

Abstract
Background
The molecular mechanism by which catecholaminergic polymorphic ventricular tachycardia (CPVT) is induced by single amino acid mutations within the cardiac ryanodine receptor (RyR2) remains elusive. Here, we investigated mutation-induced conformational defects of RyR2 using a knock-in (KI) mouse model expressing the human CPVT-associated RyR2 mutant (S2246L; Serine to Leucine mutation at the residue 2246).
Methods and Results
All KI mice we examined produced VT after exercise on a treadmill. cAMP-dependent increase in the frequency of Ca2+ sparks was more pronounced in saponin-permeabilized KI cardiomyocytes than in WT cardiomyocytes. Site-directed fluorescent labeling and quartz microbalance assays of the specific binding of DP2246 (a peptide corresponding to the 2232–2266 region: the 2246 domain) showed that DP2246 binds with the K201-binding sequence of RyR2 (1741– 2270). Introduction of S2246L mutation into the DP2246 increased the affinity of peptide binding. Fluorescence quench assays of inter-domain interactions within RyR2 showed that tight interaction of the 2246 domain/K201-binding domain is coupled with domain unzipping of the N-terminal (1-600)/central (2000–2500) domain pair in an allosteric manner. Dantrolene corrected the mutation-caused domain unzipping of the domain switch, and stopped the exercise-induced ventricular tachycardia.
Conclusions
The CPVT-linked mutation of RyR2, S2246L, causes an abnormally tight local sub-domain/sub-domain interaction within the central domain involving the mutation site, which induces defective interaction between the N-terminal and central domains. This results in an erroneous activation of Ca2+ channel in a diastolic state reflecting on the increased Ca2+ spark frequency, which then leads to lethal arrhythmia.
Keywords: ryanodine receptor, calcium, ventricular tachycardia, sarcoplasmic reticulum
Introduction
Ryanodine receptor (RyR2), the Ca2+ release channel in the cardiac sarcoplasmic reticulum (SR), plays a key role in cardiac excitation-contraction coupling.1 A considerable body of evidence shows that RyR2 function is defective in failing hearts, causing spontaneous Ca2+ leak.2 The Ca2+ leak leads to contractile dysfunction by reducing the SR Ca2+ content, and also induces delayed after-depolarization (DAD), which ultimately leads to lethal arrhythmia.2 More than 120 RyR2 mutations have been identified in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) or arrhythmogenic right ventricular cardiomyopathy (ARVC) type 2.3 RyR2 mutations are not randomly distributed, but they cluster into 3 definable regions. These domains are designated as the N-terminal domain (a.a.1–600), the central domain (a.a. 2000–2500), and the C-terminal channel-forming domain. Since a single point mutation in any of these domains has a severe impact on the channel function, these domains must play a key role in regulating the channel function of both RyR2 and RyR1.
Chiefly based on the conformational probe approach, Ikemoto and colleagues 4–6 proposed that the N-terminal domain and the central domain interact with each other to act as the implicit on/off switch that opens and closes the channel, and the interacting domain pair was designated as a “domain switch.” Parallel assays of the conformational state of RyR2 and the functional state of Ca2+ channel have shown that zipping of the interacting domains closes the channel, and unzipping opens it. A mutation in either domain weakens the inter-domain interaction and causes domain unzipping in an otherwise resting state, which results in an erroneous activation of the channel and diastolic Ca2+ leak.4–6 Consistent with the domain switch hypothesis, single particle analysis of the 3D structure of the RyR2 molecule revealed that the N-terminal and central domains (located in domains 5 and 6 of the so-called clamp region, respectively) are in a close apposition to each other. 7,8
We have previously shown that in failing hearts defective interaction between the N-terminal domain and the central domain of RyR2 induces Ca2+ leak even under the conditions of reduced SR Ca2+ load, leading to contractile dysfunction.9 In normal hearts, these domains interact with each other to produce a zipped configuration, which stabilizes the closed state of the Ca2+ channel. However, in the failing heart, this interaction becomes loose due to oxidative stress 10 or protein kinase A (PKA)-mediated hyperphosphorylation and dissociation of FKBP12.6.9 Domain unzipping destabilizes the closed state of the channel, resulting in diastolic Ca2+ leak. We have also shown that correcting the unzipped configuration to the normal zipped state by the treatment with either K201 (JTV519) 9,11 or dantrolene12 restores normal channel gating in otherwise leaky RyR2 channels of failing hearts.
Moreover, we recently demonstrated, using a transgenic mouse model in which CPVT-type R2474S mutation is knocked in the central domain of RyR2, that this mutation caused aberrant unzipping of the domain switch regions, lowering the threshold of luminal [Ca2+] for channel activation, sensitizing the channel to PKA-dependent phosphorylation, and led to CPVT. 13 Dantrolene treatment that corrects the defective inter-domain interaction prevented aberrant Ca2+ release, thereby preventing DAD and CPVT.13, 14 These findings indicate that the defective inter-domain interaction between the N-terminal and central domains, caused by single point mutation, oxidative stress and/or PKA hyperphosphorylation, destabilizes the channel, leading to lethal arrhythmia and heart failure.
In our previous study of canine RyR2, we found that the domain switch is conformationally coupled with the interaction of sub-domains (the 2114–2149 region and the 2234–2750 region) within the central domain , playing an important role in the regulation of channel function: namely, their tight interaction (sub-domain zipping) opens the Ca2+ channel and their dissociation (sub-domain unzipping) closes the channel.11 We then demonstrated several important features of the interaction: (a) diastolic SR Ca2+ leak of failing heart happens owing to erroneous zipping of these sub-domains and channel activation in an otherwise resting state, (b) specific binding of K201 to RyR2 takes place at the 2114–2149 region, indicating that this is the K201 binding domain, and (c) the binding of K201 to this domain interferes with its interaction with the 2234–2750 region, thereby correcting aberrant domain zipping and diastolic Ca2+ leak in the failing RyR2. 11
The above findings suggest the hypothesis that the S2246L (Serine to Leucine mutation at residue 2246) CPVT mutation in the 2234–2750 region (which we call ‘2246 domain’) causes an abnormal zipping between the 2246 domain and the K201 binding domain. To test this hypothesis, we used S2246L knock-in mice. We demonstrate here that the S2246L mutation introduced into the 2246 domain causes an abnormally tight interaction with its partner domain (the K201 binding domain). Importantly, this mutation-induced tight zipping of these domains is coupled in an allosteric manner with unzipping of the domain switch. This leads to several phenotypes such as hyper-sensitized channel gating to luminal [Ca2+] and to increased diastolic Ca2+ leak, the phenotypes that are commonly observed in the transgenic mouse models carrying different knock-in CPVT mutations,15–17 including our previously investigated R2474S/+ KI model.13
Methods
An expanded Methods section is available in the Online Data Supplement.
Animals
This study conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). The care of the animals and the protocols used were in accord with guidelines laid down by the Animal Ethics Committee of Yamaguchi University School of Medicine.
Peptides used and peptide synthesis
We used the 3 domain peptides (DP), as described previously 13;
DPc10: DP2460-2495
2460GFCPDHKAAMVLFLDRVYGIEVQDFLLHLLEVGFLP2495
DP2246:DP2232-2266
2232PAMRGSTPLDVAAASVMDNNELALALREPDLEKVV2266
DP2246mut:DP2232-2266mut (Serine2246 is mutated to Leucine)
2232PAMRGSTPLDVAAALVMDNNELALALREPDLEKVV2266
Analysis of local Ca2+ release events
The local Ca2+ release events were measured in saponin-permeabilized cardiomyocytes, as described previously.13 To avoid possible side effect of CaMKII on cAMP-induced change in Ca2+ sparks, CaMKII inhibitor KN-93 (1 μmol/L) was added.
Statistics
Paired or unpaired t-test was used for statistical comparison of the data between two different situations, while ANOVA with a post hoc Scheffe's test was used for statistical comparison of concentration-dependent data. Data for each different condition were obtained in the same preparation of SR or isolated cells originated from 10–16 hearts or one heart, respectively. Then, statistical comparison of data for these preparations were made. Data are expressed as means±SE. We accepted a p value less than 0.05 as statistically significant.
Results
No appreciable change in structural and functional characteristics of S2246L/+KI mice at the resting state
The hearts of wild type (WT) and knock-in (KI) mice showed no appreciable anatomical and histological abnormalities under baseline conditions (Supplemental Figure 2A), and no appreciable changes in the expression or phosphorylation levels of any of the SR proteins examined (Supplemental Figure 2B). Furthermore, echocardiography revealed no functional differences between WT mice and KI mice (Supplemental Figure 3).
Exercise or epinephrine induces ventricular tachycardia in S2246L/+ KI mice, and dantrolene prevents tachycardia
As shown in the ECG characteristics obtained from WT and KI mice by telemetry (Supplemental Table 1), there was no statistically significant difference in baseline parameters of HR, QT interval, and QTc between WT and KI mice. Ventricular tachycardia (VT) was not observed in the resting condition (without stress) in WT or KI mice. However, either treadmill exercise or epinephrine injection induced bidirectional or polymorphic VT in all KI mice we examined, but not in any of WT mice tested (Figure 1A). In KI mice, pre-treatment with daily intra-peritoneal injection of 20mg/kg dantrolene (7–10 days) prevented the VT (Figure 1B). Moreover, dantrolene significantly increased the total running distance during treadmill (Treated; 178±55.3 m, vs Untreated; 59±10 m, P<0.01). On the other hand, pre-treatment with K201 (JTV519) was without effect on the inducible VT (Figure 1B).
Figure 1.
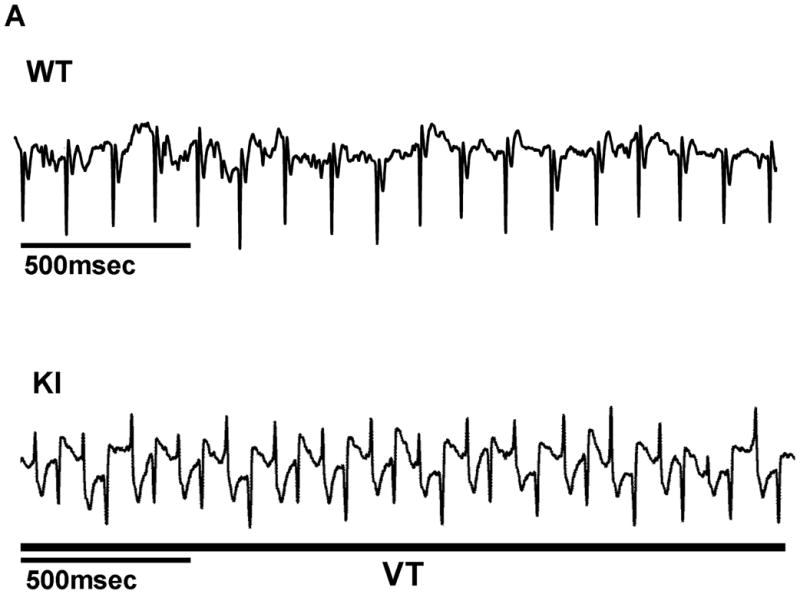

A. Representative ECG recordings in WT and KI mice. In all KI mice examined, bidirectional or polymorphic ventricular tachycardia (VT) was induced by exercise on treadmill or epinephrine injection (2 mg/kg i.p.). B, Summarized data for the effect of dantrolene or K201 on exercise-induced or epinephrine-induced VT in KI mice. Data represent means±SE of 4 hearts.
S2246L mutation reduces the threshold of SR Ca2+ load for channel activation
There was no appreciable difference in the level of PKA phosphorylation of RyR2 at Ser2808 between KI and WT cardiomyocytes, both without and with added cAMP. The added cAMP was washed away to prevent further PKA phosphorylation (Figure 2A). The frequency of Ca2+ sparks (SpF) increased with an increase of the concentration of cAMP (0.1–1 μmol/L) in both KI and WT cardiomyocytes, to a comparable degree (Figure 2B). However, because there was a considerable reduction in the SR Ca2+ content in KI cardiomyocytes, the SpF per the releasable SR Ca2+ content is significantly higher in KI cardiomyocytes compared with WT cardiomyocytes. As a result, the plot of SpF versus SR Ca2+ content shows a considerable left-shift compared with that of WT cardiomyocytes (Figure 2C). Even under the conditions where the SR Ca2+ content was progressively reduced in the presence of thapsigargin (without cAMP), an inhibitor of SERCA2a, the SR Ca2+-dependent increase of SpF was significantly higher in the KI compared with WT (Figure 2C); the increase of SpF was accompanied with the increase in both full width at half maximum (FWHM) and full duration at half maximum (FDHM) at matched SR Ca2+ content (Supplemental Table 2). The effects of cAMP on Ca2+ spark characteristics of KI cardiomyocytes are summarized in Supplemental Table 3. In the KI cardiomyocytes, the peak amplitude and FWHM decreased, and FDHM increased, suggesting a delay in RyR2 inactivation. Taken together, these data suggest that the thresholds of SR luminal [Ca2+] for channel opening and for channel inactivation decreased considerably in the KI cardiomyocytes because of the single S2246L mutation in RyR2.
Figure 2.
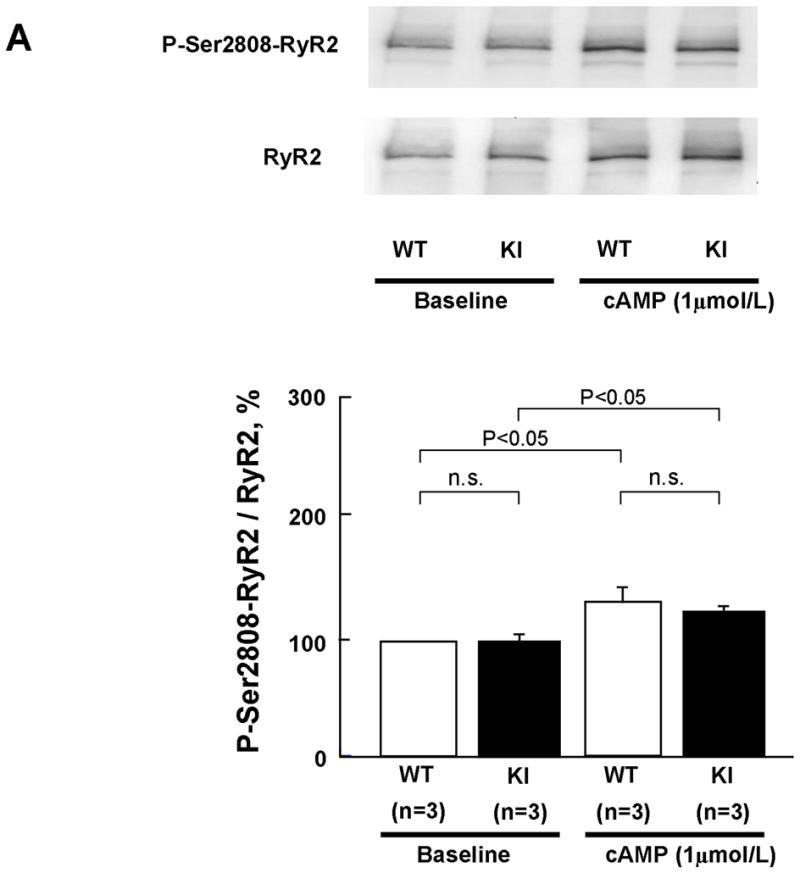





PKA phosphorylation at Ser2808 in RyR2 (P-Ser2808-RyR2) and local Ca2+ release events in saponin-permeabilized cardiomyocytes. A. Comparison of P-Ser2808-RyR2 in KI and WT cardiomyocytes. In the presence of cAMP (1 μmol/L), okadaic acid (1 μmol/L), and CaMKII inhibitor KN-93 (1 μmol/L), KI and WT cardiomyocytes were incubated in the lysis buffer, and centrifuged. Then, the supernatant fraction containing crude homogenate was used for phosphorylation assay. N: the number of cell lysate from 3–4 hearts. B, Representative line-scan images of cardiomyocytes after addition of cAMP (0.1–1 μmol/L), at 30 nmol/L [Ca2+] buffered by 0.5 mmol/L EGTA. C, The plots of SpF versus SR Ca2+ content. SR Ca2+ content was measured by addition of 10 mmol/L caffeine. To obtain the data at lower SR Ca2+ contents, thapsigargin was added to the cardiomyocytes. Then, both Ca2+ sparks and SR Ca2+ content were measured at 3, 5, and 10 minutes after the addition of thapsigargin [WT (n=4 to 5 hearts), KI (n=4 to 5 hearts)]. D, Effect of protein kinase A (PKA) phosphorylation on SpF during inhibition of SR Ca2+ uptake in WT and S2246L/+ KI saponin-permeabilized cardiomyocytes. For PKA phosphorylation of the RyR2, both cAMP (1 μmol/L) and okadaic acid (1 μmol/L) were added to the cardiomyocytes at 30 nmol/L [Ca2+] buffered by 0.5 mmol/L EGTA. Two minutes after the addition of cAMP, the added cAMP was washed away to prevent further PKA phosphorylation. Then, thapsigargin (Thap) (0.3 μmol/L) was added. One minute later, line-scanned images were obtained [WT (n=4 to 5 hearts), KI (n=4 to 5 hearts)]. E. The dependence of SpF on SR Ca2+ content in the absence or in the presence of DPc10 or dantrolene. Arrow indicates the shift of the data points in the absence or in the presence of DPc10 (50 μmol/L) or dantrolene (1 μmol/L) [WT (n=4 to 5 hearts), KI (n=4 to 5 hearts)]. F. The dependence of SpF on SR Ca2+ content in the absence or in the presence of K201 (1 μmol/L). [WT (n=4 to 5 hearts), KI (n=4 to 5 hearts)].
PKA phosphorylation modulates not only RyR2 Ca2+ release, but also SR Ca2+ uptake. Thus, we measured the SpF and SR Ca2+ content in the presence of 0.3 μmol/L thapsigargin (Figure 2D). Thapsigargin eliminated the cAMP-dependent SpF increase in both WT and KI cardiomyocytes. This suggests that the cAMP-dependent SpF increase is primarily due to the activation of SR Ca2+ uptake and subsequent increase in SR Ca2+ load.
To determine whether preventing unzipping of the domain switch (cf. Introduction) reverses the leftward shift of the SpF/SR Ca2+ content dependence seen in the KI cardiomyocytes, we used dantrolene, which was shown to bind to residues 602–620 of RyR2 and prevent both domain unzipping and abnormal Ca2+ release in failing hearts.12 Dantrolene shifted the SpF / SR Ca2+ content plot to the right resulting in a WT-like pattern (Figure 2E). However, K201, which prevented the development of heart failure,18 did not correct the leftward shift of the SpF/SR Ca2+ content dependence in KI mouse cardiomyocytes (Figure 2F; for the reason, see the Discussion section).
Probing the 2246 domain/the K201-binding domain interaction with DP2246 (a domain peptide corresponding to the 2246 domain)
In order to localize the partner domain to which DP2246 binds, we first labeled the RyR2 using the peptide-mediated site-directed methylcoumarin acetamido (MCA) labeling technique.6, 9 Then, the MCA-labeled RyR2 was subjected to tryptic digestion to analyze the degradation of the fluorescently labeled polypeptide chain by SDS-PAGE and immuno-mapping with site-specific antibodies. As shown in Figure 3A, DP2246-mediated photo-affinity labeling of the fluorescent probe MCA resulted in specific fluorescent labeling of RyR2. The presence of an excess concentration of unlabeled DP2246 (10 mmol/L) prevented DP2246-mediated MCA labeling. Ab2141 (an antibody raised against the K201 binding domain), but not non-immune IgG, prevented DP2246-mediated MCA labeling. These results indicate that DP2246 binds specifically to the K201-binding domain.
Figure 3.
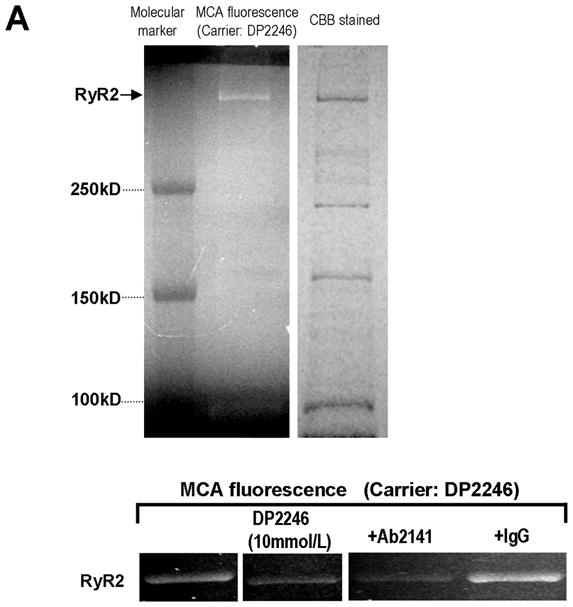

A. Site-directed fluorescence labeling of the RyR2 with MCA. A. MCA fluorescence labeling took place specifically on RyR2 (fluorescence image, left upper panel) of many SR proteins (CBB protein staining, right upper panel), by using DP2246 as the carrier. MCA fluorescence was barely detected when either an excess concentration of DP2246 (10 mmol/L) (‘cold chase’) or Ab2141 (antibody against the K201-binding domain) was added during the labeling, indicating that DP2246-mediated fluorescent labeling was site-specific. Three exogenous proteins with Mr (in kDa) 250, 150 and 100 were used as Mr markers. CBB: Coomassie Brilliant Blue.
B. Identification of the partner domain of DP2246 in the tryptic fragments of the fluorescence labeled RyR2. Using DP2246 as the site-directed carrier, MCA fluorescence labeling took place within RyR2 in SR vesicles. After tryptic digestion of the fluorescently labeled RyR2, the 60 kD fragment was detected as the shortest MCA-labeled segment of the RyR2 by Ab2141. Ab2141; antibody against K201-binding domain (epitope 2134MGKEEEKLMIRGLGDI2149). Ab12: antibody against N-terminal region (epitope6 EGEDEIQFLRTDDE19); Ab2808: antibody against phosphorylation site (epitope 2801YNRTRRISQT2810); Ab4963: antibody against C-terminal region (epitope 4959RKQYEDQLN4967). C. Identification of the partner domain of DP2246 by site-specific MCA labeling of recombinant RyR2 fragments. Of several recombinant RyR2 fragments (1–610, 741–1260, 1245–1768, 1741–2270, 2234–2750), only the fragment1741–2270 was specifically MCA-labeled.
The MCA-labeled RyR2 polypeptide chain was degraded to 210-kDa, 155-kDa, 125-kDa, and then to 60-kDa fragments, all of which were immuno-stained with Ab2141, an antibody directed against the central region (Figure 3B). Furthermore, of several recombinant RyR2 fragments, which span residues 1–2750, only the fragment 1741–2270 was specifically labeled with MCA (Figure 3C). These data suggest that DP2246 interacts with the 1741–2270 region of RyR2, which includes the previously reported 2114–2149 K201 binding domain.11
In order to further verify the DP2246-binding domain of RyR2, we used the quartz crystal microbalance (QCM) binding assay as an in vitro model of domain-domain interaction. As shown in Figure 4A, DP2246 rapidly bound to fragment 1741–2270, but not to other recombinant fragments. Ab2141, but not non-immune IgG, abolished DP2246 binding to fragment 1741–2270 (Figure 4B). This further supports the notion that DP2246 binds to the K201-binding domain (res. 2114–2149) within the 1741–2270 residue region.
Figure 4.

Screening of the binding partner of DP2246 using quartz-crystal microbalance (QCM) binding assays of recombinant RyR2 fragments. A. Time course of the binding of DP2246 to various RyR2 fragments. DP2246 was immobilized on the sensor chip surface as described in “EXPANDED MATERIALS AND METHODS”. B. Time course of the binding of DP2246 to the fragment 1741–2270 in the presence of Ab2141, an antibody against the K201 binding site, or non-immune IgG.
S2246L mutation increases the affinity of binding of DP2246 to the RyR2
As the first approach to test the hypothesis that the S2246L CPVT mutation in the 2246 domain causes an abnormal zipping between the 2246 domain and the K201 binding domain (cf. Introduction), we performed the binding assay of DP2246 and DP2246mut (DP2246 in which S2246L mutation is introduced) to RyR2 using the peptide-mediated site-directed MCA labeling technique.6, 9 The fluorescence labeling mediated by DP2246mut is also site-specific to the K201 binding domain as indicated by both cold-chase and Ab-chase experiments, but importantly the intensity of the MCA label of the RyR2 is significantly higher compared with the DP2246-mediated labeling (Figure 5A).
Figure 5.

A. Site-directed fluorescence labeling of the RyR2 with MCA by using DP2246mut as the carrier. MCA fluorescence was barely detected when either an excess concentration of DP2246/2246mut (10 mmol/L) (‘cold chase’) or Ab2141 (antibody against the K201-binding domain) was added during the labeling. B, Concentration dependence of DP2246 or DP2246mut on the binding to the RyR2. Data are means±SE of 3 experiments using 3 SR preparations from 30 hearts.
To assess the affinity of binding of DP2246 to its partner domain, we plotted the extent of peptide-mediated site-directed MCA fluorescence labeling of RyR2 as a function of the concentration of DP2246 or DP2246mut (Figure 5B). The apparent Kd of peptide binding to the K201-binding domain was 0.09 μmol/L with DP2246mut, and 0.3 μmol/L with DP2246, indicating that the S2246L mutation in the 2246 domain produced a significant enhancement of the interaction of DP2246 with its target domain, i.e. K201-binding domain.
In vivo DP2246 binding in WT and KI cardiomyocytes
To assess the peptide binding to RyR2 in the in vivo conditions, we fluorescently labeled DP2246 or DP2246mut with Oregon Green, and introduced into saponin-permeabilized cardiomyocytes. In WT cardiomyocytes, both the DP2246 binding signal and the DP2246mut binding signal were detected as green bands localized along the sarcomeres (Figure 6). As shown in the middle panel of Figure 6, DP2246mut showed higher binding affinity than DP2246 in the WT cardiomyocytes. In contrast, the fluorescence signal of both DP2246 and DP2246mut was barely detectable in the KI cardiomyocytes. The plot of the intensity of the fluorescence along the sarcomeres (see Methods) as a function of the concentration of DP2246 (or DP2246mut) (Figure 6 bottom panel) shows that in the WT cardiomyocytes, maximal binding of both peptides takes place at 3 μmol/L peptides (Kd with DP2246mut-probe, 0.08 μmol/L; Kd with DP2246-probe, 0.14 μmol/L). In the KI cardiomyocytes, however, the binding is very partial even at the maximal concentration of the peptide we tested (3 μmol/L). This suggests that the S2246L mutation leads to an abnormally strong interaction between the 2246 domain and the K201 domain, which precludes the binding of DP2246 and DP2246mut to the K201 domain of KI RyR2.
Figure 6.

Localization and the characteristics of DP2246 binding to the cellular components (SR in situ) of saponin-permeabilized WT and KI cardiomyocytes. (Top) The DP2246, fluorescently labeled with Oregon Green 514 (Molecular Probes, OR), was delivered into the cardiomyocytes. (Middle) Representative images of Oregon Green-labeled DP2246 or DP2246mut in WT and KI cardiomyocytes. (Bottom) The plot of the intensity of the fluorescence along the sarcomeres as a function of the concentration of DP2246 (open circle) /DP2246mut (closed circle)-Oregon Green. Data represent means±SE of 4 to 5 cells from 3 to 4 hearts.
The mutation-induced aberrant tight interaction of the 2246 domain/K201-binding domain in S2246L/+KI cardiomyocytes causes aberrant unzipping of the domain switch
In order to investigate whether or not the domain switch is defective in S2246L/+KI mice, we adopted the fluorescence quench technique,6, 9 which permits us to spectroscopically monitor whether the domains are zipped or unzipped.
As in our previous study,13 the N-terminal domain of the RyR2 was labeled with the fluorescent probe MCA using DPc10 as a site-directing carrier (see Suppl. Figure in Ref. 11). As shown previously, in a zipped configuration of the domain switch the attached MCA probe is inaccessible to a large size fluorescence quencher QSY-BSA conjugate; while, in an unzipped configuration, because of an opened gap between the interacting domains, the quencher becomes accessible to the attached MCA probe to quench the MCA fluorescence. In agreement with our previous study,13 the extent of fluorescence quenching (KQ: the Stern-Volmer quenching constant determined from the slope of the plot, which is a measure of the extent of domain unzipping) of the MCA increased when the WT SR was treated with a domain unzipping peptide DPc10 (Figure 7A WT, left). In the KI (S2246L/+) SR, on the other hand, the KQ showed a high value even before the addition of DPc10, which was comparable with the level that was reached by addition of DPc10 to the WT SR; and the addition of DPc10 to the KI SR produced no further increase in the KQ value (Figure 7B KI). These results suggest that S2246L mutation in the 2246 domain produced an allosteric effect on the domain switch making it unzip: the same net result as would have been produced by the domain switch mutation R2474S.
Figure 7.

Determination of the mode of inter-domain interactions in the domain switch (interacting pair of the N-terminal and central domains) and in the 2246 domain/K201 binding domain pair of WT and KI RyR2-s. The slope of the Stern-Volmer plots of the fluorescence quenching data with QSY-BSA(KQ) corresponds with the accessibility of the quencher and serves as a measure of the magnitude of domain unzipping. Effect of DPc10 or DP2246 on the KQ in WT (A) or KI (B) SR; MCA labeling was done in a site-directed manner using DPc10 or DP2246 as a carrier. The data are shown as means±SE of 5 to 6 experiments using 3 to 4 SR preparations from 30 to 40 mouse hearts.
Addition of dantrolene or K201 to the WT SR reduced the KQ, indicating that the defective, or unzipped, configuration of the domain switch that had been produced by DPc10 treatment was restored to a normal zipped configuration (Figure 7A WT). Addition of dantrolene to the KI SR also reduced the KQ, indicative of recovered normal zipped configuration (Figure 7B KI). However, K201 was without effect on KI SR (Figure 7B KI), presumably owing to the fact that the interaction between the 2246 domain and the K201 binding domain was so tight that K201 was inaccessible to the drug binding domain.
The in vitro and in vivo assays of DP2246 binding to RyR2 (Figures 5 and 6) and the fluorescence quench assay of the domain switch (Figure 7B KI) suggest that the S2246L mutation causes aberrant zipping between the two sub-domains within the central domain (namely, the 2246 domain and the K201 binding domain), and this produced unzipping of the domain switch in an allosterically coupled manner. In order to test this postulated conformational coupling between the two regions (namely, domain zipping of the 2246 domain/K201 binding domain pair, and reciprocally coupled domain unzipping of the domain switch), we performed site-specific MCA labeling of the K201 domain of the WT RyR2. We then performed the fluorescence quench assay using QSY-BSA following the same principle as described above. As shown in Figure 7A WT (right), unzipping of the domain switch by DPc10 produced zipping of the 2246 domain/K201 domain pair, as evidenced by a significant reduction in the KQ. Addition of dantrolene (50μM), which completely restored normal zipped state of domain switch (Figure 7A WT, left), produced no appreciable effect on the 2246 domain/K201 binding domain interaction (Figure 7A WT, right). However, K201 (1μmol/L) completely restored normal unzipped state of the 2246 domain/K201 binding domain pair from the DPc10-induced zipped state (Figure 7A WT, right).
In the KI RyR2, DP2246 was virtually inaccessible to the K201 binding domain because of tight interaction of the in vivo 2246 domain with the K201 binding domain as described above. For this reason we could not perform site-specific fluorescent labeling for the fluorescence quench assay of the 2246 domain/K201 binding domain interaction. However, in the light of the data shown in Figure 7B KI, we assume that the addition of DPc10 to the KI RyR2 would produce no further change, because unzipping of the domain switch and the coupled zipping of the 2246 domain/K201 binding domain pair would have already taken place.
FKBP12.6 binding to the RyR2 remains unaltered in KI RyR2 mice
According to several reports19, 20 dissociation of RyR2-bound FKBP12.6 and resultant channel de-stabilization may be involved in both heart failure and lethal arrhythmia. We examined whether the CPVT mutation produced any effect on the RyR2-bound FKBP12.6 using a pull-down assay. As shown in Supplemental Figure 4, there was no significant difference in the amount of RyR2-bound FKBP12.6 between WT and KI mice.
Discussion
A considerable body of evidence accumulated over recent years suggests that mutation-linked RyR2 defects cause SR Ca2+ leak, which triggers DAD and ultimately leads to CPVT.2 Recently, several groups developed knock-in mouse models harboring several different point mutations of RyR2 corresponding to the CPVT mutations of human patients (R4496C,15 R176Q,16 and R2474S13, 17), and have shown that these mutations produce phenotypes of CPVT similar to those of human patients. The fact that these point mutations produce almost identical phenotypes of CPVT regardless of their locations within the RyR2, suggests that a mutation introduced into a particular location produces a global impact on the channel regulation mechanism, e.g. by mediation of defective inter-domain interaction in its local area first, and then by coupled global conformational change of the RyR2. However, little work has been done in this new area of exploration.
The important new concept deduced from the present study, summarized in Figure 8, is that the S2246L mutation that is introduced into the region of RyR2 different from that of the aforementioned R2474S mutation causes an abnormally tight interaction (zipping) of a new interacting domain pair, namely two sub-domains [the 2246-domain /the K201-binding domain (2114–2149)] located in the central domain, and that this inter-subdomain interaction is coupled with unzipping of the domain switch in an allosteric manner. Thus, the S2246L mutation ends up with aberrant unzipping of the domain switch, then produced the same type of channel dysfunction and CPVT phenotypes as those produced by the R2474S mutation. This concept is supported by the following pieces of evidence. First, DP2246, a peptide corresponding to the 2246 domain (2232–2266), was found to bind with the 2114–2149 region of RyR2, the previously identified K201-binding domain.11 This suggests that there is an interaction between the 2246 domain and the K201-binding domain. As shown in the experiment with WT SR, the binding affinity of DP2246mut to RyR2 was much higher than that of DP2246, suggesting that the mutation induces a tight zipping of this interacting sub-domain pair. In support of this view, DP2246-Oregon Green was not accessible to its binding site in the KI cardiomyocytes, although it was accessible in the WT cardiomyocytes. Second, DPc10, the central domain peptide that binds to the N-terminal domain and unzips the domain switch,9, 13 induced domain zipping between the 2246 domain and its partner K201-binding domain, indicating again that reciprocal conformational changes in the two regions are tightly coupled. Importantly, in the KI RyR2, DPc10 was without effect; this is because the domain switch had already been unzipped owing to the allosteric coupling with the tightly zipped 2246 domain/K201 binding domain pair. Third, the S2246L mutation produced the identical CPVT phenotypes as those produced by the R2474S mutation. Thus, the threshold of SR Ca2+ load for channel activation (determined as an increased Ca2+ sparks) was markedly reduced in both the S2246L and the R2474S KI cardiomyocytes. It is worth noting that the same phenotype could be reproduced in the WT cardiomyocytes by experimentally unzipping the domain switch with DPc10. Thus, the present study provides a preliminary evidence for a more generalized hypothesis that critical mutation in a particular area of the RyR2 causes defective inter-domain interaction in its vicinity, which is then linked to a global conformational change that leads to channel activation by mediation of the allosteric conformational coupling mechanism. This would account for the fact that most of all the CPVT mutations in different locations of RyR2 investigated in transgenic mouse models of CPVT have produced virtually identical type of channel disorder and CPVT phenotypes.13, 15–17 One of the three mutations investigated in the literature of the transgenic mouse models of CPVT, R4496C,15 is localized in the ‘I’ domain (3772–4610), whose steady interaction with the trans-membrane channel region seems to play an important role in normal channel regulation.21, 22 On the basis of this new expanded concept, we postulated that the I-domain/channel domain interaction is coupled with the conformational changes in the domain switch and the 2246 domain/K201-binding domain pair, and mutation-caused defective inter-domain interaction in any of these regions is transmitted to the channel as a pathogenic signal.
Figure 8.

Hypothetical model showing how CPVT-associated mutation causes a drastic change in the channel function by mediation of defective inter-domain interactions within RyR2.
In the ‘normal state’ of RyR2, tight interaction between the N-terminal domain and the central domain (zipped domain switch) is coupled with a loose interaction (unzipping) between the two sub-domains of the central domain [the 2246 domain (S2246L mutable region) and the K201-binding domain], then the closed state of the channel is maintained. In the ‘destabilized state’, the S2246L mutation in the 2246 domain induces an abnormally tight interaction of the S2246 domain/K201-binding domain pair. This produces domain unzipping of the N-terminal domain/central domain pair (the domain switch), and erroneous activation of the channel results in diastolic Ca2+ leak. Addition of DPc10, which interferes with a tight interaction between the N-terminal and central domains, to the wild type RyR2 produces domain unzipping of the domain switch, mimicking the situation caused by CPVT mutation (R2474S, S2246L).
One of the most important aspects in the present study is the finding that dantrolene corrected defective unzipped state of the domain switch (Figure 7), and then inhibited exercise-induced ventricular tachycardia in the S2246L KI mice (Figure 1). Since the dantrolene binding site is localized in the 600–620 residue region of the N-terminal domain of RyR2,12 this indicates that dantrolene binding to its binding site corrected defective unzipping of the interacting N-terminal domain/central domain pair that had been produced by the S2246L mutation in the 2246 domain. As mentioned above, the mutation in the 2246 domain tightens its interaction with the K201-binding domain, and this local conformational change causes domain unzipping in the domain switch by the mechanism of conformational coupling between these two regions. Conversely, dantrolene-mediated correction of aberrant unzipped configuration of the domain switch restores a normal stabilized state of the channel and stops ventricular tachycardia, even though the drug produced virtually no effect on the 2246 domain/K201 binding domain interaction. We have previously shown in the canine heart failure model that dantrolene corrects defective unzipping between the N-terminal domain and the central domain to a normal zipped state, and prevents the development of heart failure inducible by ventricular pacing.12 Thus, it appears that defective inter-domain interaction of the domain switch is a source mechanism underlying CPVT and heart failure and provides a key target of therapeutic treatment.
Although the R2474S and S2246L mutations produce basically identical impacts on the structure and function of the RyR2 as described above, we must point out that there are delicate differences in the two aspects: (a) the response to PKA phosphorylation, and (b) pharmacological effect of K201. (a) We previously reported that the R2474S mutation caused a partial unzipping of the domain switch, and upon PKA phosphorylation at Ser2808 of RyR2, the domain switch was unzipped to a ‘full’ extent.13 The S2246L/+ KI RyR2, on the other hand, showed a nearly maximal extent of domain unzipping under baseline conditions, accompanied with increased Ca2+ spark frequency with reduced SR Ca2+ content; consequently, PKA phosphorylation produced no further domain unzipping and hence no further effect on Ca2+ spark frequency (Figure. 2D). (b) As we demonstrated previously in the studies of canine heart failure model,11 K201 binding to its binding domain interfered with the interaction between the K201-binding domain and the 2246 domain, and corrected channel dysfunctions of RyR2 of the failing heart. In the R2474S/+ KI mouse model, K201 as well as dantrolene suppressed PKA phosphorylation-dependent channel activation (Uchinoumi et al., unpublished data). In the S2246L/+ KI mouse model, however, K201 showed no appreciable corrective effect on both domain switch unzipping and channel activation (Figures 2F, 7), although dantrolene produced the expected effects. These differences outlined in (a) and (b) may be accounted for by the fact that the S2246L mutation causes an excessively tight interaction of the K201 binding domain/2246 domain pair, which results in (a) facilitated unzipping of domain switch by the conformational coupling between the two regions, and results in (b) the inaccessibility of the K201 binding domain for the drug binding owing to its tight interaction with the 2246 domain.
Taken together, conformational coupling of different regions of RyR2 may explain the fact that mutations in different areas of the receptor produce a similar phenotype. However, in view of non-identical effects of the two types of potential therapeutic agents described here, differences in the mode of inter-domain interaction in those different areas, to which dantrolene and K201 bind, may result in the observed differences in their therapeutic efficacy.
It should also be mentioned that the present studies do not include the potential contribution of CaMKII, although the use of KN-93 could be complicated by off-target effects that are unrelated to CaMKII activity, based on similar experience with KN-93 on other ion channels. A further investigation is clearly needed to assess the role of CaMKII on mutation-linked local Ca2+ events.
In conclusion, introduction of a human CPVT mutation S2246L into the mouse RyR2 induces aberrant activation of channel gating by forming abnormally tight domain-domain interaction between the two sub-domains located in the central domain: the S2246L mutable domain (res. 2232–2266) and the K201-binding domain (res. 2114–2149). This produces a defective domain unzipping between the N-terminal (res. 1–600) domain and the central (res. 2000–2500) domain owing to the allosteric conformational coupling between the two sets of interacting domain pairs. The coupled conformational changes in these regions trigger diastolic Ca2+ release and lethal arrhythmia. Dantrolene treatment corrected the defective inter-domain interaction and prevented aberrant Ca2+ release and CPVT, indicating that correction of conformational disorder of the RyR2 is a new therapeutic strategy of CPVT
Supplementary Material
Clinical Perspective.
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited disease characterized by stress- or exercise- induced polymorphic ventricular tachycardia, frequently leading to sudden cardiac death. A considerable body of evidence accumulated over recent years suggests that mutation-linked cardiac ryanodine receptor (RyR2) defects cause Ca2+ leak from sarcoplasmic reticulum, which triggers delayed afterdepolarization and ultimately leads to CPVT. However, the underlying mechanism, by which a single mutation in such a large molecule causes drastic effects on the channel function, remains unresolved. Here we report that introduction of a human CPVT mutation S2246L (Serine to Leucine mutation at residue 2246) into the mouse RyR2 induces aberrant activation of channel gating by forming abnormally tight domain-domain interaction between the S2246L mutable domain (res. 2232–2266) and the K201-binding domain (res. 2114–2149). This produces more global conformational change in the RyR2: namely, an aberrant domain unzipping between the N-terminal (res. 1–600) domain and the central (res. 2000–2500) domain owing to the allosteric conformational coupling mechanism. The coupled conformational changes in these local and global inter-domain interactions trigger diastolic Ca2+ release and lethal arrhythmia. Pharmacological correction of the defective inter-domain interactions can stop the aberrant Ca2+ release and lethal arrhythmia. These results provide a new pathogenic mechanism of CPVT and a novel therapeutic strategy against CPVT.
Acknowledgments
Sources of Funding
This work was supported by grants-in-aid for scientific research from The Ministry of Education in Japan (grant Nos. 20390226 to MY, 20590868 to TY, 20591805 to SK), Takeda Science Foundation to MY, and grant from the National Heart, Lung and Blood Institutes (HL072841 to NI). The authors declare no competing financial interests.
Footnotes
Disclosures
None.
References
- 1.Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol. 2004;37:417–429. doi: 10.1016/j.yjmcc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 2.Clusin WT. Calcium and cardiac arrhythmias: DADs, EADs, and alternans. Crit Rev Clin Lab Sci. 2003;40:337–75. doi: 10.1080/713609356. [DOI] [PubMed] [Google Scholar]
- 3.Thomas NL, Maxwell C, Mukherjee S, Williams AJ. Ryanodine receptor mutations in arrhythmia: The continuing mystery of channel dysfunction. FEBS Lett. 2010;584:2153–2160. doi: 10.1016/j.febslet.2010.01.057. [DOI] [PubMed] [Google Scholar]
- 4.Ikemoto N, Yamamoto T. Regulation of calcium release by interdomain interaction within ryanodine receptors. Front Biosci. 2002;7:d671–683. doi: 10.2741/A803. [DOI] [PubMed] [Google Scholar]
- 5.Ikemoto N. Intra-molecular domain-domain interaction: a key mechanism for calcium channel regulation of ryanodine receptors. In: Wehrens XHT, Marks AR, editors. 2004 In Ryanodine Receptors:Structure, Function and Dysfunction in Clinical Disease. Springer; New York, USA: pp. 53–65. [Google Scholar]
- 6.Yamamoto T, El-Hayek R, Ikemoto N. Postulated role of interdomain interaction within the ryanodine receptor in Ca2+ channel regulation. J Biol Chem. 2000;275:11618–11625. doi: 10.1074/jbc.275.16.11618. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z, Wang R, Zhang J, Chen SR, Wagenknecht T. Localization of a disease-associated mutation site in the three-dimensional structure of the cardiac muscle ryanodine receptor. J Biol Chem. 2005;280:37941–37947. doi: 10.1074/jbc.M505714200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang R, Chen W, Cai S, Zhang J, Bolstad J, Wagenknecht T, Liu Z, Chen SR. Localization of an NH2-terminal disease-causing mutation hot spot to the "clamp" region in the three-dimensional structure of the cardiac ryanodine receptor. J Biol Chem. 2007;282:17785–17793. doi: 10.1074/jbc.M700660200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oda T, Yano M, Yamamoto T, Tokuhisa T, Okuda S, Doi M, Ohkusa T, Ikeda Y, Kobayashi S, Ikemoto N, Matsuzaki M. Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation. 2005;111:3400–3410. doi: 10.1161/CIRCULATIONAHA.104.507921. [DOI] [PubMed] [Google Scholar]
- 10.Mochizuki M, Yano M, Oda T, Tateishi H, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J Am Coll Cardiol. 2007;49:1722–32. doi: 10.1016/j.jacc.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto T, Yano M, Xu X, Uchinoumi H, Tateishi H, Mochizuki M, Oda T, Kobayashi S, Ikemoto N, Matsuzaki M. Identification of target domains of the cardiac ryanodine receptor to correct channel disorder in failing hearts. Circulation. 2008;117:762–772. doi: 10.1161/CIRCULATIONAHA.107.718957. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi S, Yano M, Suetomi T, Ono M, Tateishi H, Mochizuki M, Xu X, Uchinoumi H, Okuda S, Yamamoto T, Koseki N, Kyushiki H, Ikemoto N, Matsuzaki M. Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing inter-domain interactions within the ryanodine receptor. J Am Coll Cardiol. 2009;53:1993–2005. doi: 10.1016/j.jacc.2009.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uchinoumi H, Yano M, Suetomi T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res. 2010;106:1413–1424. doi: 10.1161/CIRCRESAHA.109.209312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobayashi S, Yano M, Uchinoumi H, Suetomi T, Susa T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Yamamoto T, Matsuzaki M. Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/+) knock-in mouse model. Circ J. 2010;74:2579–84. doi: 10.1253/circj.cj-10-0680. [DOI] [PubMed] [Google Scholar]
- 15.Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 16.Kannankeril PJ, Mitchell BM, Goonasekera SA, Chelu MG, Zhang W, Sood S, Kearney DL, Danila CI, De Biasi M, Wehrens XH, Pautler RG, Roden DM, Taffet GE, Dirksen RT, Anderson ME, Hamilton SL. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci U S A. 2006;103:12179–12184. doi: 10.1073/pnas.0600268103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, Lederer WJ, Kass RS, Morley G, Marks AR. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–2245. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yano M, Kobayashi S, Kohno M, Doi M, Tokuhisa T, Okuda S, Suetsugu M, Hisaoka T, Obayashi M, Ohkusa T, Kohno M, Matsuzaki M. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation. 2003;107:477–84. doi: 10.1161/01.cir.0000044917.74408.be. [DOI] [PubMed] [Google Scholar]
- 19.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–76. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 20.Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayashi S, Hisamatsu Y, Yamamoto T, Kohno M, Noguchi N, Takasawa S, Okamoto H, Matsuzaki M. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131–2136. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- 21.George CH, Jundi H, Thomas NL, Scoote M, Walters N, Williams AJ, Lai FA. Ryanodine receptor regulation by intramolecular interaction between cytoplasmic and transmembrane domains. Mol Biol Cell. 2004;15:2627–2638. doi: 10.1091/mbc.E03-09-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.George CH, Jundi H, Walters N, Thomas NL, West RR, Lai FA. Arrhythmogenic mutation-linked defects in ryanodine receptor autoregulation reveal a novel mechanism of Ca2+ release channel dysfunction. Circ Res. 2006;98:88–97. doi: 10.1161/01.RES.0000199296.70534.7c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.