

Abstract
Embryonic epithelial cells serve as an ideal model to study morphogenesis where multi-cellular tissues undergo changes in their geometry, such as changes in cell surface area and cell height, and where cells undergo mitosis and migrate. Furthermore, epithelial cells can also regulate morphogenetic movements in adjacent tissues1. A traditional method to study epithelial cells and tissues involve chemical fixation and histological methods to determine cell morphology or localization of particular proteins of interest. These approaches continue to be useful and provide "snapshots" of cell shapes and tissue architecture, however, much remains to be understood about how cells acquire specific shapes, how various proteins move or localize to specific positions, and what paths cells follow toward their final differentiated fate. High resolution live imaging complements traditional methods and also allows more direct investigation into the dynamic cellular processes involved in the formation, maintenance, and morphogenesis of multicellular epithelial sheets. Here we demonstrate experimental methods from the isolation of animal cap tissues from Xenopus laevis embryos to confocal imaging of epithelial cells and simple measurement approaches that together can augment molecular and cellular studies of epithelial morphogenesis.
Protocol
Before you begin
Synthesize capped mRNA from linearized DNA template encoding the fluorescently tagged protein and purify mRNA by standard methods (AmpliCap Transcription kit; Epicentre Biotechnologies, Madison WI). mRNA should be aliquoted for single time use in 0.2 ml centrifuge tubes and stored at -80°C.
Standard methods to obtain eggs, in vitro fertilization, and removal of the egg coat have been described previously2. A procedure to obtain eggs from a Xenopus laevis female frog has been demonstrated earlier3. Fertilize eggs in vitro immediately after obtaining them from female frogs using testes previously isolated from a male frog. De-jelly eggs 20 minutes post fertilization and inject mRNA, protein, DNA, or dextran immediately to allow sufficient diffusion of microinjected reagents. Injections more than 1 hour post fertilization often fail to diffuse from the injection site.
Oocyte microinjection has been demonstrated previously4 using a pressure-valve controlled injector (PLI-100, Harvard Apparatus). Follow a similar method for injecting mRNAs in the animal hemisphere of frog embryos at the 1- to 4-cell stage. Transfer fertilized embryos to 3% Ficoll (Sigma, St. Louis MO) in 1x MBS (MBS-Ficoll) and microinject with the desired volume (2 to 4 nl) of capped mRNA encoding GFP targeted to membrane (mem-GFP) or F-actin (moe-GFP). To obtain even distribution inject mRNA at 4 equally spaced sites in the animal pole. Embryos can remain in MBS-Ficoll for several hours but need to be transferred to 1/3 x MBS before gastrulation starts.
Required materials (items marked by * are shown in Fig. 1):
Dissection stereomicroscope with fiber-optic illumination. A cooled dissection stage is recommended but optional.
Hair tools (hair knife and hair loop)*.
Forceps (Dumont #5 stainless steel; Fine Science Tools, Foster City, CA)*.
Modified Barth's Saline embryo culture media (MBS; 88 mM NaCl, 2.4 mM NaHCO3, 1 mM KCl, 0.33 mM CaCl2, 0.41 mM (CaNO3)2, 0.82 mM MgSO4, 10 mM HEPES (H3375, Sigma-Aldrich, St. Louis MO)).
Danilchik's for Amy explant culture media (DFA; 53 mM NaCl2, 5 mM Na2CO3, 4.5 mM Potassium gluconate, 32 mM Sodium gluconate, 1mM CaCl2, 1mM MgSO4). Include 0.1% Bovine serum albumin with DFA (BSA; A7906, Sigma-Aldrich). DFA must be sterile filtered but can be aliquoted in 50 ml conical tubes and stored at -20°C. Antibiotics and antimycotics (0.8 % in media; A5955, Sigma-Aldrich) should be added to freshly thawed DFA to inhibit bacterial or fungal growth.
60 or 100 mm Petri dishes*.
Large cover glass, 45 by 50 mm (12-544-F, #1.5; Fisher Scientific, Hampton, NH)*.
Small cover glass, 24 by 40 mm (12-544C, #1.5; Fisher Scientific).
Silicone grease (High Vacuum, Dow Chemical) loaded into a syringe "caulking-gun"*.
Custom acrylic chambers or nylon washers (Small Parts Inc.; Miramar, FL)*. Custom chambers can be milled in a Machine Shop from a 25 by 50 by 5 mm acrylic block to provide an inner chamber that is 1.2 cm by 2.8 cm. This chamber has a working volume of 1.68 ml. Optional nylon washers may be selected in a variety of sizes; we prefer sizes compatible with 18 mm diameter circular glass coverslips.
Diamond pencil*.
Part 1: Excision of animal cap explants
Culture embryos to the desired stage5 in 1/3 x MBS. The rate of development can be controlled with culture temperatures between 15 and 21°C so that the experiments can be done at a convenient time of the day. Room temperatures may be used but embryos will develop more quickly.
Transfer embryos to DFA.
Remove vitelline membranes using forceps.
Under the dissecting stereomicroscope, excise an animal cap explant using hair-knives and hair-loops6. Use the hair-loop to support or hold the embryo at a position and the hair-knife to make a small incision in the animal pole of the embryos. Use repeated "flick-cuts" with the hair-knife to make a 360° incision to remove the animal cap ectoderm from the embryo. With practice, one can make smooth cuts that result in minimal damage to the explant or lysed cells at the margin (Fig. 2A).
Repeat step 4 to excise 3 to 4 animal cap explants and proceed to Part 2. One must be quick while cutting explants, since they have a tendency to ball up quickly. A cooled dissection stage can slow the process or alternatively you can make fewer explants.
Part 2: Prepare chambers, load explants, and seal the chamber for long term culture
Use silicone grease to glue or seal the acrylic chamber to the large cover glass.
To reduce adhesion of explants to the glass coat the glass within the chamber with a non-adhesive substrate by incubating the chamber for 2 to 4 hours at room temperature or overnight at 4°C with 1% BSA in 1/3 x MBS (BSA-MBS).
Prepare small coverslip fragments (approx. 2 by 8 mm) using a diamond pencil. Pre-coat fragments with BSA-MBS.
Rinse BSA-MBS from the chamber and replace the media with DFA.
Excise animal cap explant(s) (Part 1; Fig. 2A) and transfer to the chamber. Position each explant using a hair-loop (with epithelial layer facing the bottom of the chamber) and gently fix the explant in place with a glass coverslip fragment held by small dabs of high vacuum grease (Fig. 2B). Be very careful while pressing the coverslip since excessive pressure can easily smash the explant. Multiple explants can be loaded into each chamber based on size of the small coverslip fragments used to hold the explants in place. With practice up to 20 explants can be loaded into the acrylic chamber described here. Fill the chambers with DFA until even with the opening at the top. Use silicone grease to seal the top of the chamber with the 24 by 40 mm cover glass. Add or remove DFA to reduce air-bubbles in the sealed chamber and blot overflow from the chamber.
This sealed chamber will allow long term culture and live-imaging of animal cap explants through the large, lower glass surface of the chamber. It is crucial to keep the lower glass surface of the chamber free of dirt, grease, or media for later imaging. We recommend preparing paper-towel bottomed Petri dishes to store chambers as they are prepared for imaging.
Part 3: High-resolution live-imaging confocal microscopy
The protocol in this section has been developed to use a confocal microscope available to our lab (Leica TCS SP5; Leica Microsystems, Bannockburn IL). Different confocal microscopes may require slight modification and we recommend users follow their respective operating instructions.
Power-up the imaging system, initialize the microscope stage, and turn on lasers appropriate for fluorophores, i.e. the Argon laser for fluorescein or EGFP and 543 HeNe laser for rhodamine or RFP.
Place the chamber housing the explants onto the microscope stage. Prior to positioning the chamber move an appropriate objective into place. We recommend using a high numerical aperture 20x air objective (0.7 n.a.) for initial imaging; we recommend using1.4 n.a. 40x or 63x oil objectives for high resolution imaging and tracking large cell-fields. (note: Once immersion oil is applied to the objective use caution when using an air objective so you do not inadvertently dip the objective in the oil.) Use small weights to hold the chamber in place.
Use bright-field or fluorescence illumination and adjust the focus until you see the apical ends of the superficial cells. Be careful not to "crash" the objective into the chamber base. Avoid including or producing air bubbles when using the oil immersion objectives. Once the samples are positioned using the "coarse" positioning mode, change to the "fine" positioning mode.
- Recommendations for tuning the confocal for live imaging:
- Laser power should be kept as low as possible: 1) to prevent bleaching, 2) to prevent phototoxic effects, and 3) to prevent any accidental triggering of cell shape change7. Laser-power requirement varies depending upon the expression level and type of molecular reporters expressed in the embryos.
- Use a scan format of 1024 by 1024 pixels for live imaging as it provides good image resolution. If you need very high frame rate (minimum is 1.3s for our system), then use scan format of 512 by 512 pixels. This will reduce the image resolution, but can still be sufficient to extract reasonable amount of information.
- Adjust the spectral range of the emission, dichroic, and excitation filters as appropriate.
- Adjust the confocal pinhole to between 1 and 1.5 Airy units.
- Photomultiplier gain must be kept at a minimum level to reduce noise yet provide sufficient signal to identify cell structures and protein localization.
- Adjust the black level offset to adjust the background to improve the quality of the images.
- Steps (1) through (6) are iteratively applied to produce the best quality image possible. Quality images should resolve the structure or localized proteins with pixel intensities over a broad dynamic range. If quantitative analyses are the goal there should be minimal numbers of zero-intensity or saturated pixels.
- Steps (1), (3), (5), and (6) need to be repeated for each additional fluorescence channel to produce the best quality image for possible.
- When imaging two or fluorescent probes it is important to ensure that there is minimal spectral bleed-through from one protein to a second protein's emission spectra. Bleed-through can be tested by reducing the laser power exciting the first protein and observing the signal produced in the wavelengths used by the second protein. Spectral bleed-through may be reduced with more restrictive choice of filters or by reducing the laser power used for excitation and increasing the gain used for detection of the first protein.
With the confocal tuned using these guidelines it should be possible to capture single images at a single focal depth in X-Y mode (Fig. 3A).
- If one is interested in collecting time-lapse movies of a dynamically occurring process such as shape change during cell contraction7, one has to choose from the following alternative confocal modes:
- X-Y-T: This mode allows one to collect a series of images over time, without changing the Z-plane. We use this mode to observe the endogenous fluctuations in the epithelial cells. Normally, we acquire one frame every 5 seconds over 20 minutes to observe endogenous variation or shape changes induced by external stimulation of epithelial cells. Choice of frame-rate is completely dependent on what one is interested in observing and how dynamic the process is.
- X-Y-Z: Using this mode, we can collect a single Z-series stack through epithelial cells. Our default settings to collect Z-series are 0.1 μm steps over a range of 5 μm. We use this mode to determine whether cytoskeletal changes are occurring only at a certain positions, such as at the level of the adherens junctions, or everywhere in the cell cortex.
- X-Z-T (Fig. 3A'): This mode is mainly used to observe rapid changes in the apical-basal organization of cell in the epithelium. This mode is popular for its utility in confirming that the X-Y-T mode is a reliable reporter of changes in the apical cell domain and that the confocal image acquisition is not drifting along the Z-axis.
- X-Y-Z-T: This combines X-Y-Z and X-Y-T modes. This mode can provide rich details on the full apical-basal dynamics of the epithelium but is slow to acquire full image stacks and can severely reduce cell viability due to photobleaching and phototoxic effects.
The image quality can be improved to a limited extent by using one or a combination of the four following approaches: 1) line averaging, 2) line accumulation, 3) frame averaging, and 4) frame accumulation. These averaging modes for image acquisition can improve image quality but will reduce cell viability and require more time to collect images.
With all the settings carefully adjusted, collect and save images, Z-series, and time-series data to an external hard drive, USB-memory stick, or network-attached server.
Once you are have completed your experiment, lower the objective for safety, clean the oil from the objective and the microscope stage with lens paper, and power-down the confocal system.
Part 4: Live-imaging of F-actin microfilaments
As has been shown in Part 3, the membrane localizing protein mem-GFP can be used to fluorescently label cell membranes. Live-imaging of sub-cellular components is achieved by using other proteins. A protein such as moesin-GFP that contains an F-actin-binding domain can be used to label F-actin in live cells. Similar strategies for live-cell imaging and collection of time-lapse sequences follow the procedures outlined in part 3. Live-imaging F-actin involves more microscopy skills than imaging mem-GFP since the structures are finer and more tightly localized than the membrane. For instance, any small bubbles in the immersion oil can produce aberration artifacts such as blurring or variable brightness in the captured image. Furthermore, any small drift in the focal or Z-plane can cause moesin-GFP labeled cortical F-actin to move out-of-focus. Thus, one should be re-double efforts to stabilize the stage, the explant, and the chamber while imaging cortical F-actin and other sub-cellular proteins. X-Y and X-Z-T images showing moesin-GFP labeled F-actin are shown as examples (Fig. 3B and B').
Part 5: Image Analysis
Images and time-lapse sequences obtained from the live-cell confocal session can be analyzed in order to test hypotheses concerning the shape changes or patterns of protein localization. The naked eye is often the best tool for qualitative analysis but can be augmented or automated by image analysis techniques to extract quantitative data. Image data from Leica confocal systems can be saved in an open format such as .TIF formatted image files or a proprietary .LIF format that retains detailed information on the microscope condition during acquisition. Both .TIF and .LIF files can be directly imported by a freely available image analysis program Image J (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/, 1997-2009) using the Bio-Formats plug-in (Kevin Eliceiri, LOCI, University of Wisconsin, Madison, WI). In the following section we offer two examples to illustrate how users might begin quantitative analysis of confocal image data.
Open any .TIF or .LIF file (Fig.4 A and A') using FILE > OPEN
- Example 1: Measure the cell area of an epithelial cell (Fig. 4B to B''').
- Select AREA in ANALYZE > SET MEASUREMENTS.
- Select polygon-selection tool from the Image J toolbar, and outline the cell.
- Open ROI Manager from ANALYZE > TOOLS > ROI MANAGER and add the outline to it by pressing [control-t], which is the shortcut for adding any new "region of interest" or ROI to the ROI Manager. It is important to add this selection and save ROI-set since you can go back at a later time and make any other parameter measurements. Sets of saved ROIs (click "SAVE") can be reloaded later for further analysis.
- One can repeat step (c) with any number of cells in an image and add the ROIs to ROI Manager. Once one has sufficient ROIs recorded, Click "MEASURE" in ROI Manager. In the RESULTS window, one can now see the AREAS against the ROIs. Image dimensions have corresponding values in measurement parameters.
- Example 2: Measure the intensity of a cell membrane (Fig. 4C to C''').
- Select "Straight line" tool from the Image J toolbar, and draw a line on the image that is perpendicular to the cell membrane of interest and crosses the membrane. This straight line selection is another type of ROI and can be add to ROI Manager as above.
- Plot relative intensities in arbitrary units by ANALYZE > PLOT PROFILE. Save this as image and as list of values by clicking SAVE and LIST > FILE > SAVE AS respectively on the PLOT window.
Various quantitative measurements can be made using Image J depending on the researchers' interests and what hypotheses are being tested. Microsoft Excel (Microsoft Corporation, Seattle, WA) or Sigma Plot (Systat Software Inc., San Jose, CA) can be used to plot data graphically. Additional examples in image analysis have been presented previously7.
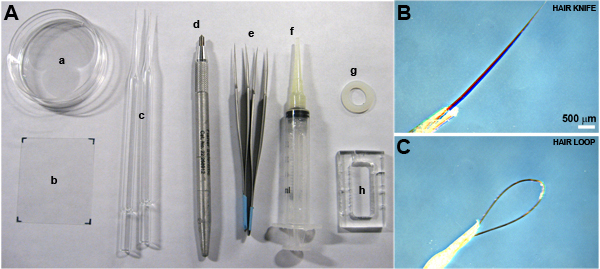 Figure 1. Required materials. A) Tools needed for microsurgery and assembly of the culture chamber. Petri dish (a), large cover slip (b), hair tools (c), diamond pencil (d), forceps (e), silicone grease "caulking gun" (f), nylon washer (g), and custom acrylic culture chamber (h). B) Close-up of the hair knife, and C) hair loop.
Figure 1. Required materials. A) Tools needed for microsurgery and assembly of the culture chamber. Petri dish (a), large cover slip (b), hair tools (c), diamond pencil (d), forceps (e), silicone grease "caulking gun" (f), nylon washer (g), and custom acrylic culture chamber (h). B) Close-up of the hair knife, and C) hair loop.
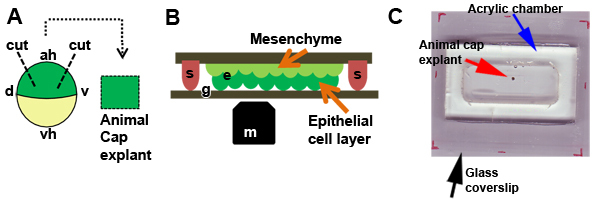 Figure 2. Excision of animal cap explants. A) Schematic of microsurgical manipulation used to remove an animal cap explant (d, dorsal; v, ventral; ah, animal hemisphere; vh, vegetal hemisphere). B) Schematic of explant (e) positioned under a coverslip fragment held in place by silicone grease (s) with the apical surface apposed to the BSA coated glass (g). The explant is positioned so the epithelial layer is closest to the microscope objective (m). C) Animal cap explant (red arrow) is cultured in DFA and housed in the acrylic chamber (blue arrow) on a large glass coverslip (black arrow) and held in place by glass coverslip fragment and silicone grease.
Figure 2. Excision of animal cap explants. A) Schematic of microsurgical manipulation used to remove an animal cap explant (d, dorsal; v, ventral; ah, animal hemisphere; vh, vegetal hemisphere). B) Schematic of explant (e) positioned under a coverslip fragment held in place by silicone grease (s) with the apical surface apposed to the BSA coated glass (g). The explant is positioned so the epithelial layer is closest to the microscope objective (m). C) Animal cap explant (red arrow) is cultured in DFA and housed in the acrylic chamber (blue arrow) on a large glass coverslip (black arrow) and held in place by glass coverslip fragment and silicone grease.
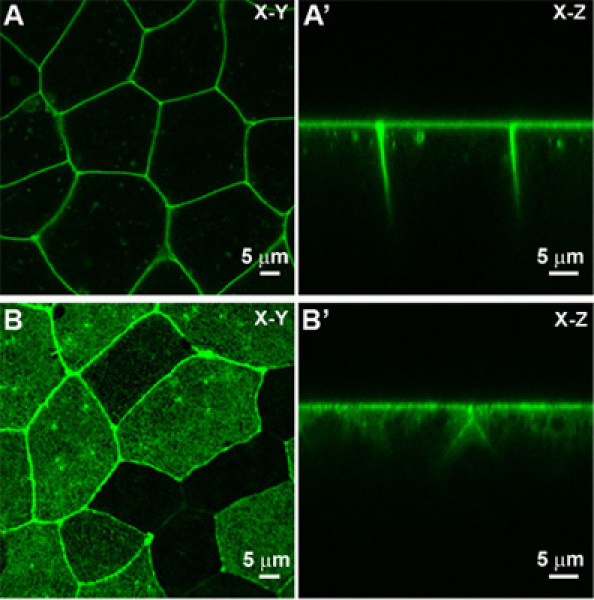 Figure 3. Epithelial cells observed using confocal microscope. A) An X-Y view and an X-Z (A') view of membrane localizing GFP (mem-GFP) labeled epithelial sheet. B) An X-Y view and X-Z view (B') of a moesin-GFP labeled F-actin apical cell cortex.
Figure 3. Epithelial cells observed using confocal microscope. A) An X-Y view and an X-Z (A') view of membrane localizing GFP (mem-GFP) labeled epithelial sheet. B) An X-Y view and X-Z view (B') of a moesin-GFP labeled F-actin apical cell cortex.
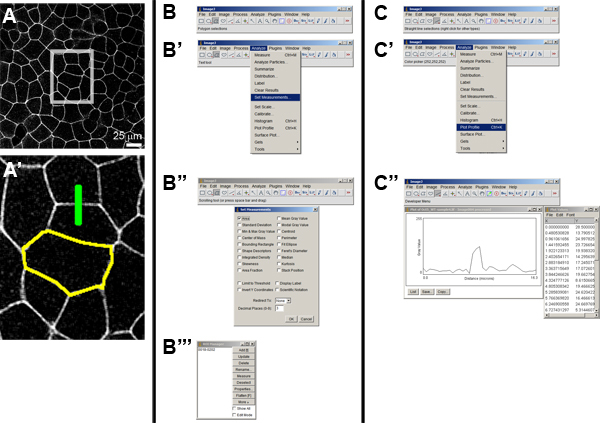 Figure 4. Image analysis using Image J. A) Original image of a membrane localizing GFP labeled cell sheet. A high resolution view (A') of the box inset in (A) shows two "regions-of-interest" or ROIs. The yellow-outline indicates the boundary of a single cell selected using the polygon tool (see panel B) and the green line indicates the path of an intensity profile across a cell-cell boundary selected using straight line tool (see panel C). B') Method to select the measurement options and choose "Area" (B''). Once the ROI is selected it can be stored in the ROI Manager (B'''). C') The method to select the measurement options for the profile took. C'') Once the straight line or segmented line tool is chosen the intensity profile along the path can be plotted with the "Plot profile" command. A list of intensity values can also be saved in a spreadsheet readable form.
Figure 4. Image analysis using Image J. A) Original image of a membrane localizing GFP labeled cell sheet. A high resolution view (A') of the box inset in (A) shows two "regions-of-interest" or ROIs. The yellow-outline indicates the boundary of a single cell selected using the polygon tool (see panel B) and the green line indicates the path of an intensity profile across a cell-cell boundary selected using straight line tool (see panel C). B') Method to select the measurement options and choose "Area" (B''). Once the ROI is selected it can be stored in the ROI Manager (B'''). C') The method to select the measurement options for the profile took. C'') Once the straight line or segmented line tool is chosen the intensity profile along the path can be plotted with the "Plot profile" command. A list of intensity values can also be saved in a spreadsheet readable form.
Discussion
We have presented experimental protocols used regularly in our lab to image dynamically changing cytoskeleton in the apical cell cortex and oscillating cell membranes of epithelial cells in Xenopus embryos. One should use these protocols as a starting point for live-imaging of epithelial cell shapes; optimization of this protocol is essential and some of the settings will change depending on the type of microscope system one has and the type of proteins one is interested in imaging. Together, the protocol steps (explant isolation, imaging and visualization of cellular and sub-cellular components, and quantification methodology) provide a comprehensive procedure to study epithelial morphogenesis.
Critical aspects to remember
It is important to prepare high-quality mRNA for micro-injection into embryos. mRNA tubes are stored on ice to avoid degradation. Once tested, the tubes are stored in -80°C in smaller aliquots to avoid multiple freeze-thaw cycles.
It is essential to inject mRNA into multiple sites to insure even distribution of mRNA over a large field of cells in the animal cap ectoderm.
Addition of antibiotic/antimycotic is essential for long term culture to discourage bacterial and fungal growth.
Care must be taken while pressing explants under the coverslip fragments since more-than-required pressure can smash the explants.
While imaging, optimal settings are essential to gather maximal data with the best possible image quality that reflects endogenous cell behaviors and protein dynamics. Choosing optimal settings comes with practice and experience.
To analyze images we strongly recommend ImageJ. ImageJ is a excellent open-source tool that can be downloaded and modified with the addition of custom-written or downloaded macros and plug-ins.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by a National Institutes of Health (R01-HD044750), a CAREER grant from the National Science Foundation, and support from the American Heart Association (Beginning Grant-in-Aid). The authors thank Davidson Lab members: H.Y. Kim, M. von Dassow and J. Zhou (for help with daily lab responsibilities), and Lin Zhang for her assistance in molecular biology and synthesizing mRNA. We acknowledge experimental efforts from all the frog labs (especially Ray Keller's and Doug DeSimone's) who have contributed to some part of this protocol, by developing and testing various methods over the years. We thank Richard Harland and John Wallingford for their kind gifts of mem-gfp and moesin-gfp plasmids respectively.
References
- Ninomiya H, Winklbauer R. Epithelial coating controls mesenchymal shape change through tissue-positioning effects and reduction of surface-minimizing tension. Nat Cell Biol. 2008;10(1):61–61. doi: 10.1038/ncb1669. [DOI] [PubMed] [Google Scholar]
- Kay BK, Peng HB. Xenopus laevis: Practical Uses in Cell and Molecular Biology. New York: Academic Press; 1991. [PubMed] [Google Scholar]
- Cross MK, Powers M. Obtaining eggs from Xenopus laevis females. J Vis Exp. 2008 doi: 10.3791/890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Au S, Pante N. Microinjection of Xenopus laevis oocytes. J Vis Exp. 2009 doi: 10.3791/1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal Tables of Xenopus laevis (Daudin) Amsterdam: Elsevier North-Holland Biomedical Press; 1967. [Google Scholar]
- Davidson LA, Ezin AM, Keller R. Embryonic wound healing by apical contraction and ingression in Xenopus laevis. Cell Motil Cytoskeleton. 2002;53(3):163–163. doi: 10.1002/cm.10070. [DOI] [PubMed] [Google Scholar]
- Joshi SD, Dassow Mvon, Davidson LA. Experimental control of excitable embryonic tissues: three stimuli induce rapid epithelial contraction. Exp Cell Res. 2010;316(1):103–103. doi: 10.1016/j.yexcr.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]