans. J Allergy Clin Immunol. 2010;125:321–327. e313. doi: 10.1016/j.jaci.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Bogardus C. Missing heritability and GWAS utility. Obesity (Silver Spring) 2009;17:209–210. doi: 10.1038/oby.2008.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Clarke AJ, Cooper DN. GWAS: heritability missing in action? Eur J Hum Genet. 2010;18:859–861. doi: 10.1038/ejhg.2010.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Heng HH. Missing heritability and stochastic genome alterations. Nat Rev Genet. 2010;11:813. doi: 10.1038/nrg2809-c3. [DOI] [PubMed] [Google Scholar]
- 236.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating Missing Heritability for Disease from Genome-wide Association Studies. Am J Hum Genet. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 238.Meaburn EL, Schalkwyk LC, Mill J. Allele-specific methylation in the human genome Implications for genetic studies of complex disease. Epigenetics. 2010;5 doi: 10.4161/epi.5.7.12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Slatkin M. Epigenetic inheritance and the missing heritability problem. Genetics. 2009;182:845–850. doi: 10.1534/genetics.109.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.So HC, Gui AH, Cherny SS, Sham PC. Evaluating the heritability explained by known susceptibility variants: a survey of ten complex diseases. Genet Epidemiol. 2011 doi: 10.1002/gepi.20579. [DOI] [PubMed] [Google Scholar]
- 241.Spencer C, Hechter E, Vukcevic D, Donnelly P. Quantifying the underestimation of relative risks from genome-wide association studies. PLoS Genet. 2011;7:e1001337. doi: 10.1371/journal.pgen.1001337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.van der Sluis S, Verhage M, Posthuma D, Dolan CV. Phenotypic complexity, measurement bias, and poor phenotypic resolution contribute to the missing heritability problem in genetic association studies. PLoS One. 2010;5:e13929. doi: 10.1371/journal.pone.0013929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Vineis P, Pearce N. Missing heritability in genome-wide association study research. Nat Rev Genet. 2010;11:589. doi: 10.1038/nrg2809-c2. [DOI] [PubMed] [Google Scholar]
- 244.Gibson G. Hints of hidden heritability in GWAS. Nat Genet. 2010;42:558–560. doi: 10.1038/ng0710-558. [DOI] [PubMed] [Google Scholar]
- 245.Craddock N, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Girirajan S, Eichler EE. Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet. 2010;19:R176–187. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat Genet. 2007;39:S37–42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- 248.Wong KK, et al. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Ionita-Laza I, et al. On the analysis of copy-number variations in genome-wide association studies: a translation of the family-based association test. Genet Epidemiol. 2008;32:273–284. doi: 10.1002/gepi.20302. [DOI] [PubMed] [Google Scholar]
- 250.Walsh KM, Bracken MB, Murk WK, Hoh J, Dewan AT. Association between reduced copy-number at T-cell receptor gamma (TCRgamma) and childhood allergic asthma: A possible role for somatic mosaicism. Mutat Res. 2010;690:89–94. doi: 10.1016/j.mrfmmm.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Cooper GM, Zerr T, Kidd JM, Eichler EE, Nickerson DA. Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nat Genet. 2008;40:1199–1203. doi: 10.1038/ng.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011 doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Handsaker RE, Korn JM, Nemesh J, McCarroll SA. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet. 2011;43:269–276. doi: 10.1038/ng.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.McCarroll SA. Copy number variation and human genome maps. Nat Genet. 2010;42:365–366. doi: 10.1038/ng0510-365. [DOI] [PubMed] [Google Scholar]
- 255.Ege MJ, et al. Gene-environment interaction for childhood asthma and exposure to farming in Central Europe. J Allergy Clin Immunol. 2011;127:138–144. e131–134. doi: 10.1016/j.jaci.2010.09.041. 144. [DOI] [PubMed] [Google Scholar]
- 256.London SJ, Romieu I. Gene by environment interaction in asthma. Annu Rev Public Health. 2009;30:55–80. doi: 10.1146/annurev.publhealth.031308.100151. [DOI] [PubMed] [Google Scholar]
- 257.Vercelli D. Gene-environment interactions in asthma and allergy: the end of the beginning? Curr Opin Allergy Clin Immunol. 2010;10:145–148. doi: 10.1097/ACI.0b013e32833653d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.von Mutius E. Gene-environment interactions in asthma. J Allergy Clin Immunol. 2009;123:3–11. doi: 10.1016/j.jaci.2008.10.046. quiz 12-13. [DOI] [PubMed] [Google Scholar]
- 259.Ober C, et al. Getting from genes to function in lung disease: a National Heart, Lung, and Blood Institute workshop report. Am J Respir Crit Care Med. 2010;182:732–737. doi: 10.1164/rccm.201002-0180PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Durham AL, Wiegman C, Adcock IM. Epigenetics of asthma. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbagen.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 261.Ho SM. Environmental epigenetics of asthma: an update. J Allergy Clin Immunol. 2010;126:453–465. doi: 10.1016/j.jaci.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kabesch M, Michel S, Tost J. Epigenetic mechanisms and the relationship to childhood asthma. Eur Respir J. 2010;36:950–961. doi: 10.1183/09031936.00019310. [DOI] [PubMed] [Google Scholar]
- 263.Kumar RK, Hitchins MP, Foster PS. Epigenetic changes in childhood asthma. Dis Model Mech. 2009;2:549–553. doi: 10.1242/dmm.001719. [DOI] [PubMed] [Google Scholar]
- 264.Pfefferle PI, Pinkenburg O, Renz H. Fetal epigenetic mechanisms and innate immunity in asthma. Curr Allergy Asthma Rep. 2010;10:434–443. doi: 10.1007/s11882-010-0147-6. [DOI] [PubMed] [Google Scholar]
- 265.Schwartz DA. Epigenetics and environmental lung disease. Proc Am Thorac Soc. 2010;7:123–125. doi: 10.1513/pats.200908-084RM. [DOI] [PubMed] [Google Scholar]
- 266.Lima JJ, Blake KV, Tantisira KG, Weiss ST. Pharmacogenetics of asthma. Curr Opin Pulm Med. 2009;15:57–62. doi: 10.1097/MCP.0b013e32831da8be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Drazen JM, et al. Pharmacogenetic association between ALOX5 promoter genotype and the response to anti-asthma treatment. Nat Genet. 1999;22:168–170. doi: 10.1038/9680. [DOI] [PubMed] [Google Scholar]
- 268.Klotsman M, et al. Pharmacogenetics of the 5-lipoxygenase biosynthetic pathway and variable clinical response to montelukast. Pharmacogenet Genomics. 2007;17:189–196. doi: 10.1097/FPC.0b013e3280120043. [DOI] [PubMed] [Google Scholar]
- 269.Lima JJ, et al. Influence of leukotriene pathway polymorphisms on response to montelukast in asthma. Am J Respir Crit Care Med. 2006;173:379–385. doi: 10.1164/rccm.200509-1412OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Tantisira KG, Lima J, Sylvia J, Klanderman B, Weiss ST. 5-lipoxygenase pharmacogenetics in asthma: overlap with Cys-leukotriene receptor antagonist loci. Pharmacogenet Genomics. 2009;19:244–247. doi: 10.1097/FPC.0b013e328326e0b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Telleria JJ, et al. ALOX5 promoter genotype and response to montelukast in moderate persistent asthma. Respir Med. 2008;102:857–861. doi: 10.1016/j.rmed.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 272.Bleecker ER, et al. Salmeterol response is not affected by beta2-adrenergic receptor genotype in subjects with persistent asthma. J Allergy Clin Immunol. 2006;118:809–816. doi: 10.1016/j.jaci.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 273.Israel E, et al. Effect of polymorphism of the beta(2)-adrenergic receptor on response to regular use of albuterol in asthma. Int Arch Allergy Immunol. 2001;124:183–186. doi: 10.1159/000053705. [DOI] [PubMed] [Google Scholar]
- 274.Tsai HJ, et al. Beta 2-adrenergic receptor polymorphisms: pharmacogenetic response to bronchodilator among African American asthmatics. Hum Genet. 2006;119:547–557. doi: 10.1007/s00439-006-0169-2. [DOI] [PubMed] [Google Scholar]
- 275.Wechsler ME, et al. Effect of beta2-adrenergic receptor polymorphism on response to longacting beta2 agonist in asthma (LARGE trial): a genotype-stratified, randomised, placebo-controlled, crossover trial. Lancet. 2009;374:1754–1764. doi: 10.1016/S0140-6736(09)61492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Cookson WOCM, Sharp PA, Faux JA, Hopkin JM. Linkage between immunoglobulin E responses underlying asthma and rhinitis and chromosome 11q. Lancet. 1989;1:1292–1295. doi: 10.1016/s0140-6736(89)92687-1. [DOI] [PubMed] [Google Scholar]
- 277.Postma DS, et al. Genetic susceptibility to asthma-bronchial hyperresponsiveness coinherited with a major gene for atopy. New Engl J Med. 1995;333:894–900. doi: 10.1056/NEJM199510053331402. [DOI] [PubMed] [Google Scholar]
- 278.Holgate ST. ADAM metallopeptidase domain 33 (ADAM33): identification and role in airways disease. Drug News Perspect. 2010;23:381–387. doi: 10.1358/dnp.2010.23.6.1437242. [DOI] [PubMed] [Google Scholar]
- 279.Holgate ST, Davies DE, Powell RM, Holloway JW. ADAM33: a newly identified protease involved in airway remodelling. Pulm Pharmacol Ther. 2005 doi: 10.1016/j.pupt.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 280.Genuneit J, et al. A multi-centre study of candidate genes for wheeze and allergy: the International Study of Asthma and Allergies in Childhood Phase 2. Clin Exp Allergy. 2009;39:1875–1888. doi: 10.1111/j.1365-2222.2009.03364.x. [DOI] [PubMed] [Google Scholar]
- 281.Morar N, Cookson WO, Harper JI, Moffatt MF. Filaggrin mutations in children with severe atopic dermatitis. J Invest Dermatol. 2007;127:1667–1672. doi: 10.1038/sj.jid.5700739. [DOI] [PubMed] [Google Scholar]