

Abstract
Sleep remains one of the least understood phenomena in biology – even its role in synaptic plasticity remains debatable. Since sleep was recognized to be regulated genetically, intense research has launched on two fronts: the development of model organisms for deciphering the molecular mechanisms of sleep and attempts to identify genetic underpinnings of human sleep disorders. In this Review, we describe how unbiased, high-throughput screens in model organisms are uncovering sleep regulatory mechanisms and how pathways, such as the circadian clock network and specific neurotransmitter signals, have conserved effects on sleep from Drosophila to humans. At the same time, genome-wide association (GWA) studies have uncovered ~14 loci increasing susceptibility to sleep disorders, such as narcolepsy and restless leg syndrome. To conclude, we discuss how these different strategies will be critical to unambiguously defining the function of sleep.
Sleep remains one of the big mysteries in biology. As a state that seemingly freezes all productive activity and puts animals in danger of being caught by predators, sleep must serve an important purpose because it has survived many years of evolution. Nevertheless, the function of sleep and the molecular processes that produce the need to sleep both remain elusive (Frank, 2006; Mignot, 2008). In the past decade, researchers have made progress in addressing fundamental questions regarding sleep, and several clinical centers have even established sleep as an independent medical discipline. Major advances include the identification of molecules regulating sleep (Allada and Siegel, 2008; Andretic et al., 2008a; Cirelli, 2009; Crocker and Sehgal, 2010), and the realization that sleep disorders are extremely common and numerous. These disorders include insomnia, breathing disturbances during sleep (i.e., sleep apnea), movement disorders during sleep (i.e., Restless leg syndrome, Periodic leg movements) and sleep-wake state dissociation disorders (i.e., narcolepsy, Rapid Eye Movement (REM) sleep Behavior Disorder, sleep walking).
It is now clear that sleep is genetically controlled. Although environmental factors can impact the duration and intensity of sleep, genetic regulation is borne out by the heritability of sleep traits (Ambrosius et al., 2008; De Gennaro et al., 2008), the identification of specific genetic polymorphisms that affect these traits (Maret et al., 2005; Tafti et al., 2003), and the existence of familial sleep disorders. Genetic model systems - zebrafish, fruit flies and worms- were recently developed for studying sleep, and they are starting to reveal the molecular underpinnings of sleep (Allada and Siegel, 2008; Andretic et al., 2008a; Cirelli, 2009; Crocker and Sehgal, 2010). Some researchers may question the relevance of these model organisms for mammalian sleep. However, we contend that the function and regulation of sleep are likely conserved through evolution, and thus, it would be strange to restrict sleep research to only a few species. For example, some would argue that the worm sleep model, which consists of developmental periods of low activity (i.e., quiescence), is dramatically different from human sleep, but we note that characteristics of sleep vary greatly even among different mammalian species. Indeed, the genetic model systems for studying sleep may not recapitulate all aspects of human sleep, but the prediction is that some key features will be conserved. As we describe in this Review, molecular and genetic studies in these models systems are, in fact, beginning to uncover regulatory mechanisms underlying sleep, which are conserved from worms to mammals.
Molecular insights from animal models
The idea of using model systems to understand a biological process of interest is clearly not new. However, until about a decade ago, studies of sleep were primarily restricted to a few mammalian and avian species. This restriction was partially because sleep was defined on the basis of altered brain electrical activity, recorded through electroencephalograms (EEGs), and this definition was not easily applied to other animals. EEGs reveal three major states of behavior: wake, Rapid Eye Movement (REM) sleep, and Non-REM (NREM sleep). In humans, REM and NREM sleep occur in 90-minute cycles through a night of sleep. NREM sleep is divided into stages 1–3, which together with REM constitute the normal “sleep architecture.” Furthermore, human sleep is mostly consolidated into a single period during the night. This phenomenon is observed in only a few other mammals that, compared with humans, have less consolidated sleep and wake periods, which alternate during the day and night. Slow wave sleep is the deepest stage of sleep, and this occurs during stage 3 of NREM. Many brain areas are active during REM sleep; thus, the quiescence in neural activity typically associated with sleep actually occurs during NREM sleep.
Although the EEG definition of sleep, which is based upon electrical activity patterns at the cortical level, precluded its study in animals that do not have a well defined cortex, pioneering efforts of a few researchers identified sleep-like states in several species of fish, reptiles, amphibians, and even some invertebrates, such as cockroach, bees, and octopus (Campbell and Tobler, 1984). These researchers proposed specific behavioral criteria to define sleep, but such practice was not widely accepted. What eventually changed the field was the realization that other fields had made rapid progress using simple animal models (Hendricks et al., 2000b). In particular, circadian biology was often cited as an example of a field in which molecular mechanisms identified in flies and fungi turned out to be conserved in humans (Hendricks et al., 2000b). Thus, sleep researchers developed simple animal models using primarily the criteria of a sleeplike state proposed originally by Campbell and Tobler (Campbell and Tobler, 1984). According to these criteria, a sleeplike state is: (1) A reversible state during which voluntary movements do not occur; (2) Controlled by a circadian clock; (3) Accompanied by an increase in arousal threshold, such that stronger sensory stimuli are required to elicit a response from the animal; (4) Controlled by a homeostatic system that ensures adequate levels of the state. It is well known that sleep deprivation is followed by a compensatory increase in sleep, or sleep rebound, which reflects the essential nature of sleep.
We now know that fish and fruit flies display periods of rest at night, which satisfy behavioral and physiological criteria for sleep (Hendricks et al., 2000a; Prober et al., 2006; Shaw et al., 2000; Yokogawa et al., 2007). Likewise, criteria for sleep are met by a quiescent state in worms –lethargus- although this occurs during development in conjunction with larval molts rather than as a 24-hour rhythm in adults (Raizen et al., 2008). Interestingly, the larval molts, and therefore lethargus, are regulated by the worm ortholog of the circadian clock gene, period (per), which regulates the timing of sleep in other organisms. This raises the intriguing possibility that lethargus is a primordial sleep state regulated by genes of the circadian clock but occurring in a developmental context. Synapses are formed during lethargus (Hallam and Jin, 1998; White et al., 1978), which is also consistent with a proposed function of sleep.
With genetic model systems now available, assays for sleep have shifted from measuring cortical electrical activity (EEGs) to direct monitoring of rest and activity behavior. Video recordings can monitor many behavior states relatively easily, whereas “beam-break assays” can monitor locomotor activity (Hendricks et al., 2000a; Prober et al., 2006; Shaw et al., 2000; Yokogawa et al., 2007). Electrophysiological recordings of fly brains have revealed how the fly sleep state correlates with specific electrophysiological characteristics (Nitz et al., 2002; van Swinderen et al., 2004), but such recordings are clearly not practical for high throughput, or even day-to-day experiments. Even in the mouse (the preferred mammalian model for genetic approaches), researchers are starting to rely upon measurements of behavior to assay sleep instead of electrophysiological measurements (Pack et al., 2007).
Importantly, these behavioral assays, used in different model systems, are corroborating a role for sleep-regulating molecules identified through more traditional approaches, and they are also identifying new components. Here we review the major classes of molecules identified thus far, focusing particularly on the findings derived from the newer models for sleep- fish, flies and worms. For more details on the molecular analysis in mammals, we direct the reader to two excellent reviews (Andretic et al., 2008a; Cirelli, 2009)
Neurotransmitter/neuropeptide systems
Regulation of sleep by various neurotransmitters was discovered, before the advent of modern genetic technologies, through pharmacological methods. Adenosine has long been touted as a major sleep-promoting molecule that acts primarily in the mammalian basal forebrain. Although there have been some challenges to this idea, the hypothesis nonetheless prevails (Bjorness and Greene, 2009). Wake-promoting effects of caffeine are thought to be mediated by its antagonistic action on adenosine receptors (Basheer et al., 2004). Indeed, mice mutant for the A2A adenosine receptor show deficits in their response to caffeine (Huang et al., 2005). However, mutants of other adenosine receptors show limited effects on sleep. Bjorness et al. (2009) found that disrupting the A1 receptor only in the central nervous system reduces slow wave activity (an electrophysiological measure thought to reflect sleep drive) in response to sleep restriction, but it has no effect on baseline sleep. These findings suggest that adenosine is one of many neurotransmitters that regulate sleep, rather than being the dominant regulator (Bjorness and Greene, 2009). Adenosine and caffeine have similar effects on Drosophila sleep as they do on mammalian sleep, and the Drosophila response to caffeine is attenuated by decreased signaling through the dopamine D1 receptor or reduced protein kinase A (PKA) activity (Andretic et al., 2008b; Wu et al., 2009). Surprisingly, the single known adenosine receptor in Drosophila is not required for wake-promoting effects of caffeine (Wu et al., 2009). Although this may be indicative of different mechanisms driving the response to caffeine (perhaps the inhibition of a phosphodiesterase, another known target of caffeine), one cannot exclude the possibility that other, unidentified adenosine receptors exist in Drosophila.
Other neurotransmitters implicated in mammalian sleep are: histamine, dopamine, acetylcholine, and norepinephrine, all of which promote wakefulness; and, GABA (gamma-Aminobutyric acid), which promotes sleep (Andretic et al., 2008a; Cirelli, 2009). Effects of serotonin are somewhat complicated; although it suppresses REM sleep, its effects on NREM are unclear and may even be stimulatory (Crocker and Sehgal, 2010). Genetic analysis in the mouse generally supports roles for these neurotransmitters in regulating sleep, although their effects are sometimes small and complicated, perhaps due to redundancy and compensation.
In Drosophila, dopamine and octopamine, which acts similarly to norepinephrine, have robust wake-promoting effects, whereas GABA and serotonin promote sleep (Agosto et al., 2008; Andretic et al., 2005; Crocker and Sehgal, 2008; Yuan et al., 2006). Analysis of the cellular circuitry underlying these effects is starting to reveal some interesting features. Dopamine invokes two different types of arousal, a startle response and normal wakefulness, and these are mediated by the same receptor but in different cellular loci (Lebestky et al., 2009). Wake-promoting octopamine is released by neurons in the dorsal part of the fly brain, and it acts through the octopamine receptor OAMB located in neuroendocrine cells that produce Drosophila insulin-like-peptide (Dilp2) (Crocker et al., 2010). Many sleep-related effects of serotonin and dopamine are mediated by anatomical structures called the mushroom bodies (MBs), which are also independently implicated in sleep (Joiner et al., 2006; Pitman et al., 2006). Thus, the dopamine D1 receptor acts in mushroom bodies to modulate the response to caffeine and to prevent learning impairments induced by sleep deprivation, whereas serotonin acts through the d5-HT1A receptor in mushroom bodies to promote sleep (Andretic et al., 2008b; Seugnet et al., 2008; Yuan et al., 2006). Finally, a major target of sleep-promoting GABA is the large ventral lateral neurons (lLNvs) (Parisky et al., 2008). These are best known for their expression of circadian clock genes, although they do not appear to have a function in free-running circadian rhythms (Nitabach and Taghert, 2008). Instead, the lLNvs promote arousal in response to light (Shang et al., 2008; Sheeba et al., 2008). GABA signaling, through the Resistance to dieldrin (Rdl) receptor, likely inhibits these neurons, allowing sleep to occur.
Pharmacological studies in zebrafish have also implicated many of the neurotransmitters that regulate sleep in flies and mammals (Rihel et al., 2010). These studies highlight the power of the fish system for identifying small molecules that affect sleep via high-throughput screens. Small molecules can be added to the water used to house the fish, allowing easy delivery and access. In addition, many different populations of fish, each treated with a different compound, can be assayed simultaneously through video recording. Through such a screen, Rihel et al. identified wake-promoting effects of β-adrenergic agonists, which is consistent with the Drosophila and mammalian data discussed above. Interestingly, as in Drosophila, selective serotonin reuptake inhibitors (SSRIs) decreased wake in zebrafish. These pharmacological approaches, together with the ease of high-throughput screening in flies and fish, may allow for more clear-cut answers regarding the role of individual sleep-regulating components.
Neuropeptides also play a large role in the regulating sleep, the best known being the hypocretins/orexins (Sakurai, 2007). These neuropeptides underlie the sleep disorder, narcolepsy, as described below in the section on human sleep genes. A sleep-regulating role for hypocretins is conserved in zebrafish (Faraco et al., 2006; Prober et al., 2006). Although orthologs of these molecules have not been found in flies, a different neuropeptide may function in an analogous fashion (Parisky et al., 2008). This peptide, pigment-dispersing factor (PDF), is secreted by central clock cells, ventral lateral neurons, in the fly brain. The small LNVs drive circadian rhythms in constant darkness, but the lLNVs are required for light-mediated arousal, which appears to depend upon PDF (Parisky et al., 2008). Thus, PDF may function in flies as hypocretin does in mammals, as a wake-promoting peptide secreted by neurons whose activity is suppressed during sleep by inhibitory neurotransmitters such as GABA (Figure 1).
Figure 1. An example of a conserved mechanism underlying sleep.
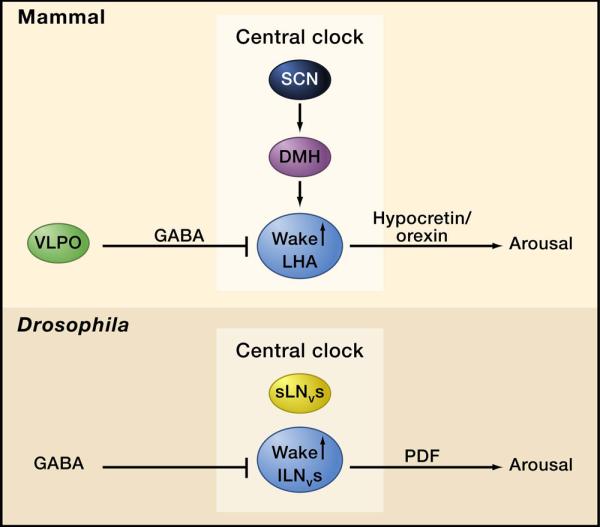
In both Drosophila and mammals, an arousal promoting peptide (PDF and hypocretin, respectively) is secreted by cells within, or in the vicinity of, the central clock network. In mammals, hypocretin-producing neurons in the lateral hypothalamus receive circadian inputs from the central clock in the suprachaismatic nucleus (SCN) via the Dorsomedial Hypothalamus (DMH). They are inhibited by GABAergic inputs from the ventrolateral preoptic (VLPO) area. In Drosophila, the large ventral lateral neurons (lLNvs) are part of the clock network although they are not required for free-running circadian rhythms. Instead they mediate light-driven arousal, at least in part through the release of PDF. As in mammals, GABAergic inputs to these neurons promote sleep.
Some molecules are required to regulate sleep under specific conditions. For instance, Drosophila sex peptide, which is secreted in the male seminal fluid, accounts for the decreased sleep in females following copulation (Isaac et al., 2010). Whereas virgin females display a robust afternoon siesta, similar to that seen in male flies, mated females have less daytime sleep presumably because they need to perform more foraging and to identify sites for egg-laying. As in flies, gender differences in mammalian sleep depend upon gonad function (Zimmerman et al., 2006).
Steroid hormones regulate many biological processes, and sleep is no exception. The Drosophila steroid hormone, ecdysone, promotes sleep (Ishimoto and Kitamoto, 2010), as does the naturally occurring neuroactive steroid, 3alpha,5alpha-tetrahydrodeoxycorticosterone, in mammals (Muller-Preuss et al., 2002). Although the relevance of such regulation is not known, the mechanism of action in mammals could involve stimulation of GABA receptors (Muller-Preuss et al., 2002).
Intracellular signaling molecules
Given the important role for neuropeptides and neurotransmitters, it is not surprising that signaling molecules acting downstream of neurotransmitters/neuropeptides also influence sleep. For example, the protein kinase A (PKA)/CREB pathway promotes wakefulness in Drosophila, and CREB promotes wake in mammals (Graves et al., 2003; Hendricks et al., 2001). We know that octopamine, and perhaps also PDF, acts through PKA to increase wake (Crocker and Sehgal, 2008; Mertens et al., 2005). In addition, some dopamine receptors signal through cyclic adenosine monophosphate (cAMP), so PKA could also contribute to wake-promoting effects of dopamine. It is important to note that, although pan-neuronal expression of PKA promotes wake, there are specific subsets of neurons in the fly brain where PKA actually drives sleep (Joiner et al., 2006). In these neurons, it may be activated by a sleep-promoting molecule like serotonin.
The cyclic guanosine monophosphate (cGMP) kinase promotes sleep in flies and worms (Raizen et al., 2008), and it also regulates mammalian sleep (Langmesser et al., 2009). Upstream signals of this kinase have not yet been identified, but an intriguing possibility is that nitric oxide (NO) is involved because NO is a known activator of cGMP and is independently implicated in sleep regulation. The current model is that neuronal NO, produced by inducible nitric oxide synthase, signals through adenosine to promote recovery sleep following sleep deprivation (Kalinchuk et al., 2010).
Another sleep-regulating pathway is the Extracellular signal-regulated kinases/Mitogen-activated protein kinase (ERK/MAPK) pathway, at least partly in response to Epidermal Growth Factor (EGF) signaling (Foltenyi et al., 2007). In Drosophila, increased EGF signaling activates ERK to promote sleep. The relevant EGF signals originate in the region of the pars intercerebralis, close to the neuroendocrine cells that mediate wake-promoting effects of octopamine. Thus, the pars intercerebralis is a hypothalamus-like structure that contains sleep and wake promoting cells in close proximity. The target of EGF action, as measured by activated ERK signaling, is in the ventral part of the fly brain. In addition, ATF-2, a transcription factor activated by MAPK in response to cellular stress, promotes sleep through its action in lLNVs (Shimizu et al., 2008). Importantly, a role for EGF signaling in the rest:activity is conserved across species. LIN-3, an EGF-like molecule in worms, promotes behavioral quiescence by activating diacylglycerol and phospholipase C-γ (Van Buskirk and Sternberg, 2007). In mammals, Transforming Growth Factor alpha (TGFα), which signals through the EGF receptor, causes a cessation of locomotor activity (Kramer et al., 2001).
Ion channels and channel-regulating proteins
Although a role for ion channels in sleep is predictable (because channels are required to regulate neural activity), it is surprising that channels and channel-regulators are the most prominent class of molecules identified through unbiased screens for sleep-regulating genes. Of the channels, voltage-gated potassium channels are turning out to have a major function in promoting sleep. A genetic screen in Drosophila by Cirelli et al. (2005) identified sleep-inhibiting effects of mutations in the Shaker potassium channel. Cirelli and colleagues then showed that Hyperkinetic, a regulatory subunit of Shaker, also influences sleep levels in flies (Bushey et al., 2007). The rebound response to sleep deprivation is unaltered in Shaker mutants, indicating that the effects are specific for baseline sleep (Cirelli et al., 2005). In an independent forward genetic screen, Koh et al. (2008) identified sleepless (sss) as another sleep-promoting gene. The sss gene product is a small, GPI-anchored protein that regulates levels and activity of the Shaker channel. SSS facilitates activation of Shaker and may also target Shaker to the appropriate compartment in the cell (Wu et al., 2010). However, the reduction in daily sleep is greater in the sss mutant than in Shaker and, in addition, the sss mutation reduces the increased sleep (i.e., rebound) following sleep deprivation (Koh et al., 2008). It is possible that misregulated Shaker in sss mutants has a stronger effect than loss of Shaker in Shaker mutants (Dean et al., 2011). Alternatively, SSS may also regulate other sleep-relevant channels. The hypothesized structure for SSS resembles that of some toxins, and so it could be an endogenous toxin-like molecule that regulates channel activity (Wu et al., 2010).
The effects that Shaker channels have on sleep are also conserved in mammals (Douglas et al., 2007). A mammalian sss ortholog has not been found yet in part because the limited coding region of the small sss gene makes bioinformatic analyses quite difficult. However, genes that share motifs with sss are found in the mammalian genome, and the protein product of one such gene, lynx1, also regulates a channel, the nicotinic acetylcholine receptor (Miwa et al., 2006). Thus, it is likely that a SSS equivalent exists in mammals.
Other studies have also pointed to the importance of channels in sleep. The zebrafish screen described above found that blockers of the ether-a-go-go-related gene (ERG) potassium channel increase waking activity (Rihel et al., 2010). Interestingly, a human syndrome that causes insomnia, in addition to other pathologies, is associated with autoantibodies to voltage-gated potassium channels (Josephs et al., 2004).
Circadian Clock genes
It is well-known that the circadian clock regulates sleep, and thus, one might expect clock mutants to exhibit sleep phenotypes. On the other hand, circadian and homeostatic controls may be, at least partially, independent systems, and thus, mutants could be specific for one system or the other. For instance, the sss mutants have normal clock function. Similarly, circadian clock mutants may disrupt the consolidation of sleep (so that it does not occur in a major block of time at night), but they need not alter the total amount of sleep, which is a measure of homeostatic regulation.
The major known clock mechanism in eukaryotes consists of cyclically expressed core clock proteins that negatively regulate their own transcription in an autoregulatory loop (Zheng and Sehgal, 2008). Typically, one of these negative regulators is critical for determining the circadian period. Transcriptional activation of the negative regulators requires two proteins that usually function as a dimer and are essential for the amplitude of the rhythm. In Drosophila, loss of the transcriptional activators, Clock (Clk) and cycle (cyc), decreases total sleep time (Hendricks et al., 2003). However, loss of the repressors, period (per) and timeless (tim,) has little to no effect on sleep. In mammals, sleep phenotypes have been reported for the activators, Clock and BMAL1, and also for repressors Per and Cryptochrome (Cry) (Laposky et al., 2005; Naylor et al., 2000; Wisor et al., 2002). In addition, a circadian gene that acts as a co-repressor in mammalian and fly clocks alters total sleep time in humans (discussed below). As to whether these effects of the clock genes are an output of the circadian clock or represent functions independent of timekeeping is not clear. It has been suggested that effects of the mammalian clock genes on sleep occur not in the central clock (the suprachiasmatic nucleus), but rather in other parts of the brain (Franken and Dijk, 2009). Definitive conclusions will require selective rescue of the two phenotypes - circadian and sleep- through tissue-specific expression. If both phenotypes are rescued through expression in the same subset of cells, they likely represent the same type of regulation.
Metabolic factors
People with diabetes show a high incidence of sleep disorders, and short sleep times have correlated with obesity, leading to the notion that one physiological process influences the other (Adamantidis and de Lecea, 2008). Little is known about this connection on a molecular level, but it is actively under investigation in model systems.
When flies are starved, they forage for food, a response that can be assayed in the laboratory as increased activity. Under these starvation conditions, sleep is suppressed, but the drive to sleep does not accumulate. In addition, learning deficits that can be caused by sleep deprivation also do not accumulate. (Keene et al., 2010; Thigman et al, 2010). Sleep suppression occurs, at least in part, through the activity of the CLK and CYC proteins. Thus, Clk and cyc mutants show decreased sleep during normal conditions, but are unable to further suppress sleep when starved (Keene et al., 2010). Whereas mutating Clk and cyc causes sleep deficits in response to a metabolic stress, brummer (bmm) and Lipid storage droplet 2 (lsd2) appear to be primarily metabolic mutants that also affect sleep (Thimgan et al., 2010). The bmm mutants are resistant to starvation and display an increased homeostatic response, measured as increased rebound, to sleep deprivation; conversely, lsd2 mutants are sensitive to starvation and display decreased sleep homeostasis. Based upon these findings, it is tempting to propose models for the interaction between sleep and metabolism, but that is premature at this point. It is entirely possible that this apparent interaction reflects shared molecular pathways between sleep and metabolism rather than mutual regulation.
Immune genes
A long-standing hypothesis posits that immune genes promote sleep (Imeri and Opp, 2009). The immune protein NF-κB is upregulated in sleep-deprived mammals (Chen et al., 1999), and sleep is usually increased during illness (Imeri and Opp, 2009). Recent studies in Drosophila reveal that immune genes, such as Relish (encoding the Drosophila NF-κB) and anti-bacterial peptides, are upregulated following sleep deprivation (Williams et al., 2007). Although Relish mutants show only minimal changes in total sleep, it appears that Relish is required to increase sleep in response to injury or infection (Kuo et al., 2010). Thus, some immune genes may promote sleep under pathological conditions, which could include prolonged sleep deprivation. Increased sleep under injury or infection supports a role for sleep in facilitating recovery (Imeri and Opp, 2009). At the same time, mutating some immune genes, such as TAK1 (TGFβ activated kinase), alters baseline sleep in Drosophila, and anti-inflammatory compounds, such as glucocorticoids and NSAIDs, increase daytime activity in zebrafish, indicating that immune/inflammatory signaling also affects baseline rest:activity levels (Rihel et al., 2010; Williams et al., 2007).
Key features to emerge from model organism studies
It is clear that mechanisms regulating sleep are conserved across species (see Table 1 for a summary of the mechanisms conserved in flies and mammals). Another striking observation from these studies is that there do not appear to be dedicated sleep genes. Even the most unbiased approaches have not, to date, uncovered genes that function specifically to regulate sleep. Thus, in contrast to circadian clocks, in which the key components are proteins dedicated to timekeeping, sleep is regulated by genes that are involved in normal neuronal function. Many such genes are also important for circadian rhythms, but they function downstream of the clock to relay time-of-day signals.
Table 1.
Signaling Mechanisms that Regulate Sleep across Species
| Molecule | Drosophilaa | Mammalsa |
|---|---|---|
| Dopamine | ↑ Wake | ↑ Wake |
| GABA | ↑ Sleep | ↑ Sleep |
| Norepinephrine (octopamine) | ↑ Wake | ↑ Wake |
| Serotonin | ↑ Sleep | ↑ REM sleep NREM sleep? |
| cAMP/cAMP signaling | ↑ Wake | ↑ Wake |
| EGF signaling | ↑ Sleep | ↑ Sleep |
| Voltage-gated K+ channels (Shaker) | ↑ Sleep | ↑ Sleep |
Worms and fish are not included because many of these pathways have not yet been investigated in these systems.
In case of sleep, the homeostat itself- the component that drives the need to sleep-may be derived from changes in routine neural function. Models for sleep homeostasis, which invoke changes in adenosine or glycogen, fit with this idea, suggesting that excess neural activity leads to altered levels of a molecule that signals an energy imbalance and promotes sleep (Scharf et al., 2008). These models naturally support energy-restoration functions for sleep. On the other hand, the information gleaned from the genetic analysis of sleep is also perfectly consistent with a role for sleep in synaptic plasticity. Indeed, a synaptic function for sleep is supported by both the mutant Drosophila lines discussed above and the molecular screens searching for genes regulated as a function of sleep state. For example, molecules and proteins that mediate recovery from neuronal hyperactivity (e.g., Homer1) are upregulated by sleep loss (Maret et al., 2007; Nelson et al., 2004). Presumably, this contributes to synaptic scaling during sleep, as discussed below. We will return to speculating about sleep function after discussing genes implicated in human sleep and sleep disorders.
Genetic Factors Underlying Circadian Rhythm Disorders
It is now well-established that Familial Advanced Sleep Phase Syndrome (FASPS) is caused by mutations in human clock-related genes (Toh et al., 2001; Xu et al., 2005). Individuals with this autosomal dominant trait have normal sleep architecture, and a lifelong tendency to wake and sleep at very early times (i.e., 1:00 a.m.– 3:00 a.m. and 6:00–8:00 p.m., respectively). Melatonin and temperature rhythms are advanced by 4–6 hours, and the free running period (i.e., the period of rhythms observed in organisms in the absence of any environment clues, thereby indicating endogenous clock time) has been measured as 1 hour shorter than in controls. Underlying mutations in two pedigree populations point to defects in phosphorylation of PER2 as the core issue, with mutations identified in both PER2 and Casein Kinase 1 genes (CK1δ).
These findings correspond well with circadian mutations in other organisms. For example, in Drosophila and the Syrian hamster, mutations in the CK1ε kinase (encoded by the Drosophila doubletime (dbt) gene and Syrian hamster tau gene), lead to deficient phosphorylation of PER and changes in circadian period. Mouse modeling studies of the FASPS mutations revealed a few surprises in the molecular pathways of these components. These include the probable action of an unknown kinase, complex interactions of phosphorylation with nuclear entry and retention, and transcriptional and posttranscriptional regulation of clock proteins (Mignot and Takahashi, 2007)
Diurnal preferences (i.e., a tendency to prefer mornings versus evenings) are heritable (Drennan et al., 1991). Thus, genetic variants affecting circadian regulation are segregating in the general population. Variable results have been obtained from genome wide or candidate gene association studies attempting to identify these variants, suggesting that the next step is to greatly increase sample size (e.g., 100,000 individuals) for genome wide association (GWA) studies. Another option is to resequence candidate loci in individuals or family clusters with phenotypes at the extremes of the normal range. Further, subjective diurnal preference scores are likely less amenable to genetic analysis than more objective physiological measures of circadian phase.
Genetic Factors Regulating EEG and the Sleep Homeostat
Genetic analysis of selected EEG traits and the sleep homeostat is an active field in model systems and in humans. In mice, spectral features of the EEG are altered by a deficiency in a single enzyme involved in short chain fatty acid metabolism (ACADS) (Tafti et al., 2003), as well as by Homer1a. The latter was identified through Quantitative Trait Locus (QTL) analysis and mapping studies in inbred mice (Maret et al., 2007). This illustrates the strong effect that genetic background has on the effect size of any identified gene, which complicates the extrapolation of loci identified in model organisms to potential human phenotypes.
In humans, a variation in the PER3 gene was recently associated with differences in EEG markers of sleep homeostasis after sleep deprivation and with behavioral consequences of this deprivation (Viola et al., 2007). Although this polymorphism was not implicated in related studies by other groups (Goel et al., 2009), the original finding suggests that circadian genes are involved in sleep homeostasis, consistent with studies in mice and Drosophila (Mignot, 2008).
Average sleep duration at night is a poorly defined phenotype, and so not surprisingly, GWA studies have yielded neither significant (~p<10–8) nor reproducible signals in studies with limited sample sizes (up to 10,000 individuals). However, a recent screen for circadian gene mutations in pedigrees with habitual short sleep duration identified a mutation in a critical basic helix-loop-helix domain of DEC2 (a repressor of CLOCK/BMAL1activity) (He et al., 2009). Studies of this mutation in animal models showed that it causes increased daily wakefulness in transgenic mice, as well as dominant effects in transgenic flies. This again illustrates the role of circadian clock proteins in the regulation of sleep and suggests that a viable approach to identifying additional sleep-regulatory genes may be systematic mutation screening, exome sequencing, or whole genome sequencing in subjects with extreme sleep phenotypes segregating in pedigrees.
Genetics of Human Narcolepsy-cataplexy
Narcolepsy affects the control of sleep and wakefulness, resulting in excessive daytime sleepiness, symptoms of dissociated REM sleep (e.g., sleep paralysis and dream-like “hypnagogic” hallucinations at sleep onset), disrupted nocturnal sleep, and cataplexy (i.e., brief episodes of muscle weakness triggered by emotions). In animal models, the full disease can be produced by disrupting hypocretin (orexin) neurotransmission either by defects in the hypocretin receptor 2 or the hypocretin ligand gene (Chemelli et al., 1999; Lin et al., 1999). By contrast, the disease in humans is sporadic, resulting in the majority of cases from a loss of ~70,000 hypothalamic neurons producing hypocretin, rather than single gene mutations (Chabas et al., 2003). Concordance is low (35%) in monozygotic twins, but recurrence risk in first degree relatives is increased by 20–40 fold, although still low overall (0.9–2.3%), suggesting that the disease results from an interaction of environmental factors on a susceptible genetic background.
The destruction of hypocretin cells is almost certainly an autoimmune event, as both Human Leukocyte Antigen (HLA) and T cell receptor (TCR) variants have strong effects on predisposition. Expressed on immune cells, HLA class II antigens present processed foreign peptides to T cells by engaging the TCR. Nearly all narcolepsy/hypocretin deficiency cases carry two specific and tightly linked class II gene alleles: DQA1*01:02 and DQB1*06:02. (HLA genes are extremely polymorphic, and alleles are grouped in broader subtypes based on sequence similarities, as noted by the first two digits; the next two digits represent minor amino acid variations among these broader families). However, these are also common alleles across ethnic groups (12–38%) and thus, are not sufficient to cause disease (Mignot et al., 2001). Other HLA class II alleles also modulate susceptibility, notably DQB1*03:01 (susceptibility) and DQB1*06:01, DQB1*05:01, and DQA1*01 (non-DQA1*01:02) (protective). Analysis of the binding pockets and dimerization activities of these variants indicates that even minor changes in the peptide binding pockets may determine risk and that some protective alleles may act by reducing the availability of the disease-associated DQA1*01:02/ DQB1*06:02 heterodimer (Hong et al., 2007; Mignot et al., 2001).
The TCR alpha gene (TCRA) is also an important susceptibility factor for narcolepsy. TCR initiates an immune response when it interacts with peptide-bound HLA antigens. Like the immunoglobulin loci, the TCRA locus undergoes somatic cell recombination between 46 functional variable (V) and 49 functional joining (J) segments. A SNP variant (rs1154155C) in the J segment region of the locus shows significant association in Caucasians and other ethnic groups (Hallmayer et al., 2009), and this association has been replicated in multiple studies. We hypothesize that the DQB1*0602/ DQA1*0102 HLA heterodimer interacts with a TCR idiotype in which a specific VJ recombinant is associated with the presence of rs1154155 (directly or indirectly). In the context of a selected antigenic trigger, this could then lead to further immune reaction ending in the destruction of hypocretin-producing cells.
Most autoimmune diseases have an array of strong HLA susceptibility factors. Thus, perhaps one of the most intriguing aspects of the autoimmune process in narcolepsy is the remarkable specificity of both the susceptibility loci and the autoimmune target, as destruction appears highly selective toward hypocretin cells. In addition, typical markers of an autoimmune process disappear rapidly after the disease destroys hyocretin cells, if they are ever present at all (Overeem et al., 2008). Increased autoantibodies against Tribbles homolog 2 (TRIB2) near disease onset were recently suggested by several groups (Lim and Scammell, 2010) but not confirmed in samples collected later (Dauvilliers et al., 2010). Upper airway infections, such as strep throat and flu, have been implicated as environmental triggers of narcolepsy. Higher rates of diagnosed strep throat infections and high titers of Anti Streptolysin O (marking strep infections) near disease onset have been reported (Longstreth et al., 2009). Additionally, H1N1 vaccines containing the AS03 adjuvant and H1N1 infections are also implicated as rare triggers of narcolepsy (Dauvilliers et al., 2010). These triggers may either act directly by contributing important epitopes or non-specifically through reactivation of dormant T cell clones, superantigen activity, or permeabilizing the blood brain barrier (for example by fever) and thereby facilitating immune cell entry (Dauvilliers et al., 2010).
Additional narcolepsy susceptibility loci have been identified through GWA studies. A SNP marker located between and decreasing expression of carnitine palmitoyltransferase 1B (CPT1B) and Choline Kinase B (CHKB) was associated with narcolepsy in a Japanese sample, and the association was replicated in a second sample of Japanese but not in Caucasians (Miyagawa et al., 2008). Both genes are plausible REM sleep regulatory candidates. CHKB metabolizes choline, the precursor of acetylcholine, a regulator of REM sleep. Likewise, CPT1B is part of the carnitine shuttle, transporting long-chain fatty acyl-CoAs from the cytoplasm into the mitochondria. CPT1B is also a rate-controlling enzyme of the beta-oxidation pathway in mitochondria, a pathway involved in the regulation of theta oscillations during REM sleep (Tafti et al., 2003). The possibility that this polymorphism modulates REM sleep independently of hypocretin cell loss was recently bolstered by the finding of an association in Essential Hypersomnia, a milder form of narcolepsy typically not associated with hypocretin deficiency (Miyagawa et al., 2009). A direct effect of this polymorphism on REM sleep would suggest that decreased mitochondrial beta oxidation (a process that occurs primarily in the periphery) is associated with increased REM, linking REM sleep with energy homeostasis.
A role for purinergic receptors in narcolepsy was identified by a GWA study in Caucasians, followed by fine mapping in multiple ethnic groups (Kornum et al., 2011). Purinergic signaling plays a key role in immune regulation. The SNP rs2305795, located in the 3' untranslated region of the purinergic receptor gene P2Y11, decreases the receptor's expression in peripheral mononuclear cells and is significantly associated with narcolepsy susceptibility. The P2Y11 receptor also modulates immune cell chemotaxis and cell death induced by ATP, suggesting immune modulatory effects.
Genetics of Other Sleep Disorders
Restless Leg Syndrome
Restless legs syndrome (RLS) is a common disorder characterized by an uncomfortable and intrusive urge to move the lower limbs. Symptoms manifest during rest, are worse in the evening, and improve with movement. Periodic leg movements in sleep are often also present. Numerous studies have shown that iron deficiency in the brain and reduced dopaminergic neuronal activity are critical pathophysiological factors (Salas et al., 2010).
RLS has a strong genetic component, with up to 60% of cases reporting affected family members, and high concordance (83%) reported in monozygotic twins. Attempts to identify RLS genes through linkage analysis in families have identified neither specific mutations nor specific genes, although three linked genomic regions have been replicated (reviewed in (Trenkwalder et al., 2009)). By contrast, studies using a GWA design have been fruitful, (Stefansson et al., 2007; Winkelmann, 2011; Winkelmann et al., 2007), revealing a surprising role for developmental regulatory factors. These transcription factors likely affect spinal cord regulation of sensory perception and locomotor pattern generation and may also interact with brain iron homeostastis.
The MEIS1 locus is the most important RLS susceptibility gene. Variants near exon 9, a region with high interspecies conservation, have shown the strongest association with RLS, displaying odds ratio greater than 2 (Winkelmann et al., 2007). More recently, an extended study also found an additional, independent association 1.9 MB away from MEIS1, in a region likely to also regulate MEIS1 expression (Winkelmann, 2011) (Table 2). MEIS1 is strongly expressed in dopaminergic neurons of the substantia nigra, (where studies have reported lower iron levels in RLS cases), in the spinal cord, and in the red nucleus, a region that regulates coordination of limb movement and which also contains lower iron levels in RLS. MEIS1 is part of a Hox transcriptional regulatory network that specifies motor neuron pool identity and thus the pattern of target-muscle connectivity, suggesting a key link to the pathophysiology of RLS within the spinal cord.
Table 2.
Human Susceptibility Loci for Sleep and Sleep Disorders
| Genes | Pathology | Experimental Design | Associated SNP, Allele, or Mutation | Allelic Odds Ratio | Comments |
|---|---|---|---|---|---|
| DQB1 and DQA1 (forming the DQ heterodimer) | Narcolepsy/hypocertin deficiency | Candidate gene | Main predisposing effect is DQB1*06:02–DQA1*01:02; Secondary predisposing effects: DQB1*03:01; Secondary protective effects: DQA1*01, DQB1*05, or DQB1*06 that are not DQA1*01:02, DQB1*06:02. | OR0602 = 8.8 (Caucasians) | Effects conserved across African Americans, Asians, and Caucasians. Most effects in these loci are dominant mediated by DQ01*06:02; very few cases are DQB1*06:02 negative. Almost all subjects are DQ1*0:102, an allele in tight linkage with DQB1*06:02. |
| CPT1B/CHKB | Narcolepsy/hypocrerin deficiency Essential hypersomnia | GWAS | rs5770917C (affect expression) | OR= 1.8 (Japanese only) | Association is still tentative. Identified in Japanese narcolepsy patients, replicated in Koreans. The association is not significant in European populations or those of African descent. Did not replicate in a Chinese narcolepsy sample. Also associated with hypersomnia in Japan. Loci have roles in beta-oxidation and acetylcholine synthesis, potentially modulating rapid eye movement (REM) sleep. |
| TCRA | Narcolepsy/hypocretin deficiency | GWAS | rs1154155C (may modify TCRJ usage or sequence) | OR= 1.7 (all ethnic groups) | Identified in Caucasian narcolepsy patients and replicated across ethnic groups (Asians and African Americans). Independently replicated in European and Chinese narcolepsy patients and in Japanese cases with HLA (human leukocyte antigen)-positive essential hypersomnia. This suggests the involvement of a specific T cell receptor on narcolepsy patients, possibly interacting with the DQ locus. |
| P2RY11 | Narcolepsy/hypocretin deficiency | GWAS | rs2305795A (affect expression) | OR= 1.3 | Identified in Caucasians, with replication across ethnic groups; not yet replicated independently; immunomodulator y function or reduced ATP-induced apoptosis of immune cells. |
| HPER2 | Familial advanced sleep phase syndrome | Candidate gene sequencing | S662G (removal of a functional phosphorylation site) | n.a. (fully penetrant) | Autosomal-dominant transmission; validation in in vitro and mice models. |
| CK1d | Familial Advanced Sleep Phase Syndrome | Candidate gene sequencing | T44A (reduced kinase activity of the enzyme) | n.a. (fully penetrant) | Autosomal-dominant transmission; validation in in vitro and mice models |
| DEC2 | Familial short Sleep | Candidate gene sequencing | P385R (reduced Clk/B mal1-mediated transactivation by DEC2) | n.a. (fully penetrant) | Autosomal-dominant transmission (allele dosage model); validation in in vitro and mice models. |
| MEIS1(a) | Restless leg syndrome | GWAS | rs6710341A-rs12469063G haplotype | OR = 2.0 (Caucasians) | Identified in Caucasians; replicated by multiple studies in Caucasians; decreased expression in restless leg syndrome (RLS); function still unknown in mice. MEIS1 functions in CNS and motor neuron development. |
| MEIS1/ETAA1 (b) | Restless leg syndrome | GWAS | rs6747972A | OR= 1.2 (Caucasians) | Independent association; intergenic region on chromosome 2p14 located 1.3 MB dowstream of MEIS1; likely regulates MES1 or ETAA1 expression. |
| BTBD9 | Restless leg syndrome | GWAS | rs9296249T | OR =1.7 (Caucasians) | Replicated by multiple studies in Caucasians; also associated with periodic leg movements during sleep independent of RLS; risk allele may be associated with decreased ferritin (more prominent in women than in men); allele dosage model; involvement in RLS unknown. |
| MAP2K5/SKOR1(LBXC OR1) | Restless leg syndrome | GWAS | rs1026732G | OR= 1.5 (Caucasians) | Most likely LBXCOR1 (SKOR1); recessive effect; allele dosage model; gene has a function in the development of the CNS/spinal cord/dorsal horn; involvement in RLS unknown. |
| PTPRD | Restless leg syndrome | GWAS | rs4626664T, rs1975197A | OR= 1.4 (Caucasians) | Replicated independently in Caucasian populations; allele dosage model; involvement in RLS unknown. In principle, it functions during the development of the CNS/motorneuron and in axon guidance. |
| TOX3, noncoding BC034767 RNA | Restless leg syndrome | rs3104767G | OR =1.3 (Caucasians) | TOX3 is a well-known breast cancer susceptibility gene, but it associates with a different SNP. Genome-wide significant but no clear functional data or independent replication. | |
| NOS1 | Restless leg syndrome | Case-control association in RLS linkage region | rs7977109A | OR = 0.76 (Caucasians) | Not yet replicated; involvement in RLS unknown; suggested to modulate the dopaminergic neurotransmission; different variants across the gene were associated in two case-control studies. |
Variants in BTBD9 (BTB (POZ) domain containing 9) have also repeatedly shown association with RLS, with allelic odds ratios between 1.5–1.8. Notably, in an Icelandic cohort (Stefansson et al., 2007), SNP associations were strongly tied to the presence of periodic limb movements (i.e., the repetitive cramping or jerking of the legs during sleep), implying that this locus confers risk specifically for the motor component of RLS. Serum ferritin levels were also found to vary by genotype, potentially underlying the iron deficiency associated with RLS. Little is known of the function of BTBD9 in mammals; however, in Drosophila, proteins containing the BTB (POZ) domain have important roles in metamorphosis and limb pattern formation. These proteins have wide-ranging functions, making assignment of a specific function to BTBD9 difficult.
A third RLS locus surrounds the MAP2K5 and SKOR1 (LBXCOR1) genes, but linkage disequilibrium has prevented identification of the relevant gene (Winkelmann et al., 2007). SKOR1 (SKI family transcriptional corepressor 1) has appealing links to RLS, as it is expressed selectively in a subset of dorsal horn interneurons in the developing spinal cord, which relay pain and touch. The SKOR1/Lbx1 pathway is essential to the generation of a GABAergic versus glutamatergic phenotype in these cells (Cheng et al., 2005), and this locus may contribute to the sensory component of the phenotype by affecting modulation of sensory and pain inputs.
Extended fine mapping and GWA studies have identified two additional loci (Table 2). The PTPRD (protein tyrosine phosphatase receptor type delta) locus emerged through fine mapping of a suggestive GWA signal in a linkage region on chromosome 9p (Schormair et al., 2008). Although no mutations were identified among patients, this is still an excellent candidate, with established roles in axon guidance and termination of motor neurons during embryonic development in the mouse. Most recently, a large linkage disequilibrium block containing TOX3 (a breast cancer susceptibility locus) and untranslated BC034767 was associated with susceptibility in an extended RLS sample, although a role for these two transcripts in RLS pathogenesis is not yet known (Winkelmann, 2011).
A role for neuronal nitric oxide synthase (NOS1) in RLS was also suggested through finemapping of a region on chromosome 12q, which was first identified through linkage analysis in population based RLS cases and controls (Winkelmann et al., 2008), although no mutations were detected in RLS1-linked familiy members. The NOS1 gene is an appealing candidate for underlying specific symptoms of the syndrome because nitric oxide acts as an atypical neurotransmitter in the central nervous system with roles in pain perception, control of sleep-wake regulation, and modulation of dopaminergic activity (Winkelmann et al., 2008),.
Taken together, results to date suggest that RLS is characterized by an imbalance of the spinal circuitry gating sensory integration and controlling locomotor outputs. This imbalance may be developmental and is mostly the result of genetic polymorphisms in transcription factors. Descending influences, for example descending dopaminergic projections, cognitive influences and the effect of iron deficiency further destabilizes the circuitry.
Hypersomnias, insomnia, and parasomnias
Table 2 provides a summary of the various sleep disorders for which some genetic data are available. Many additional sleep disorders and parasomnias (i.e., sleep disorders that involve abnormal and unnatural movements or behaviors) show genetic effects or familial clustering, but no specific genes are yet implicated. Insomnia runs in families and has higher concordance in monozygotic twins, but this heterogeneous phenotype will require large samples and potentially EEG-based endophenotypes for genetic mapping. Individuals with Morvan's syndrome, a disorder associated with insomnia, have autoantibodies to potassium channels suggesting a potentially conserved mechanism, given the role of these channels in sleep in model organisms.
In the general population, Idiopathic Hypersomnia or isolated sleepiness, is another poorly defined and heterogeneous phenotype. Candidate loci identified (Gottlieb et al., 2007) in a low density GWA study await confirmation. The distinction between narcolepsy without cataplexy and idiopathic hypersomnia is difficult. These may lie on a continuum with narcolepsy-cataplexy, as the frequency of DQB1*0602 (40%) in patients with Idiopathic Hypersomnia is intermediate between that in the general population (12–38%) and in narcolepsy cataplexy (70%–100%). Although measurements of hypocretin in cerebrospinal fluid are typically within the normal range for narcolepsy without cataplexy, some patients show low levels in the narcolepsy-cataplexy range in association with DQB1*0602 (Mignot et al., 2002). Hypocretin levels are within normal range for idiopathic hypersomnia, with rare exceptions. As mentioned above, a narcolepsy polymorphism located between CPT1B and CHKB may be associated with both narcolepsy and hypersomnia in Japanese cohorts.
Kleine Levin syndrome (KLS) is a rare disorder primarily affecting adolescent males. Characterized by recurring episodes of profound hypersomnia and cognitive and behavioral changes, it typically attenuates and disappears in adulthood. Recent studies suggest that genetic factors confer susceptibility. Ashkenazi Jewish heritage is often reported, suggesting a potential founder effect. Furthermore, 5 out of 105 KLS patients reported an affected family member. This suggests the action of a major susceptibility gene that potentially controls the response after exposure to an unknown environmental trigger, such as an infection. An HLA association with KLS was suggested, but this finding has not been replicated in subsequent studies (Arnulf et al., 2008).
Features of dissociated REM sleep, such as sleep paralysis and hypnagogic hallucinations are highly heritable and frequent in the general population, particularly with insufficient sleep. Sleep paralysis shows high concordance in monozygotic twins, and autosomal dominant transmission has been reported (reviewed (Mignot, 1997)). In REM sleep behavior disorder, inhibition of motor pathways in REM sleep is lost, allowing robust and potentially dangerous motor activity in response to dream content. As with other features of dysregulated REM sleep, REM sleep behavior disorder is common in narcolepsy, but it also occurs in the population and in Parkinson's disease. REM sleep behavior disorder is often an early sign of neurodegenerative disorders, particularly Parkinson's disease (Massicotte-Marquez et al., 2008). The extent of heredity in REM sleep behavior disorder has not been established, but a variety of single gene defects and HLA-DR/DQ are implicated in the development of Parkinson's disease.
NREM sleep parasomnias, including sleepwalking, sleep talking, and night terrors, typically occur during slow wave sleep. Prevalence is high in children, but rarely requires medical intervention and typically disappears during adulthood. Sleepwalking may be present in up to 20% of children, and is present in up to 3% of adults. Sleepwalking, sleep talking, enuresis (i.e., bed-wetting), bruxism (i.e., grinding teeth), and night terrors have substantial genetic effects, and also co-occur, suggesting some common genetic susceptibilities (Hublin et al., 2001).
Sleep Disordered Breathing and Obstructive Sleep Apnea
Obstructive sleep apnea is a highly prevalent disorder characterized by intermittent upper-airway collapse, which impairs ventilation and disrupts sleep (White, 2005). Numerous genetically influenced or physiologic factors can contribute to upper-airway collapse, including anatomical features (e.g., craniofacial features), reduced dilator muscle activity during sleep, decreased end-expiratory lung volume, ventilatory control instability, and sleep-state instability, although obesity may outweigh these other predispositions.
Candidate gene and small GWA studies of this complex phenotype (which use the apnea hypopnea index as phenotype), have not led to consistently replicated findings apart from an association with Apolipoprotein E allele e4 (APOE e4), which has been variably replicated (for example (Gottlieb et al., 2004)). APOE e4 is well known to be associated with Alzheimer's disease. The association with sleep apnea remains controversial, as samples differed by age, ethnicity, and body mass index, as well as screening methodology. The association may also be confounded by interactions with cognitive decline, which affects symptom reporting. However, APOE e4 could predispose an individual to sleep apnea through multiple mechanisms, including lowering levels of choline acetyltransferase and reducing neuromuscular activation of the upper airway dilator muscles. Furthermore, differential lipid binding to β-amyloid or Tau proteins could lead to plaque formation in respiratory centers. Based on the range of interacting physiologic traits that contribute to susceptibility to sleep apnea, future studies will likely need to use endophenotypes to reduce heterogeneity in order to identify underlying loci.
Overlap between human sleep genes and those found in model organisms
Studies of sleep in humans have focused on specific sleep disorders rather than variations in amount of sleep. To the extent that these disorders can be modeled in animals, mechanisms appear to be conserved as highlighted by narcolepsy symptoms resulting from hypocretin system defects in humans, dogs, or rodents. Animal models do not exist for many other human sleep-related behaviors (RLS, parasomnias, etc) and thus comparisons cannot be made; however, conserved effects of the circadian genes are well-documented. For instance, alleles of per in flies, which shorten the circadian period, result in a phase advance in light-dark cycles, similar to that seen in humans with ASPS (Marrus et al., 1996). In addition, Dec2 is associated with sleep phenotypes in humans, mice, and flies (He et al, 2009). Finally, sleep-modulatory drugs used in humans act through many of the neurotransmitter systems discussed in the section on animal models, and they typically have similar behavioral effects in these animals, including flies.
Sleep function: Insights from model organisms
It is difficult to address sleep function in humans through experimentation, but studies in model organisms suggest a role for sleep in supporting cognitive function (Diekelmann and Born, 2010; Poe et al., 2010) through the promotion of synaptic plasticity. As noted above, the worm model for sleep is based upon a developmental stage associated with neural changes, and many of the identified sleep-regulatory genes function in plasticity. Several sleep-regulating loci, including PKA and CREB, are among the major components required for learning and memory (Bailey et al., 1996). In addition, their effects on sleep are mediated in tissues important for the consolidation of memory. Thus, effects of PKA on Drosophila sleep are exerted largely, although not entirely, in the Drosophila mushroom bodies, well-known for their function in learning and memory (Joiner et al., 2006). Moreover, the sleep-consolidating effects of serotonin, involved in synaptic facilitation in Aplysia (Bailey et al., 1996), are mediated by the 5-HT1A receptor in the mushroom bodies (Yuan et al., 2006).
Although sleep is regulated by brain structures important for learning and memory, the reverse is also true: sleep deprivation impacts memory and the sites of memory formation. Learning impairments induced by sleep deprivation of Drosophila can be rescued by the expression of the dopamine D1 receptor in mushroom bodies (Seugnet et al., 2008). Similarly, sleep deprivation of mice impairs learning by impacting gene expression in the hippocampus (Vecsey et al., 2009), most notably, through increases in phosphodiesterase 4 (PDE4). Thus, these molecules that regulate both sleep and plasticity may well provide the mechanistic link between these two processes.
The critical question then is, how does sleep promote the consolidation of memory, or what specifically is its role in synaptic plasticity? Whereas some argue that sleep promotes synaptic potentiation, others suggest that sleep is required for synaptic depression. In support of the latter, molecular markers of potentiation appear to be high in rats during wake and low during sleep (Vyazovskiy et al., 2008). A similar study conducted in Drosophila also reported that levels of key synaptic proteins increase with wake and decline with sleep (Gilestro et al., 2009). This model for sleep function postulates that increased synaptic activity during wake is followed by increased sleep in order to downscale, and thereby normalize neural connectivity. Indeed, flies maintained in social conditions, which presumably increase potentiation, sleep more than those kept in isolation (Ganguly-Fitzgerald et al., 2006). The effect of social experience on sleep is mediated by specific genes in circadian clock neurons (Donlea et al., 2009).
Although the emphasis is definitely on synaptic plasticity, hints of other functions for sleep have also arisen from studies on model organism. The induction of chaperone proteins can abrogate the lethal effects of sleep loss, suggesting that sleep normally promotes activity of such chaperones (Shaw et al., 2002). A general function for sleep in curbing stress is supported by the effects of sleep deprivation on endoplasmic reticulum (ER) stress, as BiP (a marker of ER stress) increases during sleep deprivation in mice and flies (Naidoo et al., 2007; Naidoo et al., 2005). BiP levels also determine the extent of recovery sleep in Drosophila, suggesting that cellular stress influences the need for sleep (Naidoo et al., 2007). As noted above, sleep is also implicated in recovery from injury and infection.
Regardless of which hypothesis turns out correct and which sleep function stands the test of time, it is clear that model organisms are blazing a trail in the sleep field. In contrast, human genetic studies have made strides in our understanding of a few selected neurological sleep disorders, such as narcolepsy and restless leg syndrome, but have not yet shed light on the genetic basis of sleep homeostasis or the need for sleep, the two most burning questions in sleep research today. This is likely to change once a large sample of subjects, each phenotyped for sleep variables, has been subjected to genetic analysis that could include whole genome sequencing. Combined genetic and EEG analyses in large samples are also likely to assist in this search as they may offer more objective and discrete sleep related phenotypes. We do not expect that all aspects of sleep regulation will be conserved across evolution, or that sleep functions will be exactly the same in all species. However, we predict that some unifying principles will emerge (Mignot, 2008); indeed, we believe that they have already started to make an appearance.
Acknowledgements
We would like to thank laboratory members for helpful discussions, Juliette Faraco for assistance in writing this review and Julie Williams for comments on the role of immune genes. Our work on sleep is supported by P01 AG017628 (Sehgal laboratory) and NS23724 (Mignot laboratory).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Adamantidis A, de Lecea L. Sleep and metabolism: shared circuits, new connections. Trends Endocrinol Metab. 2008;19:362–370. doi: 10.1016/j.tem.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Agosto J, Choi JC, Parisky KM, Stilwell G, Rosbash M, Griffith LC. Modulation of GABAA receptor desensitization uncouples sleep onset and maintenance in Drosophila. Nat Neurosci. 2008;11:354–359. doi: 10.1038/nn2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allada R, Siegel JM. Unearthing the phylogenetic roots of sleep. Curr Biol. 2008;18:R670–R679. doi: 10.1016/j.cub.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosius U, Lietzenmaier S, Wehrle R, Wichniak A, Kalus S, Winkelmann J, Bettecken T, Holsboer F, Yassouridis A, Friess E. Heritability of sleep electroencephalogram. Biol Psychiatry. 2008;64:344–348. doi: 10.1016/j.biopsych.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Andretic R, Franken P, Tafti M. Genetics of sleep. Annu Rev Genet. 2008a;42:361–388. doi: 10.1146/annurev.genet.42.110807.091541. [DOI] [PubMed] [Google Scholar]
- Andretic R, Kim YC, Jones FS, Han KA, Greenspan RJ. Drosophila D1 dopamine receptor mediates caffeine-induced arousal. Proc Natl Acad Sci U S A. 2008b;105:20392–20397. doi: 10.1073/pnas.0806776105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andretic R, van Swinderen B, Greenspan RJ. Dopaminergic modulation of arousal in Drosophila. Curr Biol. 2005;15:1165–1175. doi: 10.1016/j.cub.2005.05.025. [DOI] [PubMed] [Google Scholar]
- Arnulf I, Lin L, Gadoth N, File J, Lecendreux M, Franco P, Zeitzer J, Lo B, Faraco JH, Mignot E. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol. 2008;63:482–493. doi: 10.1002/ana.21333. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci U S A. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basheer R, Strecker RE, Thakkar MM, McCarley RW. Adenosine and sleep-wake regulation. Prog Neurobiol. 2004;73:379–396. doi: 10.1016/j.pneurobio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Bjorness TE, Greene RW. Adenosine and sleep. Curr Neuropharmacol. 2009;7:238–245. doi: 10.2174/157015909789152182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorness TE, Kelly CL, Gao T, Poffenberger V, Greene RW. Control and function of the homeostatic sleep response by adenosine A1 receptors. J Neurosci. 2009;29:1267–1276. doi: 10.1523/JNEUROSCI.2942-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushey D, Huber R, Tononi G, Cirelli C. Drosophila Hyperkinetic mutants have reduced sleep and impaired memory. J Neurosci. 2007;27:5384–5393. doi: 10.1523/JNEUROSCI.0108-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SS, Tobler I. Animal sleep: a review of sleep duration across phylogeny. Neurosci Biobehav Rev. 1984;8:269–300. doi: 10.1016/0149-7634(84)90054-x. [DOI] [PubMed] [Google Scholar]
- Chabas D, Taheri S, Renier C, Mignot E. The genetics of narcolepsy. Annu Rev Genomics Hum Genet. 2003;4:459–483. doi: 10.1146/annurev.genom.4.070802.110432. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Chen Z, Gardi J, Kushikata T, Fang J, Krueger JM. Nuclear factor-kappaB-like activity increases in murine cerebral cortex after sleep deprivation. Am J Physiol. 1999;276:R1812–1818. doi: 10.1152/ajpregu.1999.276.6.R1812. [DOI] [PubMed] [Google Scholar]
- Cheng L, Samad OA, Xu Y, Mizuguchi R, Luo P, Shirasawa S, Goulding M, Ma Q. Lbx1 and Tlx3 are opposing switches in determining GABAergic versus glutamatergic transmitter phenotypes. Nat Neurosci. 2005;8:1510–1515. doi: 10.1038/nn1569. [DOI] [PubMed] [Google Scholar]
- Cirelli C. The genetic and molecular regulation of sleep: from fruit flies to humans. Nat Rev Neurosci. 2009;10:549–560. doi: 10.1038/nrn2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C, Bushey D, Hill S, Huber R, Kreber R, Ganetzky B, Tononi G. Reduced sleep in Drosophila Shaker mutants. Nature. 2005;434:1087–1092. doi: 10.1038/nature03486. [DOI] [PubMed] [Google Scholar]
- Crocker A, Sehgal A. Octopamine regulates sleep in drosophila through protein kinase A-dependent mechanisms. J Neurosci. 2008;28:9377–9385. doi: 10.1523/JNEUROSCI.3072-08a.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker A, Sehgal A. Genetic analysis of sleep. Genes Dev. 2010;24:1220–1235. doi: 10.1101/gad.1913110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker A, Shahidullah M, Levitan IB, Sehgal A. Identification of a neural circuit that underlies the effects of octopamine on sleep:wake behavior. Neuron. 2010;65:670–681. doi: 10.1016/j.neuron.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauvilliers Y, Montplaisir J, Cochen V, Desautels A, Einen M, Lin L, Kawashima M, Bayard S, Monaca C, Tiberge M, et al. Post-H1N1 narcolepsy-cataplexy. Sleep. 2010;33:1428–1430. doi: 10.1093/sleep/33.11.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gennaro L, Marzano C, Fratello F, Moroni F, Pellicciari MC, Ferlazzo F, Costa S, Couyoumdjian A, Curcio G, Sforza E, et al. The electroencephalographic fingerprint of sleep is genetically determined: a twin study. Ann Neurol. 2008;64:455–460. doi: 10.1002/ana.21434. [DOI] [PubMed] [Google Scholar]
- Dean T, Xu R, Joiner W, Sehgal A, Hoshi T. Drosophila QVR/SSS modulates the activation and C-type inactivation kinetics of Shaker K+ channels. J Neuro. 2011 doi: 10.1523/JNEUROSCI.0502-11.2011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–126. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- Donlea JM, Ramanan N, Shaw PJ. Use-dependent plasticity in clock neurons regulates sleep need in Drosophila. Science. 2009;324:105–108. doi: 10.1126/science.1166657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas CL, Vyazovskiy V, Southard T, Chiu SY, Messing A, Tononi G, Cirelli C. Sleep in Kcna2 knockout mice. BMC Biol. 2007;5:42. doi: 10.1186/1741-7007-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drennan MD, Klauber MR, Kripke DF, Goyette LM. The effects of depression and age on the Horne-Ostberg morningness-eveningness score. J Affect Disord. 1991;23:93–98. doi: 10.1016/0165-0327(91)90096-b. [DOI] [PubMed] [Google Scholar]
- Faraco JH, Appelbaum L, Marin W, Gaus SE, Mourrain P, Mignot E. Regulation of hypocretin (orexin) expression in embryonic zebrafish. J Biol Chem. 2006;281:29753–29761. doi: 10.1074/jbc.M605811200. [DOI] [PubMed] [Google Scholar]
- Foltenyi K, Greenspan RJ, Newport JW. Activation of EGFR and ERK by rhomboid signaling regulates the consolidation and maintenance of sleep in Drosophila. Nat Neurosci. 2007;10:1160–1167. doi: 10.1038/nn1957. [DOI] [PubMed] [Google Scholar]
- Frank MG. The mystery of sleep function: current perspectives and future directions. Rev Neurosci. 2006;17:375–392. doi: 10.1515/revneuro.2006.17.4.375. [DOI] [PubMed] [Google Scholar]
- Franken P, Dijk DJ. Circadian clock genes and sleep homeostasis. Eur J Neurosci. 2009;29:1820–1829. doi: 10.1111/j.1460-9568.2009.06723.x. [DOI] [PubMed] [Google Scholar]
- Ganguly-Fitzgerald I, Donlea J, Shaw PJ. Waking experience affects sleep need in Drosophila. Science. 2006;313:1775–1781. doi: 10.1126/science.1130408. [DOI] [PubMed] [Google Scholar]
- Gilestro GF, Tononi G, Cirelli C. Widespread changes in synaptic markers as a function of sleep and wakefulness in Drosophila. Science. 2009;324:109–112. doi: 10.1126/science.1166673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel N, Banks S, Mignot E, Dinges DF. PER3 polymorphism predicts cumulative sleep homeostatic but not neurobehavioral changes to chronic partial sleep deprivation. PLoS One. 2009;4:e5874. doi: 10.1371/journal.pone.0005874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb DJ, DeStefano AL, Foley DJ, Mignot E, Redline S, Givelber RJ, Young T. APOE epsilon4 is associated with obstructive sleep apnea/hypopnea: the Sleep Heart Health Study. Neurology. 2004;63:664–668. doi: 10.1212/01.wnl.0000134671.99649.32. [DOI] [PubMed] [Google Scholar]
- Gottlieb DJ, O'Connor GT, Wilk JB. Genome-wide association of sleep and circadian phenotypes. BMC Med Genet. 2007;8(Suppl 1):S9. doi: 10.1186/1471-2350-8-S1-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves LA, Hellman K, Veasey S, Blendy JA, Pack AI, Abel T. Genetic evidence for a role of CREB in sustained cortical arousal. J Neurophysiol. 2003;90:1152–1159. doi: 10.1152/jn.00882.2002. [DOI] [PubMed] [Google Scholar]
- Hallam SJ, Jin Y. lin-14 regulates the timing of synaptic remodelling in Caenorhabditis elegans. Nature. 1998;395:78–82. doi: 10.1038/25757. [DOI] [PubMed] [Google Scholar]
- Hallmayer J, Faraco J, Lin L, Hesselson S, Winkelmann J, Kawashima M, Mayer G, Risch N, Mignot E. Narcolepsy is strongly associated with the TCR alpha locus. Nature Genetics. 2009 doi: 10.1038/ng.372. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Jones CR, Fujiki N, Xu Y, Guo B, Holder JL, Jr., Rossner MJ, Nishino S, Fu YH. The transcriptional repressor DEC2 regulates sleep length in mammals. Science. 2009;325:866–870. doi: 10.1126/science.1174443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks JC, Finn SM, Panckeri KA, Chavkin J, Williams JA, Sehgal A, Pack AI. Rest in Drosophila is a sleep-like state. Neuron. 2000a;25:129–138. doi: 10.1016/s0896-6273(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Hendricks JC, Lu S, Kume K, Yin JC, Yang Z, Sehgal A. Gender dimorphism in the role of cycle (BMAL1) in rest, rest regulation, and longevity in Drosophila melanogaster. Journal of Biological Rhythms. 2003;18:12–25. doi: 10.1177/0748730402239673. [DOI] [PubMed] [Google Scholar]
- Hendricks JC, Sehgal A, Pack AI. The need for a simple animal model to understand sleep. Progress in Neurobiology. 2000b;61:339–351. doi: 10.1016/s0301-0082(99)00048-9. [DOI] [PubMed] [Google Scholar]
- Hendricks JC, Williams JA, Panckeri K, Kirk D, Tello M, Yin JC, Sehgal A. A non-circadian role for cAMP signaling and CREB activity in Drosophila rest homeostasis. Nature Neuroscience. 2001;4:1108–1115. doi: 10.1038/nn743. [DOI] [PubMed] [Google Scholar]
- Hong SC, Lin L, Lo B, Jeong JH, Shin YK, Kim SY, Kweon Y, Zhang J, Einen M, Smith A, et al. DQB1*0301 and DQB1*0601 modulate narcolepsy susceptibility in Koreans. Hum Immunol. 2007;68:59–68. doi: 10.1016/j.humimm.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Huang ZL, Qu WM, Eguchi N, Chen JF, Schwarzschild MA, Fredholm BB, Urade Y, Hayaishi O. Adenosine A2A, but not A1, receptors mediate the arousal effect of caffeine. Nat Neurosci. 2005;8:858–859. doi: 10.1038/nn1491. [DOI] [PubMed] [Google Scholar]
- Hublin C, Kaprio J, Partinen M, Koskenvu M. Parasomnias: co-occurrence and genetics. Psychiatr Genet. 2001;11:65–70. doi: 10.1097/00041444-200106000-00002. [DOI] [PubMed] [Google Scholar]
- Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nat Rev Neurosci. 2009;10:199–210. doi: 10.1038/nrn2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac RE, Li C, Leedale AE, Shirras AD. Drosophila male sex peptide inhibits siesta sleep and promotes locomotor activity in the post-mated female. Proc Biol Sci. 2010;277:65–70. doi: 10.1098/rspb.2009.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto H, Kitamoto T. The steroid molting hormone Ecdysone regulates sleep in adult Drosophila melanogaster. Genetics. 2010;185:269–281. doi: 10.1534/genetics.110.114587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner WJ, Crocker A, White BH, Sehgal A. Sleep in Drosophila is regulated by adult mushroom bodies. Nature. 2006;441:757–760. doi: 10.1038/nature04811. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Silber MH, Fealey RD, Nippoldt TB, Auger RG, Vernino S. Neurophysiologic studies in Morvan syndrome. J Clin Neurophysiol. 2004;21:440–445. doi: 10.1097/00004691-200411000-00008. [DOI] [PubMed] [Google Scholar]
- Kalinchuk AV, McCarley RW, Porkka-Heiskanen T, Basheer R. Sleep deprivation triggers inducible nitric oxide-dependent nitric oxide production in wake-active basal forebrain neurons. J Neurosci. 2010;30:13254–13264. doi: 10.1523/JNEUROSCI.0014-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene AC, Duboue ER, McDonald DM, Dus M, Suh GS, Waddell S, Blau J. Clock and cycle limit starvation-induced sleep loss in Drosophila. Curr Biol. 2010;20:1209–1215. doi: 10.1016/j.cub.2010.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K, Joiner WJ, Wu MN, Yue Z, Smith CJ, Sehgal A. Identification of SLEEPLESS, a sleep-promoting factor. Science. 2008;321:372–376. doi: 10.1126/science.1155942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornum BR, Kawashima M, Faraco J, Lin L, Rico TJ, Hesselson S, Axtell RC, Kuipers H, Weiner K, Hamacher A, et al. Common variants in P2RY11 are associated with narcolepsy. Nat Genet. 2011;43:66–71. doi: 10.1038/ng.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, Yang FC, Snodgrass P, Li X, Scammell TE, Davis FC, Weitz CJ. Regulation of daily locomotor activity and sleep by hypothalamic EGF receptor signaling. Science. 2001;294:2511–2515. doi: 10.1126/science.1067716. [DOI] [PubMed] [Google Scholar]
- Kuo TH, Pike DH, Beizaeipour Z, Williams JA. Sleep triggered by an immune response in Drosophila is regulated by the circadian clock and requires the NFkappaB Relish. BMC Neurosci. 2010;11:17. doi: 10.1186/1471-2202-11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmesser S, Franken P, Feil S, Emmenegger Y, Albrecht U, Feil R. cGMP-dependent protein kinase type I is implicated in the regulation of the timing and quality of sleep and wakefulness. PLoS One. 2009;4:e4238. doi: 10.1371/journal.pone.0004238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laposky A, Easton A, Dugovic C, Walisser J, Bradfield C, Turek F. Deletion of the mammalian circadian clock gene BMAL1/Mop3 alters baseline sleep architecture and the response to sleep deprivation. Sleep. 2005;28:395–409. doi: 10.1093/sleep/28.4.395. [DOI] [PubMed] [Google Scholar]
- Lebestky T, Chang JS, Dankert H, Zelnik L, Kim YC, Han KA, Wolf FW, Perona P, Anderson DJ. Two different forms of arousal in Drosophila are oppositely regulated by the dopamine D1 receptor ortholog DopR via distinct neural circuits. Neuron. 2009;64:522–536. doi: 10.1016/j.neuron.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim AS, Scammell TE. The trouble with Tribbles: do antibodies against TRIB2 cause narcolepsy? Sleep. 2010;33:857–858. doi: 10.1093/sleep/33.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Longstreth WT, Jr., Ton TG, Koepsell TD. Narcolepsy and streptococcal infections. Sleep. 2009;32:1548. doi: 10.1093/sleep/32.12.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret S, Dorsaz S, Gurcel L, Pradervand S, Petit B, Pfister C, Hagenbuchle O, O'Hara BF, Franken P, Tafti M. Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci U S A. 2007;104:20090–20095. doi: 10.1073/pnas.0710131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret S, Franken P, Dauvilliers Y, Ghyselinck NB, Chambon P, Tafti M. Retinoic acid signaling affects cortical synchrony during sleep. Science. 2005;310:111–113. doi: 10.1126/science.1117623. [DOI] [PubMed] [Google Scholar]
- Marrus SB, Zeng H, Rosbash M. Effect of constant light and circadian entrainment of perS flies: evidence for light-mediated delay of the negative feedback loop in Drosophila. EMBO Journal. 1996;15:6877–6886. [PMC free article] [PubMed] [Google Scholar]
- Massicotte-Marquez J, Decary A, Gagnon JF, Vendette M, Mathieu A, Postuma RB, Carrier J, Montplaisir J. Executive dysfunction and memory impairment in idiopathic REM sleep behavior disorder. Neurology. 2008;70:1250–1257. doi: 10.1212/01.wnl.0000286943.79593.a6. [DOI] [PubMed] [Google Scholar]
- Mertens I, Vandingenen A, Johnson EC, Shafer OT, Li W, Trigg JS, De Loof A, Schoofs L, Taghert PH. PDF receptor signaling in Drosophila contributes to both circadian and geotactic behaviors. Neuron. 2005;48:213–219. doi: 10.1016/j.neuron.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Mignot E. Genetics of narcolepsy and other sleep disorders. Am J Hum Genet. 1997;60:1289–1302. doi: 10.1086/515487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot E. Why we sleep: the temporal organization of recovery. PLoS Biol. 2008;6:e106. doi: 10.1371/journal.pbio.0060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot E, Lammers GJ, Ripley B, Okun M, Nevsimalova S, Overeem S, Vankova J, Black J, Harsh J, Bassetti C, et al. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Arch Neurol. 2002;59:1553–1562. doi: 10.1001/archneur.59.10.1553. [DOI] [PubMed] [Google Scholar]
- Mignot E, Lin L, Rogers W, Honda Y, Qiu X, Lin X, Okun M, Hohjoh H, Miki T, Hsu S, et al. Complex HLA-DR and -DQ interactions confer risk of narcolepsy-cataplexy in three ethnic groups. Am J Hum Genet. 2001;68:686–699. doi: 10.1086/318799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot E, Takahashi JS. A circadian sleep disorder reveals a complex clock. Cell. 2007;128:22–23. doi: 10.1016/j.cell.2006.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa JM, Stevens TR, King SL, Caldarone BJ, Ibanez-Tallon I, Xiao C, Fitzsimonds RM, Pavlides C, Lester HA, Picciotto MR, et al. The prototoxin lynx1 acts on nicotinic acetylcholine receptors to balance neuronal activity and survival in vivo. Neuron. 2006;51:587–600. doi: 10.1016/j.neuron.2006.07.025. [DOI] [PubMed] [Google Scholar]
- Miyagawa T, Honda M, Kawashima M, Shimada M, Tanaka S, Honda Y, Tokunaga K. Polymorphism located between CPT1B and CHKB, and HLADRB1*1501-DQB1*0602 haplotype confer susceptibility to CNS hypersomnias (essential hypersomnia) PLoS One. 2009;4:e5394. doi: 10.1371/journal.pone.0005394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagawa T, Kawashima M, Nishida N, Ohashi J, Kimura R, Fujimoto A, Shimada M, Morishita S, Shigeta T, Lin L, et al. Variant between CPT1B and CHKB associated with susceptibility to narcolepsy. Nat Genet. 2008;40:1324–1328. doi: 10.1038/ng.231. [DOI] [PubMed] [Google Scholar]
- Muller-Preuss P, Rupprecht R, Lancel M. The effects of the neuroactive steroid 3 alpha,5 alpha-THDOC on sleep in the rat. Neuroreport. 2002;13:487–490. doi: 10.1097/00001756-200203250-00026. [DOI] [PubMed] [Google Scholar]
- Naidoo N, Casiano V, Cater J, Zimmerman J, Pack AI. A role for the molecular chaperone protein BiP/GRP78 in Drosophila sleep homeostasis. Sleep. 2007;30:557–565. doi: 10.1093/sleep/30.5.557. [DOI] [PubMed] [Google Scholar]
- Naidoo N, Giang W, Galante RJ, Pack AI. Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. J Neurochem. 2005;92:1150–1157. doi: 10.1111/j.1471-4159.2004.02952.x. [DOI] [PubMed] [Google Scholar]
- Naylor E, Bergmann BM, Krauski K, Zee PC, Takahashi JS, Vitaterna MH, Turek FW. The circadian clock mutation alters sleep homeostasis in the mouse. Journal of Neuroscience. 2000;20:8138–8143. doi: 10.1523/JNEUROSCI.20-21-08138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SE, Duricka DL, Campbell K, Churchill L, Krueger JM. Homer1a and 1bc levels in the rat somatosensory cortex vary with the time of day and sleep loss. Neurosci Lett. 2004;367:105–108. doi: 10.1016/j.neulet.2004.05.089. [DOI] [PubMed] [Google Scholar]
- Nitabach MN, Taghert PH. Organization of the Drosophila circadian control circuit. Curr Biol. 2008;18:R84–93. doi: 10.1016/j.cub.2007.11.061. [DOI] [PubMed] [Google Scholar]
- Nitz DA, van Swinderen B, Tononi G, Greenspan RJ. Electrophysiological correlates of rest and activity in Drosophila melanogaster. Curr Biol. 2002;12:1934–1940. doi: 10.1016/s0960-9822(02)01300-3. [DOI] [PubMed] [Google Scholar]
- Overeem S, Black JL, 3rd, Lammers GJ. Narcolepsy: immunological aspects. Sleep Med Rev. 2008;12:95–107. doi: 10.1016/j.smrv.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pack AI, Galante RJ, Maislin G, Cater J, Metaxas D, Lu S, Zhang L, Von Smith R, Kay T, Lian J, et al. Novel method for high-throughput phenotyping of sleep in mice. Physiol Genomics. 2007;28:232–238. doi: 10.1152/physiolgenomics.00139.2006. [DOI] [PubMed] [Google Scholar]
- Parisky KM, Agosto J, Pulver SR, Shang Y, Kuklin E, Hodge JJ, Kang K, Liu X, Garrity PA, Rosbash M, et al. PDF cells are a GABA-responsive wake-promoting component of the Drosophila sleep circuit. Neuron. 2008;60:672–682. doi: 10.1016/j.neuron.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman JL, McGill JJ, Keegan KP, Allada R. A dynamic role for the mushroom bodies in promoting sleep in Drosophila. Nature. 2006;441:753–756. doi: 10.1038/nature04739. [DOI] [PubMed] [Google Scholar]
- Poe GR, Walsh CM, Bjorness TE. Cognitive neuroscience of sleep. Prog Brain Res. 2010;185:1–19. doi: 10.1016/B978-0-444-53702-7.00001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prober DA, Rihel J, Onah AA, Sung RJ, Schier AF. Hypocretin/orexin overexpression induces an insomnia-like phenotype in zebrafish. J Neurosci. 2006;26:13400–13410. doi: 10.1523/JNEUROSCI.4332-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raizen DM, Zimmerman JE, Maycock MH, Ta UD, You YJ, Sundaram MV, Pack AI. Lethargus is a Caenorhabditis elegans sleep-like state. Nature. 2008;451:569–572. doi: 10.1038/nature06535. [DOI] [PubMed] [Google Scholar]
- Rihel J, Prober DA, Arvanites A, Lam K, Zimmerman S, Jang S, Haggarty SJ, Kokel D, Rubin LL, Peterson RT, et al. Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science. 2010;327:348–351. doi: 10.1126/science.1183090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci. 2007;8:171–181. doi: 10.1038/nrn2092. [DOI] [PubMed] [Google Scholar]
- Salas RE, Gamaldo CE, Allen RP. Update in restless legs syndrome. Curr Opin Neurol. 2010;23:401–406. doi: 10.1097/WCO.0b013e32833bcdd8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf MT, Naidoo N, Zimmerman JE, Pack AI. The energy hypothesis of sleep revisited. Prog Neurobiol. 2008;86:264–280. doi: 10.1016/j.pneurobio.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schormair B, Kemlink D, Roeske D, Eckstein G, Xiong L, Lichtner P, Ripke S, Trenkwalder C, Zimprich A, Stiasny-Kolster K, et al. PTPRD (protein tyrosine phosphatase receptor type delta) is associated with restless legs syndrome. Nat Genet. 2008;40:946–948. doi: 10.1038/ng.190. [DOI] [PubMed] [Google Scholar]
- Seugnet L, Suzuki Y, Vine L, Gottschalk L, Shaw PJ. D1 receptor activation in the mushroom bodies rescues sleep-loss-induced learning impairments in Drosophila. Curr Biol. 2008;18:1110–1117. doi: 10.1016/j.cub.2008.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Griffith LC, Rosbash M. Light-arousal and circadian photoreception circuits intersect at the large PDF cells of the Drosophila brain. Proc Natl Acad Sci U S A. 2008;105:19587–19594. doi: 10.1073/pnas.0809577105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Cirelli C, Greenspan RJ, Tononi G. Correlates of sleep and waking in Drosophila melanogaster. Science. 2000;287:1834–1837. doi: 10.1126/science.287.5459.1834. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Tononi G, Greenspan RJ, Robinson DF. Stress response genes protect against lethal effects of sleep deprivation in Drosophila. Nature. 2002;16:287–291. doi: 10.1038/417287a. [DOI] [PubMed] [Google Scholar]
- Sheeba V, Fogle KJ, Kaneko M, Rashid S, Chou YT, Sharma VK, Holmes TC. Large ventral lateral neurons modulate arousal and sleep in Drosophila. Curr Biol. 2008;18:1537–1545. doi: 10.1016/j.cub.2008.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Shimoda M, Yamaguchi T, Seong KH, Okamura T, Ishii S. Drosophila ATF-2 regulates sleep and locomotor activity in pacemaker neurons. Mol Cell Biol. 2008;28:6278–6289. doi: 10.1128/MCB.02242-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Rye DB, Hicks A, Petursson H, Ingason A, Thorgeirsson TE, Palsson S, Sigmundsson T, Sigurdsson AP, Eiriksdottir I, et al. A genetic risk factor for periodic limb movements in sleep. N Engl J Med. 2007;357:639–647. doi: 10.1056/NEJMoa072743. [DOI] [PubMed] [Google Scholar]
- Tafti M, Petit B, Chollet D, Neidhart E, de Bilbao F, Kiss JZ, Wood PA, Franken P. Deficiency in short-chain fatty acid beta-oxidation affects theta oscillations during sleep. Nat Genet. 2003;34:320–325. doi: 10.1038/ng1174. [DOI] [PubMed] [Google Scholar]
- Thimgan MS, Suzuki Y, Seugnet L, Gottschalk L, Shaw PJ. The perilipin homologue, lipid storage droplet 2, regulates sleep homeostasis and prevents learning impairments following sleep loss. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, Fu YH. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001;291:1040–1043. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- Trenkwalder C, Hogl B, Winkelmann J. Recent advances in the diagnosis, genetics and treatment of restless legs syndrome. J Neurol. 2009;256:539–553. doi: 10.1007/s00415-009-0134-9. [DOI] [PubMed] [Google Scholar]
- Van Buskirk C, Sternberg PW. Epidermal growth factor signaling induces behavioral quiescence in Caenorhabditis elegans. Nat Neurosci. 2007;10:1300–1307. doi: 10.1038/nn1981. [DOI] [PubMed] [Google Scholar]
- van Swinderen B, Nitz DA, Greenspan RJ. Uncoupling of brain activity from movement defines arousal States in Drosophila. Curr Biol. 2004;14:81–87. [PubMed] [Google Scholar]
- Vecsey CG, Baillie GS, Jaganath D, Havekes R, Daniels A, Wimmer M, Huang T, Brown KM, Li XY, Descalzi G, et al. Sleep deprivation impairs cAMP signalling in the hippocampus. Nature. 2009;461:1122–1125. doi: 10.1038/nature08488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola AU, Archer SN, James LM, Groeger JA, Lo JC, Skene DJ, von Schantz M, Dijk DJ. PER3 polymorphism predicts sleep structure and waking performance. Curr Biol. 2007;17:613–618. doi: 10.1016/j.cub.2007.01.073. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nature Neuroscience. 2008;11:200–208. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- White DP. Pathogenesis of obstructive and central sleep apnea. Am J Respir Crit Care Med. 2005;172:1363–1370. doi: 10.1164/rccm.200412-1631SO. [DOI] [PubMed] [Google Scholar]
- White JG, Albertson DG, Anness MA. Connectivity changes in a class of motoneurone during the development of a nematode. Nature. 1978;271:764–766. doi: 10.1038/271764a0. [DOI] [PubMed] [Google Scholar]
- Williams JA, Sathyanarayanan S, Hendricks JC, Sehgal A. Interaction between sleep and the immune response in Drosophila: a role for the NFkappaB relish. Sleep. 2007;30:389–400. doi: 10.1093/sleep/30.4.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelmann J. Genome-wide association study identifies novel Restless Legs Syndrome susceptibility loci on 2p14 and 16q12.1. PLoS Genetics. 2011 doi: 10.1371/journal.pgen.1002171. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelmann J, Lichtner P, Schormair B, Uhr M, Hauk S, Stiasny-Kolster K, Trenkwalder C, Paulus W, Peglau I, Eisensehr I, et al. Variants in the neuronal nitric oxide synthase (nNOS, NOS1) gene are associated with restless legs syndrome. Mov Disord. 2008;23:350–358. doi: 10.1002/mds.21647. [DOI] [PubMed] [Google Scholar]
- Winkelmann J, Schormair B, Lichtner P, Ripke S, Xiong L, Jalilzadeh S, Fulda S, Putz B, Eckstein G, Hauk S, et al. Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat Genet. 2007;39:1000–1006. doi: 10.1038/ng2099. [DOI] [PubMed] [Google Scholar]
- Wisor JP, O'Hara BF, Terao A, Selby CP, Kilduff TS, Sancar A, Edgar DM, Franken P. A role for cryptochromes in sleep regulation. BMC Neurosci. 2002;3:20. doi: 10.1186/1471-2202-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MN, Ho K, Crocker A, Yue Z, Koh K, Sehgal A. The effects of caffeine on sleep in Drosophila require PKA activity, but not the adenosine receptor. J Neurosci. 2009;29:11029–11037. doi: 10.1523/JNEUROSCI.1653-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MN, Joiner WJ, Dean T, Yue Z, Smith CJ, Chen D, Hoshi T, Sehgal A, Koh K. SLEEPLESS, a Ly-6/neurotoxin family member, regulates the levels, localization and activity of Shaker. Nature Neuroscience. 2010;13:69–75. doi: 10.1038/nn.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, Saigoh K, Ptacek LJ, Fu YH. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature. 2005;434:640–644. doi: 10.1038/nature03453. [DOI] [PubMed] [Google Scholar]
- Yokogawa T, Marin W, Faraco J, Pezeron G, Appelbaum L, Zhang J, Rosa F, Mourrain P, Mignot E. Characterization of sleep in zebrafish and insomnia in hypocretin receptor mutants. PLoS Biol. 2007;5:e277. doi: 10.1371/journal.pbio.0050277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, Joiner WJ, Sehgal A. A sleep-promoting role for the Drosophila serotonin receptor 1A. Curr Biol. 2006;16:1051–1062. doi: 10.1016/j.cub.2006.04.032. [DOI] [PubMed] [Google Scholar]
- Zheng X, Sehgal A. Probing the relative importance of molecular oscillations in the circadian clock. Genetics. 2008;178:1147–1155. doi: 10.1534/genetics.107.088658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman JE, Rizzo W, Shockley KR, Raizen DM, Naidoo N, Mackiewicz M, Churchill GA, Pack AI. Multiple mechanisms limit the duration of wakefulness in Drosophila brain. Physiol Genomics. 2006;27:337–350. doi: 10.1152/physiolgenomics.00030.2006. [DOI] [PubMed] [Google Scholar]