

Abstract
This is the first study to show that cigarette smoking induced the LKB1/PEA 3/ΔNp63-dependent transcriptional regulation of inflammatory molecules, such as COX-2/PTGS-2. Using mainstream smoke extract (MSE) and sidestream smoke extract (SSE) as modeling tools for primary and secondhand smoking, we found that both MSE and SSE downregulated protein levels for LKB1, while upregulated protein levels for PEA 3 and COX-2 in a dose-dependent manner. Using the endogenous ChIP analysis, we further found that the C/EBPβ, NFκB, NF-Y (CHOP), PEA 3 (ETS) and ΔNp63 proteins bound to the specific area (-550 to -130) of the COX-2 promoter, while forming multiple protein complexes in lung cancer cells exposed to MSE and SSE. Our results define a novel link between various transcription factors occupying the COX-2 promoter and cellular response to cigarette smoke exposure bringing a new component, ΔNp63α, showing a critical role for cooperation between various chromatin components in regulation of COX-2 expression and, therefore strengthening the central role of inflammatory process in tumorigenesis of epithelial cells, especially after cigarette smoke exposure (both primary and secondhand).
1. Introduction
Tobacco smoke contains over 4,700 chemical components that have been implicated in the etiology of oxidative stress-related diseases e.g., chronic obstructive pulmonary disease, Parkinson's disease, Alzheimer disease, asthma, cancer and cardiovascular disease [1–7]. Epidemiological studies support the notion that lung cancers are directly caused by cigarette primary and secondhand smoking [1–3]. Carcinogen exposure and chronic inflammation are two important events in tumor development, and both have been implicated in the development of many human epithelial cancers [4–6]. Exposure to external factors including cigarette smoking, infectious agents, dietary carcinogens and hormonal imbalances could injure the tissue (e.g., lung) and lead to chronic inflammation [5, 6]. At the cellular and molecular levels, cigarette smoking might induce oxidative stress and DNA damage, implicated in the etiology of cancer and resulting in modulation of reactive oxygen species (ROS) production and the cell's own antioxidant defenses, therefore leading to activation of numerous signaling pathways underlying apoptosis and autophagy [8–10].
Epithelial/mesenchymal transition (EMT) and increased cell motility/migratory/invasive phenotype were also found to occur during the development and progression of lung epithelial cancers [11–13]. Thus, the understanding of mechanisms underlying these processes (apoptosis, angiogenesis, cell migration, invasiveness) in lung cancer would assist development of new therapeutic strategies [5, 6, 13].
Studies of genetic mechanisms underlying lung cancer, along with other human cancers, demonstrated that tobacco exposure is causing inactivation of tumor suppressor genes via genetic/epigenetic changes affecting many cellular processes [1, 13, 14]. LKB1 tumor suppressor gene (also known as serine/threonine kinase-11, STK11) is capable to regulate other protein's function by phosphorylation, thereby affecting cell proliferation and survival [15–21]. Smoking has been linked to human epithelial cancers, which overexpress proteins implicated in inflammatory signaling pathways [e.g., NFκB, cyclin D1, cyclooxygenase (COX)-2 (also known as PTGS-2, prostaglandin-endoperoxide synthase-2)] [7, 22–32].
We previously showed that that LKB1 physically and functionally associates with PEA3 leading to the PEA3 phosphorylation and subsequent PEA3 protein degradation via proteasomedependent pathway [16]. We also showed that the downregulated PEA3 expression and activity leads to a subsequent downregulation of COX-2/PTGS-2 expression [16]. We further showed that cells expressing mutant LKB1 deficient of kinase activity failed to downregulate PEA3 and activate COX-2 transcription, while increased cell invasiveness compared to cells with wild-type LKB1 [16]. Similarly, LKB1 knockdown by siRNA dramatically increased migration/invasiveness shown by lung cancer cells in vitro. However, lung cancer cells transfected with PEA3siRNA displayed decreased invasiveness, while the PEA3 forced expression resulted in decrease of epithelial markers and increase of mesenchymal markers suggesting that PEA3 stabilization due to LKB1 inactivation leads to a greater cancer cell invasiveness [16].
In the current study, we provide the first evidence that cigarette smoke treatment [mainstream smoke extract (MSE) and sidestream smoke extract (SSE)] of cultured lung normal and cancer cells decreased LKB1 expression and elevated expression of both PEA3 (polyomavirus enhancer activator 2, also known as E1A enhancer binding protein 4) factor and COX-2/ PTGS-2 inflammatory signaling molecule. We showed that cigarette smoking affects the molecular processes underlying EMT of lung cancer/epithelial cells and involving LKB1 inactivation and PEA3/ΔNp63-mediated regulation of COX-2/PTGS-2. We defined a novel LKB1/PEA3/ΔNp63α molecular pathway leading to COX-2/PTGS-2 regulation in human lung cancer cells exposed to mainstream and sidestream smokes therefore linking ΔNp63α to tumorigenesis and inflammation processes as a new biomarker for oxidative stress and DNA damage [33].
2. Results
LKB1 is downregulated in lung tumor samples from patients affected by smoking. Recent report shows that normal human lung epithelial tissues overexpress LKB1, while in primary lung tumors (squamous cell carcinomas), LKB1 levels were dramatically downregulated [15, 16]. We previously showed that LKB1 overexpression led to a binding to and subsequent phosphorylation of the PEA3 transcription factor followed by downregulation of PEA3 and in turn inhibition of COX-2/PTGS-2 expression [16]. In this study, we examined whether cigarette smoking exposure of lung normal and cancer cells affect the expression levels of LKB1, PEA3 and COX-2. We exposed normal human bronchiolar epithelial (NHBE) cells and H1299 lung cancer cells to 0.5% MSE for 48 h, and found that LKB1 protein levels were downregulated, while levels for PEA3 and COX-2/PTGS-2 were upregulated in both cell lines upon MSE exposure (Fig. 1A). We found that H1299 cells exposed to MSE in dose-dependent manner displayed altered expression of LKB1, PEA3 and COX-2/PTGS-2 (Fig. 1B). We further examined the effect of SSE on the protein levels of LKB1, PEA3 and COX-2/PTGS-2 in H1299 cells. We then found that 1% SSE added to cells for 48 h decreased the LKB1 protein levels, while upregulated the PEA3 and COX-2 protein levels (Fig. 2). We thus observed that both MSE and SSE decreased LKB1 protein levels in lung cancer cells.
PEA3 physically associates with the COX-2 specific transcription factors in lung cancer cells upon cigarette smoke exposure. We further examined molecular mechanisms underlying LKB1 downregulation and its effects on PEA3 transcriptional regulation of COX-2/PTGS-2 in CSE-exposed lung cancer cells and lung normal epithelial cells. We thus tested whether various putative transcription factors involved in regulation of COX-2 expression in lung cancer cells upon cigarette smoke exposure. First, we defined the consensus sequences for the potential transcription factors in the COX-2 promoter sequence (Sup. Material) using the TFSEARCH web-engine (http://mbs.cbrc.jp/rese-arch/db/TFSEARCH.html).
The following cis-regulatory elements were found in the 1,700 bp COX-2 promoter sequence: C/EBPβ, NFκB, NF-Y, PEA3 and p63 (Fig. 3).
We then accessed whether these transcription factors endogenously bind to the COX-2 promoter in lung cancer cells after cigarette smoke exposure. We found that both MSE and SSE induced binding of the C/EBPβ, NFκB, NF-YA, PEA3, p53 and p63 proteins to COX-2 promoter evidenced by ChIP assay followed by RT-PCR assay given rise to a 420 bp product (Fig. 4). Since lung cancer cells were previously shown to predominantly express ΔNp63α isoform of p63 (reviewed in ref. 34), the antibody that exclusively recognizes the ΔNp63 was used in these experiments [34, 35].
Previous reports pointed out the possibility for LKB1 to form protein-protein complexes with p53 (reviewed in ref. 36). We therefore tested whether p53 homolog ΔNp63α is forming protein complexes with LKB1 and the other transcription factors occupying the COX-2 promoter in lung cancer cells after cigarette smoke exposure. We found that, indeed, ΔNp63α formed protein-protein complexes with LKB1 in untreated cells (Fig. 5A), while the amount of these ΔNp63/LKB1 protein complexes dramatically decreased upon cigarette smoke exposure (Fig. 5A). In addition, the ΔNp63α protein associated with C/EBPβ, NFκB, NF-YA and PEA3 in cells exposed to both MSE and SSE (Fig. 5A).
We previously showed that the PEA3 ETS domain interacted with the LKB1 kinase domain pLexA-LKB1-KD (1–300) in vitro [16]. We also showed that the LKB1/ PEA3 protein complexes formed in transfected cells and endogenously in H1299 lung cancer cells found exclusively at the nucleus [16]. To further understand the effect of molecular interactions between LKB1 and PEA3 in lung cancer cells exposed to cigarette smoke extract, we performed immunoprecipitation analysis of the LKB1/ PEA3 protein-protein complexes. We thus found that, while LKB1 is downregulated in lung cancer cells upon cigarette smoke exposure by both MSE and SSE (Figs. 1 and 2), the formation of protein complexes between LKB1 and PEA3 is dramatically diminished during cellular response to cigarette smoke (Fig. 5B).
Cigarette smoking exposure induces invasiveness, foci formation and survival of lung cancer cells. Our previous results supported the notion that the PEA3 overexpression can mediate increased cell migration/cell invasiveness of lung cancer cells potentially through EMT-dependent mechanism [16].
We further examined whether MSE or SSE exposure leads to change of cellular characteristics of lung cancer cells and whether siRNA knockdown of PEA3 or COX-2 expression would affect these potential changes. A427 lung cancer cells with altered LKB1 protein kinase16 were transiently transfected with scramble siRNA (Fig. 6A–C, samples 1, 4 and 7), PEA3 siRNA(Fig. 6A–C, samples 2, 5 and 8) and COX-2/PTGS-2 (Fig. 6A–C, samples 3, 6 and 9) for 48 h. Cells then were exposed to control medium (sample 1–3), 1% SSE (samples 4–6), 0.5% MSE (samples 7–9). We then tested the ability of A427 cells to migrate in Matrigel, form foci/clones in soft agar, and undergo apoptosis/survival after cigarette smoke exposure. We found that both SSE and MSE increased cell migration/cell invasiveness (Fig. 6A, samples 4 and 7), cellular ability to form foci/ clones in soft agar (Fig. 6B, samples 4 and 7) and cell survival (Fig. 6C, samples 4 and 7). However, siRNA against PEA3 (samples 5 and 8) and COX-2 (samples 6 and 9) dramatically modulated this increase in cell migration, foci formation and cell survival (Fig. 6A–C).
3. Discussion
Cigarette primary and secondhand smoking is a direct cause of lung cancer associated with inactivation of tumor suppressor genes (e.g., LKB1) via genetic/epigenetic changes therefore affecting many cellular processes including oxidative and DNA damage, inflammatory processes, cell migration and invasiveness. Search and “assessment of novel biomarkers” and therapeutic targets affected by oxidative stress/DNA damage implicated in the cellular response to tobacco-induced pathologies “may have critical clinical utility for the formulation of novel therapeutic options” [33].
This is the first study to be undertaken to support a notion that cigarette smoking is an initiating event for loss of LKB1 expression/function and the LKB1-mediated transcriptional regulation of inflammatory molecules, such as COX-2/PTGS- 2. We hypothesized that cigarette smoking affects the molecular processes underlying EMT of lung cancer/epithelial cells, therefore linking together inactivation of LKB1 tumor suppressor and PEA3-mediated transcriptional regulation of inflammatory signaling molecules (e.g., COX-2/PTGS-2).
Using MSE and SSE as modeling tools for primary and secondhand smoking, we found that both MSE and SSE downregulated protein levels for LKB1, while upregulated protein levels for PEA3 and COX-2/PTGS-2. We further found that MSE and SSE induced these changes in a dose-dependent manner. We next determined that the COX-2/PTGS-2 promoter sequence contains many regulatory sequences recognized by key transcriptional regulators, such as C/EBPβ, NFκB, NF-YA, PEA3 (ETS), STAT and p53.
In addition to several p53 consensus sequences (-1,446 to -1,431; 1,283 to -1,266; -986 to -969 and -941 to -928), we defined a couple of p63 responsive elements (RE) in the COX-2 promoter sequence located at the positions -460 to -441 and -200 to -182.
As a member of the p53 gene family, p63 regulates downstream target gene expression by binding to sequence-specific response elements similar to those of p53 (reviewed in ref. 37). However, p63RE was shown to be distinct from the canonical p53RE, suggesting that p53 preferentially binds to the RRR CAT GYY Y sequence, whereas p63 preferentially recognizes RRR CGT GYY Y (reviewed in ref. 37). Using the endogenous ChIP analysis of the COX-2/PTGS-2 promoter in lung cancer cells upon cigarette smoke exposure (MSE and SSE), we found that the C/EBPβ, NFκB, NF-YA, PEA3 (ETS) and ΔNp63 proteins bound to the narrow area of the COX-2/PTGS-2 promoter spanning from -550 to -130 upstream of the transcription start site (Fig. 3). We then showed that after cigarette smoke exposure (MSE and SSE) of lung cancer cells, these transcription factors C/EBPβ, NFκB, NF-YA, PEA3 (ETS) and ΔNp63 formed protein complexes. While levels of ΔNp63, p-ΔNp63 (reviewed in ref. 35), C/EBPβ and PEA3 increased, the levels of NFκB and NF-YA remained the same after cigarette smoke exposure.
Several transcription factors (C/EBPβ, NFκB, STAT3, p53 and PEA3) shown to activate COX-2 expression were previously reported playing critical roles in gene expression during lung cancer cellular response to tobacco smoking exposure [21–24, 27, 28, 36–41]. Moreover, PEA3 was found to be activated by cigarette smoke exposure and regulate MMP-1 transcription by binding to a smoke response region in the distal MMP-1 promoter suggesting that PEA3 function has implications for smoking-related cancer, heart disease and emphysema [30]. Furthermore, by association with other chromatin/transcription regulators, PEA3 was shown to activate Twist expression and promote cell migration/cell invasiveness in human cancer cells suggesting its role in cancer metastasis [11, 42, 43]. PEA3 along with CBP/p300 was also shown to be a key transcriptional regulator of many genes (MMP-1, 2, 7 and 9, COX-2/ PTGS-2) implicated in inflammation and cell invasiveness [44–47].
The current study defines a novel link between various transcription factors occupying the COX-2/PTGS-2 promoter and cellular response to cigarette smoke exposure bringing a new component, ΔNp63α, showing a critical role for cooperation between various chromatin components in regulation of COX-2/ PTGS-2 expression and, therefore strengthening the central role of inflammatory process and oxidative stress in tumorigenesis of epithelial cells, especially after cigarette smoke exposure (both primary and secondhand).
Numerous signaling pathways are implicated in cellular response to oxidative and genotoxic stress potentially induced by smoke exposure. For example, the ROS-induced interplay between the DNA-damage and oxidative stress through an activation of ATM-dependent phosphorylation of p53 family members and TSG tumor suppressor via the PARP1-LKB1-AMPK-mTOR metabolic pathway known to stimulate autophagy [48–50].
Furthermore, as a substrate for ATM-dependent phosphorylation, ΔNp63α was shown serving as a pro-survival factor by upregulating a glutathione peroxidase (GPX2) to reduce the p53-dependent oxidative stress-induced apoptotic response [51]. p63 was also shown to transcriptionally regulate REDD1 that implicates ROS in the p53-dependent DNA damage response and in p63-mediated regulation of epithelial differentiation [52]. ROS were found to activate p53 family members (p53, p63, and p73) inducing the expression of ferredoxin reductase (FDXR), which in turn sensitizes cells to ROS-mediated apoptosis [53].
As a target for p53 family proteins, COX-2 was shown producing oxidative damage and affecting the stress-induced cellular senescence [54]. Hyperoxia was also shown to induce the cellular senescence through the p53-LKB1-AMPK pathway [55].
ROS were further shown to stimulate cancer cell growth by regulating AMPK-COX-2 pathway [56]. Sp1/Sp3-dependent transcriptional regulation of COX-2 was shown playing an essential role in the modulation of COX-2 expression that mediates neuronal homeostasis and survival by preventing DNA damage [57]. Furthermore, oxidative stress was shown to induce cGMPprotein kinase-mediated thioredoxin peroxidase 1 transcription through PEA3, AP-1, c-Myc and c-Jun transcription factors [58].
Accumulated data strongly suggest that continuous (chronic) upregulation of pro-inflammatory mediators (e.g., TNFalpha, IL-1beta, IL-6, COX-2, NOS-2) are induced during the aging process due to an age-related redox imbalance that activates many pro-inflammatory signaling pathways, including the NFκB signaling pathway [59]. Both ROS and pro-inflammatory genes (e.g., COX-2) were found contributing to the expansion of cellular inflammatory responses and reduce the expression of genes required to maintain synaptic structure and function ultimately leading to progressive dysfunction, apoptosis and/or necrosis and brain cell death [60]. Pro-inflammatory genes shown playing a role in neurodegeneration (e.g., Alzheimer disease) are transiently activated by the heterodimeric oxygen-sensitive protein-protein complexes between NFκB and HIF-1α (reviewed in ref. 62).
Many cellular responses to tobacco smoke, such as oxidative stress/DNA damage, EMT, altered adhesion-mediating signaling pathways and altered protein degradation, chromatin modifications/epigenetic changes, angiogenesis and autophagy/apoptosis complement the inflammatory/neoplastic processes as the key underlying mechanisms in both chronic obstructive pulmonary disease, cardiovascular disease, lung cancer, aging and age-related diseases [4–13, 24, 28, 49, 54–56, 59, 60]. Thus, understanding the cellular and molecular mechanisms underlying these processes will provide novel venues for devising therapeutic strategies against smoke-related diseases.
4. Materials and Methods
Preparation of CSE
Mainstream smoke extract (MSE) and sidestream smoke extract (SSE) made from research-grade cigarettes (2R4F, from Tobacco Health Research, University of Kentucky, Louisville) contain nicotine: 0.85 mg/cigarette and tar: 9.70 mg/cigarette as previously described [61]. SSE was collected from the burning end of the cigarettes without puffing at the rate of 200 ml/min and MSE was collected with 35 ml/min puff per 2 sec using the opposite end of two smoking machines (MasterFlex Pump Systems, Cole-Parmer Instrument). Briefly, the smoke of 20 cigarettes for MSE and 40 cigarettes for SSE was bubbled into each flask containing 20 ml of pre-warmed phosphate buffer saline. The aqueous smoke extract was filtered through 0.22 μm pore syringe filter to remove large particles. The smoke bubbled into MSE flask was acidic and that into SSE flask appeared to be basic, therefore the pH of each solution was adjusted to 7.4. The solution was aliquoted and kept frozen at -80°C until use. The concentration of SSE was monitored at the absorbance of 1 at A230 was considered 100%. The concentration of MSE solution was considered 100% [61]. MSE and SSE were used to imitate cigarette primary smoking and secondhand smoking, respectively.
Cell cultures and transfections
Human lung cancer cell lines (A427 and H1299) and normal human bronchiolar epithelial (NHBE) cells were purchased from the American Type Culture Collection (ATCC) were grown in the recommended media. The 200 pmol/six-well plate of scramble siRNA, siRNA against PEA3 (sc-36205) and COX-2 (sc-29279) were purchased from Santa Cruz Biotechnology and were transiently introduced into cells for 24 h using FuGENE-6 (Roche Molecular Biochemicals) as previously described [16]. Cells were then exposed to control medium or various concentrations of both MSE and SSE for indicated periods of time [61].
Chromatin immunoprecipitation (ChIP) assay
H1299 cells (2 x 106) were transfected exposed to control medium, MSE and SSE. ChIP was done using a ChIP assay kit (Upstate Cell Signaling Solutions). H1299 cells (2 x 106) were cross-linked with 1% formaldehyde and ChIP was performed as previously described [16, 35]. After reversing the cross-links, chromatin was purified using the QIAquick PCR Purification kit (Qiagen, Inc.) and samples were eluted with 30 μl of elution buffer. Chromatin was immunoprecipitated with 1 μg of primary antibodies against PEA3 (clone 1A2G3, sc-130661, Santa Cruz Biotechnology), NFκB p65 subunit (ab7970, Abcam), ΔNp63, C/EBP/β (#3087, Cell Signaling Technology) and NF-YA (ab6558, Abcam). Immunoprecipitates were used as templates for PCR amplification of the COX-2 promoter with the following primers: COX-2, sense primer, (-550) 5'-TCC CGA CGT GAC TTC CTC GA (-530) and antisense primer, (-150) 5'-GGA GAG GAG GGA AAA ATT TG-3' (-130) yielding the PCR product of 420 bp, which was amplified as follows: 94°C for 1 minute, 55°C for 1 minute and 72°C for 30 seconds using Platinum Taq I DNAa polymerase (Invitrogen) for 37 cycles [16].
Immunoblotting and immunoprecipitation
We used primary antibodies against PEA3 (clone 1A2G3, sc-130661) and ΔNp63(clone (3F306): sc-71825) from Santa Cruz Biotechnology; against LKB1 (clone 27D10, ab#3050) and C/EBP/β (#3087) from Cell Signaling Technology; NFκB p65 subunit (ab7970) and NF-YA (ab6558) from Abcam; against b-actin (Sigma), COX-2/PTGS-2 (Cayman Chemical) or ΔNp63 (Ab-1, EMD) for immunoblotting analysis and immunoprecipitation [16, 35]. Immunoblots were scanned and quantification was carried out by Image Quant software version 3.3 (Molecular Dynamics). Values were expressed as percentage of a control sample (defined as 100%). 5 x 105 cells per well were plated into six-well plates and exposed to control medium, MSE and SSE. Cells were lysed in buffer [50 mmol/l Tris (pH 7.5), 150 mmol/l NaCl, 5 μg/ml aprotinin, pepstatin, 1% Nonidet P-40, 1 mmol/l EDTA, 0.25% deoxycholate]. For immunoprecipitation, total cell lysates (500 μl) were precipitated with 5 μg of the indicated primary antibodies overnight at 4°C followed by incubation with anti-rabbit or anti-mouse immunoglobulins (IgG)-coupled to agarose beads (Sigma) and washed with lysis buffer before being resolved by SDS-PAGE [16, 35].
Cell invasion/Matrigel assay
Cells (1 x 104) in 0.5 ml of serum-free MEM were added to each well of 24-well/8-μm pore invasion membrane chambers coated with Matrigel (BD Discovery Labware). The lower chambers contained 10% fetal bovine serum (FBS) in MEM to serve as a chemoattractant. Cells were allowed to migrate or invade over the course of 48 h. Chambers were fixed with 100% methanol for 2 min, stained with 0.5% crystal violet for 2 min, rinsed in water and examined under a bright-field microscope. Values for invasion and migration were obtained by counting five fields per membrane (20x objective) and represented the average of three independent experiments done over multiple days [16, 61].
Colony formation assay
Soft agar colony formation assays were carried out in six-well dishes. Cells (1 x 104) suspended in 2 ml of 0.36% bactoagar (Becton Dickinson) with growth medium (RPMI 1640 supplemented with 10% FBS) were added on a base layer of 0.72% bactoagar containing culture medium [61]. The plates were incubated at 37°C in a 5% CO2 incubator for 3 weeks. Colonies were counted under low magnification (x100).
MTT survival assay
The number of cells in each well after treatment was estimated using the 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (ATCC) as previously described [35, 61]. MTT labeling reagent (final concentration, 0.5 mg/ml) was added to cells in 96-well culture plates (final volume, 100 μl culture medium/well) and incubated for 4 h at 37°C in a humidified atmosphere of 10% CO2. Cells were then solubilized overnight and the samples were quantified at 570 nm using a microtiter plate reader (Bio-Rad Laboratories).
Note
Supplementary materials can be found at: Supplementary materials can be found at: http://www.landesbioscience.com/supplement/RatovitskiOMCL3-5-Sup.pdf.
Figure 1.
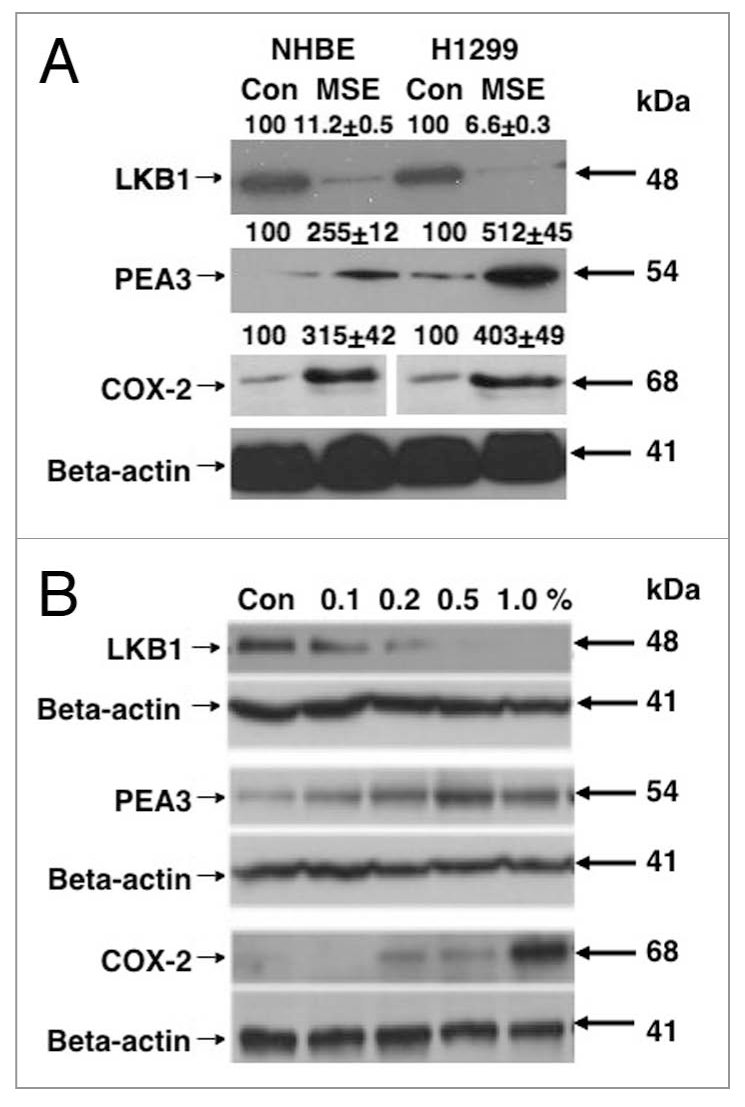
MSE affects the levels for LKB1, PEA 3 and PTGS-2/COX-2 in normal lung and lung cancer cells. (A) NHBE normal lung and H1299 lung cancer cells were exposed to 0.5% MSE for 48 h. (B) H1299 lung cancer cells were incubated with MSE in a dose-dependent manner (Con, control, 0.2, 0.5, 1.0 and 2% MSE) for 24 h. Total lysates were analyzed by immunoblotting with antibodies against LKB1, PEA 3, PTGS-2/COX-2 and b-actin. Immunoblots were scanned and quantified by Image Quant software version 3.3 and normalized for the b-actin protein levels. Median data expressed as percentages of data obtained from control sample representing three independent experiments were shown above blot images. Statistical analysis was performed using a student t-test [16].
Figure 2.
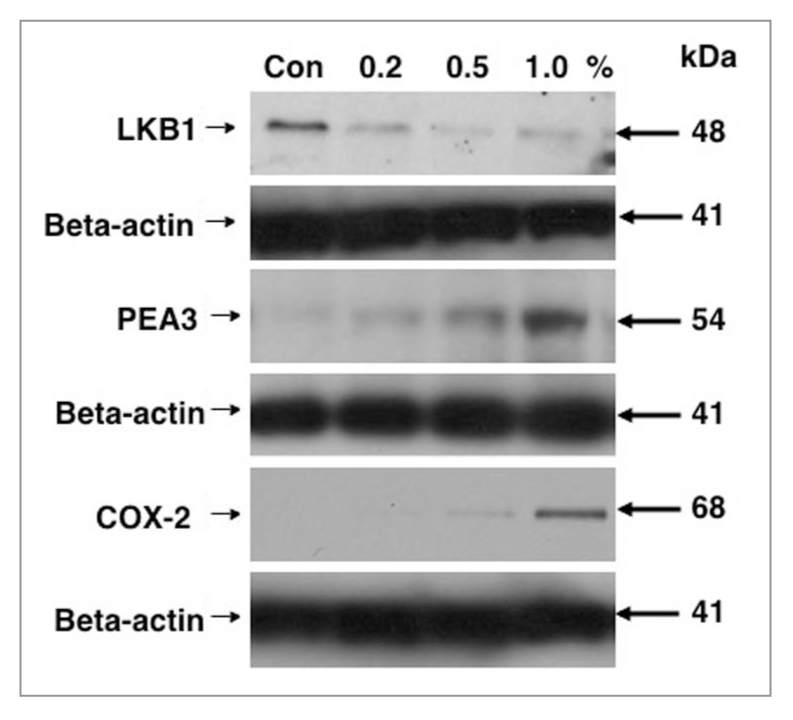
SSE affects the levels for LKB1, PEA 3 and COX-2/PTGS-2 in lung cancer cells. H1299 cells were incubated with SSE in a dose-dependent manner (Con, control, 0.2, 0.5 and 1.0% SSE) for 48 h. Total lysates were analyzed by immunoblotting with antibodies against LKB1, PEA 3, COX-2/ PTGS-2 and b-actin. Immunoblots were scanned and quantified by Image Quant software version 3.3 and normalized for the b-actin protein levels. Median data expressed as percentages of data obtained from control sample representing three independent experiments were shown above blot images. Statistical analysis was performed using a student t-test [16].
Figure 3.

Schematic structure of COX-2/PTGS-2 promoter. Sequence of the human 1,700 bp COX-2/PTGS-2 promoter was found in the UCSC human genome website and certain potential transcription factor cis-elements were defined using the TFSEA RCH website. The transcription factor sequences are shown in boxes. The TSS (transcription start site) is shown in bold and shadow capital letter and underlined. Coding sequence is shown in italics.
Figure 4.

Cigarette smoke exposure induces binding of endogenous transcription factors to the COX-2/PTGS-2 promoter. ChIP assay of the binding of NF-YA, ΔNp63α or phospho-ΔNp63α and PEA 3 to COX-2/ PTGS-2 promoter using indicated antibodies. A427 lung cancer cells (expressing LKB1 with altered protein kinase ability) were exposed to control medium, 1% SSE and 0.5% MSE for 24 h. PCR was used to amplify regions of the COX-2/PTGS-2 promoter around the PEA 3-binding sites. Negative control (normal rabbit and normal mouse IgGs) was used to confirm the binding specificity (data not shown).
Figure 5.
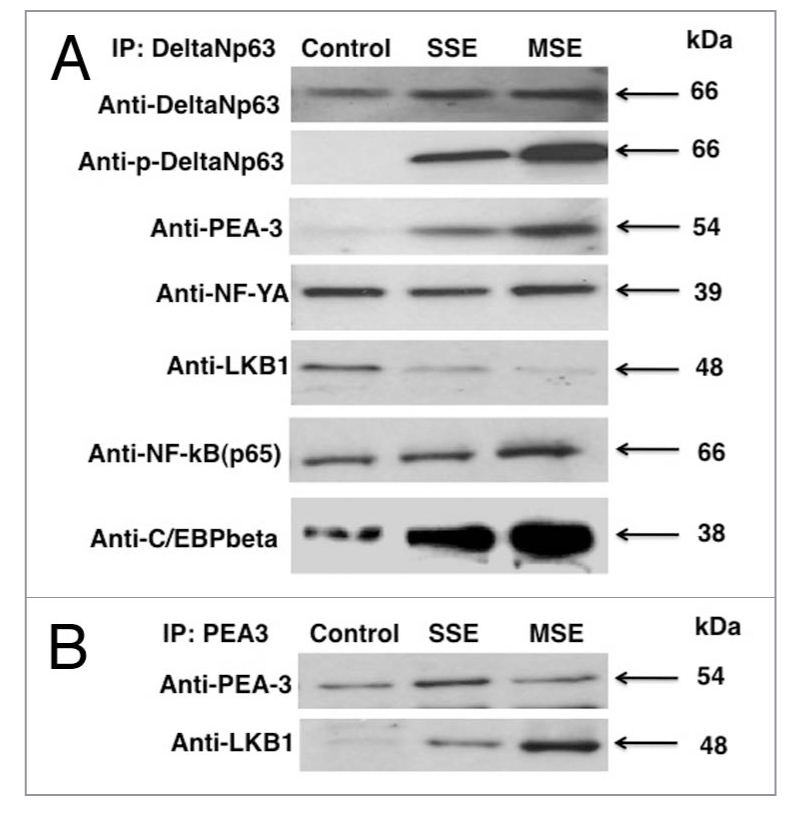
Transcription factors form complexes in lung cancer cells upon cigarette smoke exposure. A427 cells were exposed to control medium, 1% SSE and 0.5% MSE for 24 h. (A) Immunoprecipitation (IP) was performed with Ab-1 antibody exclusively recognizing ΔNp63α protein. Complexes were detected with indicated antibodies. (B) Immunoprecipitation (IP) was performed with PEA 3 antibody. Complexes were detected with indicated antibodies.
Figure 6.
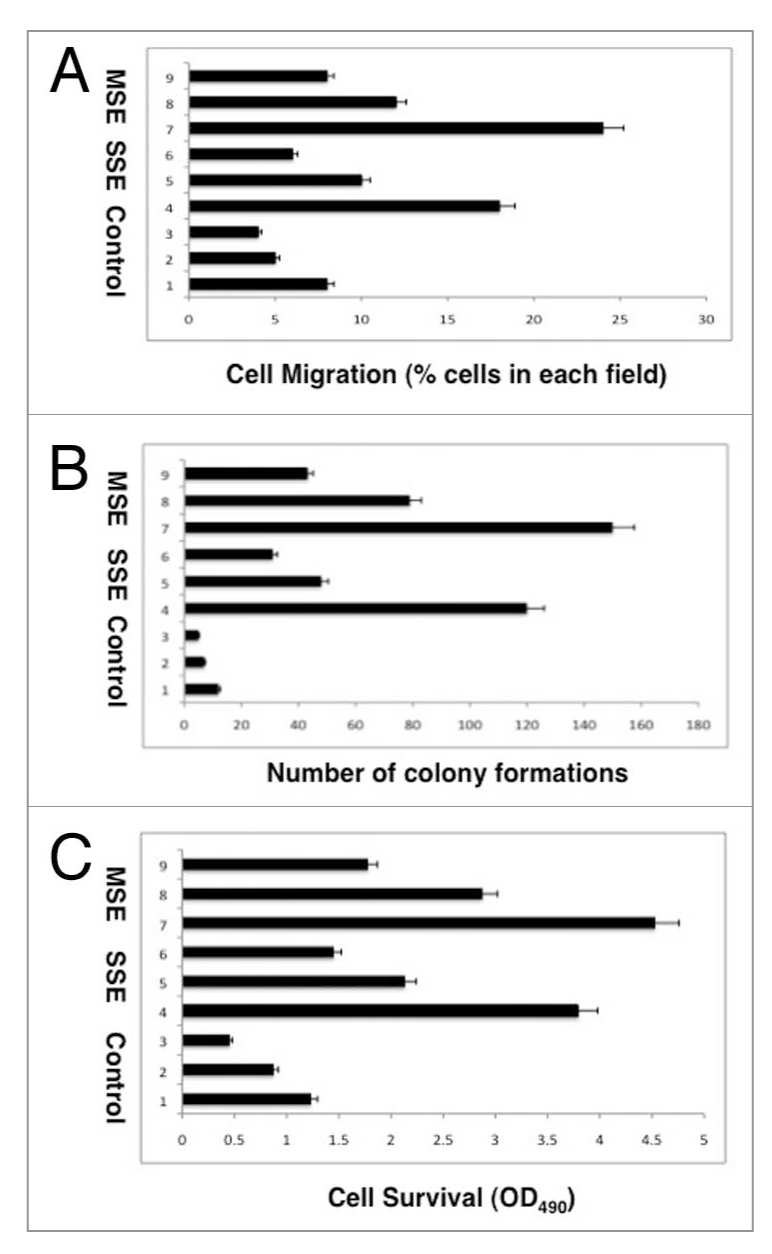
Cigarette smoke affects the cellular characteristics of lung cancer cells. (A) Migration assay. (B) Soft agar growth assay. (C) MTT Survival Assay. A427 cells were transiently transfected with scramble siRNA (samples 1, 4 and 7), PEA 3 siRNA (samples 2, 5 and 8) and COX-2/ PTGS-2 (samples 3, 6 and 9) for 48 h. Cells then were exposed to control medium (sample 1–3), 1% SSE (samples 4–6), 0.5% MSE (samples 7–9). Experiments were performed in triplicate.
Acknowledgements
This work was in part supported by grant #082469 from Flight Attendant Medical Research Institutions.
References
- 1.Hecht SS. Tobacco smoke carcinogens and lung cancer. Journal of the National Cancer Institute. 1999;91(14):1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 2.Boffetta P. Human cancer from environmental pollutants: the epidemiological evidence. Mutation Research. 2006;608(2):157–162. doi: 10.1016/j.mrgentox.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Zhou W, Heist RS, Liu G, et al. Second hand smoke exposure and survival in early-stage non-small-cell lung cancer patients. Clinical Cancer Research. 2006;12(23):7187–7193. doi: 10.1158/1078-0432.CCR-06-1460. [DOI] [PubMed] [Google Scholar]
- 4.Cianchi F, Cortesini C, Bechi P, et al. Up-regulation of cyclooxygenase 2 gene expression correlates with tumor angiogenesis in human colorectal cancer. Gastroenterology. 2001;121(6):1339–1347. doi: 10.1053/gast.2001.29691. [DOI] [PubMed] [Google Scholar]
- 5.De Marzo AM, Platz EA, Sutcliffe S, et al. Inflammation in prostate carcinogenesis. Nature Reviews Cancer. 2007;7(4):256–269. doi: 10.1038/nrc2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao H, Rahman I. Current concepts on the role of inflammation in COPD and lung cancer. Current Opinion in Pharmacology. 2009;9(4):375–383. doi: 10.1016/j.coph.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elahi MM, Kong YX, Matata BM. Oxidative stress as a mediator of cardiovascular disease. Oxidative Medicine and Cellular Longevity. 2009;2(5):259–269. doi: 10.4161/oxim.2.5.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxidative Medicine and Cellular Longevity. 2010;3(1):23–34. doi: 10.4161/oxim.3.1.10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maiuri MC, Tasdemir E, Criollo A, et al. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death and Differentiation. 2009;16(1):87–93. doi: 10.1038/cdd.2008.131. [DOI] [PubMed] [Google Scholar]
- 10.Essick EE, Sam F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxidative Medicine and Cellular Longevity. 2010;3(3):168–177. doi: 10.4161/oxim.3.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Yoshino I, Kometani T, Shoji F, et al. Induction of epithelial-mesenchymal transition-related genes by benzo[a]pyrene in lung cancer cells. Cancer. 2007;110(2):369–374. doi: 10.1002/cncr.22728. [DOI] [PubMed] [Google Scholar]
- 13.Lee JM, Yanagawa J, Peebles KA, Sharma S, Mao JT, Dubinett SM. Inflammation in lung carcinogenesis: new targets for lung cancer chemoprevention and treatment. Critical Reviews in Oncology/Hematology. 2008;66(3):208–217. doi: 10.1016/j.critrevonc.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu G, Zhou W, Christiani DC. Molecular epidemiology of non-small cell lung cancer. Seminars in Respiratory and Critical Care Medicine. 2005;26(3):265–272. doi: 10.1055/s-2005-871983. [DOI] [PubMed] [Google Scholar]
- 15.Sanchez-Cespedes M, Parrella P, Esteller M, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Research. 2002;62(13):3659–3662. [PubMed] [Google Scholar]
- 16.Upadhyay S, Liu C, Chatterjee A, et al. LKB1/STK11 suppresses cyclooxygenase-2 induction and cellular invasion through PEA3 in lung cancer. Cancer Research. 2006;66(16):7870–7879. doi: 10.1158/0008-5472.CAN-05-2902. [DOI] [PubMed] [Google Scholar]
- 17.Ghaffar H, Sahin F, Sanchez-Cepedes M, et al. LKB1 protein expression in the evolution of glandular neoplasia of the lung. Clinical Cancer Research. 2003;9(8):2998–3003. [PubMed] [Google Scholar]
- 18.Gurumurthy S, Hezel AF, Berger JH, Bosenberg MW, Bardeesy N. LKB1 deficiency sensitizes mice to carcinogen-induced tumorigenesis. Cancer Research. 2008;68(1):55–63. doi: 10.1158/0008-5472.CAN-07-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji H, Ramsey MR, Hayes DN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448(7155):807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 20.Scott KD, Nath-Sain S, Agnew MD, Marignani PA. LKB1 catalytically deficient mutants enhance cyclin D1 expression. Cancer Research. 2007;67(12):5622–5627. doi: 10.1158/0008-5472.CAN-07-0762. [DOI] [PubMed] [Google Scholar]
- 21.Zeng PY, Berger SL. LKB1 is recruited to the p21/WAF1 promoter by p53 to mediate transcriptional activation. Cancer Research. 2006;66(22):10701–10708. doi: 10.1158/0008-5472.CAN-06-0999. [DOI] [PubMed] [Google Scholar]
- 22.Dalwadi H, Krysan K, Heuze-Vourc’h N, et al. Cyclooxygenase-2-dependent activation of signal transducer and activator of transcription 3 by interleukin-6 in non-small cell lung cancer. Clinical Cancer Research. 2005;11(21):7674–7682. doi: 10.1158/1078-0432.CCR-05-1205. [DOI] [PubMed] [Google Scholar]
- 23.Hida T, Kozaki KI, Muramatsu H, et al. Cyclooxygenase-2 inhibitor induces apoptosis and enhances cytotoxicity of various anticancer agents in non-small cell lung cancer cell lines. Clinical Cancer Research. 2000;6(5):2006–2011. [PubMed] [Google Scholar]
- 24.Moodie FM, Marwick JA, Anderson CS, et al. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-κB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB Journal. 2004;18(15):1897–1899. doi: 10.1096/fj.04-1506fje. [DOI] [PubMed] [Google Scholar]
- 25.Subbaramaiah K, Norton L, Gerald W, Dannenberg AJ. Cyclooxygenase-2 is overexpressed in HER-2/neu-positive breast cancer: evidence for involvement of AP-1 and PEA3. The Journal of Biological Chemistry. 2002;277(21):18649–18657. doi: 10.1074/jbc.M111415200. [DOI] [PubMed] [Google Scholar]
- 26.Tsujii M, Kawano S, Dubois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(7):3336–3340. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimäki A. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Research. 1998;58(22):4997–5001. [PubMed] [Google Scholar]
- 28.Yang SR, Chida AS, Bauter MR, et al. Cigarette smoke induces proinflammatory cytokine release by activation of NF-κB and posttranslational modifications of histone deacetylase in macrophages. American Journal of Physiology. 2006;291(1):L46–L57. doi: 10.1152/ajplung.00241.2005. [DOI] [PubMed] [Google Scholar]
- 29.Baglole CJ, Maggirwar SB, Gasiewicz TA, Thatcher TH, Phipps RP, Sime PJ. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-κB family member RelB. The Journal of Biological Chemistry. 2008;283(43):28944–28957. doi: 10.1074/jbc.M800685200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mercer BA, Wallace AM, Brinckerhoff CE, D’Armiento JM. Identification of a cigarette smoke-responsive region in the distal MMP-1 promoter. American Journal of Respiratory Cell and Molecular Biology. 2009;40(1):4–12. doi: 10.1165/rcmb.2007-0310OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu ESL, Shin VY, Ye YN, Luo JC, Wu WKK, Cho CH. Cyclooxygenase-2 in cancer cells and macrophages induces colon cancer cell growth by cigarette smoke extract. European Journal of Pharmacology. 2005;518(1):47–55. doi: 10.1016/j.ejphar.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 32.Carpagnano GE, Spanevello A, Palladino GP, et al. Cigarette smoke and increased COX-2 and survivin levels in exhaled breath condensate of lung cancer patients: how hot is the link? Lung Cancer. 2010;67(1):108–113. doi: 10.1016/j.lungcan.2009.03.033. [DOI] [PubMed] [Google Scholar]
- 33.Maiese K. Marking the onset of oxidative stress: biomarkers and novel strategies. Oxidative Medicine and Cellular Longevity. 2009;2(1, article 1) doi: 10.4161/oxim.2.1.8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hibi K, Trink B, Patturajan M, et al. AIS is an oncogene amplified in squamous cell carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(10):5462–5467. doi: 10.1073/pnas.97.10.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y, Chuang AY, Romano RA, et al. Phospho-ΔNp63α/NF-Y protein complex transcriptionally regulates DDIT3 expression in squamous cell carcinoma cells upon cisplatin exposure. Cell Cycle. 2010;9(2):328–338. doi: 10.4161/cc.9.2.10432. [DOI] [PubMed] [Google Scholar]
- 36.Hiroumi H, Dosaka-Akita H, Yoshida K, et al. Expression of E1AF/PEA3, an Ets-related transcription factor in human non-small-cell lung cancers: its relevance in cell motility and invasion. International Journal of Cancer. 2001;93(6):786–791. doi: 10.1002/ijc.1410. [DOI] [PubMed] [Google Scholar]
- 37.Howe LR, Crawford HC, Subbaramaiah K, Hassell JA, Dannenberg AJ, Brown AMC. PEA3 Is up-regulated in response to Wnt1 and activates the expression of cyclooxygenase-2. The Journal of Biological Chemistry. 2001;276(23):20108–20115. doi: 10.1074/jbc.M010692200. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Borchert GL, Phang JM. Polyoma enhancer activator 3, an Ets transcription factor, mediates the induction of cyclooxygenase-2 by nitric oxide in colorectal cancer cells. The Journal of Biological Chemistry. 2004;279(18):18694–18700. doi: 10.1074/jbc.M308136200. [DOI] [PubMed] [Google Scholar]
- 39.Connors SK, Balusu R, Kundu CN, Jaiswal AS, Gairola CG, Narayan S. C/EBPβ-mediated transcriptional regulation of bcl-xl gene expression in human breast epithelial cells in response to cigarette smoke condensate. Oncogene. 2009;28(6):921–932. doi: 10.1038/onc.2008.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsurutani J, Castillo SS, Brognard J, et al. Tobacco components stimulate Akt-dependent proliferation and NFκB-dependent survival in lung cancer cells. Carcinogenesis. 2005;26(7):1182–1195. doi: 10.1093/carcin/bgi072. [DOI] [PubMed] [Google Scholar]
- 41.Anto RJ, Mukhopadhyay A, Shishodia S, Gairola CG, Aggarwal BB. Cigarette smoke condensate activates nuclear transcription factor-κB through phosphorylation and degradation of IκBα: correlation with induction of cyclooxygenase-2. Carcinogenesis. 2002;23(9):1511–1518. doi: 10.1093/carcin/23.9.1511. [DOI] [PubMed] [Google Scholar]
- 42.Han JA, Kim JI, Ongusaha PP, et al. p53-mediated induction of Cox-2 counteracts p53- or genotoxic stress-induced apoptosis. EMBO Journal. 2002;21(21):5635–5644. doi: 10.1093/emboj/cdf591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Launoit Y, Baert JL, Chotteau-Lelievre A, et al. The Ets transcription factors of the PEA3 group: transcriptional regulators in metastasis. Biochimica et Biophysica Acta. 2006;1766(1):79–87. doi: 10.1016/j.bbcan.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 44.Qin L, Liu Z, Chen H, Xu J. The steroid receptor coactivator-1 regulates Twist expression and promotes breast cancer metastasis. Cancer Research. 2009;69(9):3819–3827. doi: 10.1158/0008-5472.CAN-08-4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang XQ, Li H, Van Putten V, Winn RA, Heasley LE, Nemenoff RA. Oncogenic K-ras regulates proliferation and cell junctions in lung epithelial cells through induction of cyclooxygenase-2 and activation of metalloproteinase-9. Molecular Biology of the Cell. 2009;20(3):791–800. doi: 10.1091/mbc.E08-07-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang R, Wang X, Lin F, Gao P, Dong K, Zhang HZ. shRNA-targeted Cyclooxygenase (COX)-2 inhibits proliferation, reduces invasion and enhances chemosensitivity in laryngeal carcinoma cells. Molecular and Cellular Biochemistry. 2008;317(1-2):179–188. doi: 10.1007/s11010-008-9847-9. [DOI] [PubMed] [Google Scholar]
- 47.Dahl KDC, Zeineldin R, Hudson LG. PEA3 is necessary for optimal epidermal growth factor receptor-stimulated matrix metalloproteinase expression and invasion of ovarian tumor cells. Molecular Cancer Research. 2007;5(5):413–421. doi: 10.1158/1541-7786.MCR-07-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lynch CC, Crawford HC, Matrisian LM, McDonnell S. Epidermal growth factor upregulates matrix metalloproteinase-7 expression through activation of PEA3 transcription factors. International Journal of Oncology. 2004;24(6):1565–1572. [PubMed] [Google Scholar]
- 49.Jorgensen ED, Zhao H, Traganos F, Albino AP, Darzynkiewicz Z. DNA damage response induced by exposure of human lung adenocarcinoma cells to smoke from tobacco- and nicotine-free cigarettes. Cell Cycle. 2010;9(11):2170–2176. doi: 10.4161/cc.9.11.11842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alexander A, Cai SL, Kim J, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(9):4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang Q, Wu YT, Tan HL, Ong CN, Shen HM. A novel function of poly(ADP-ribose) polymerase-1 in modulation of autophagy and necrosis under oxidative stress. Cell Death and Differentiation. 2009;16(2):264–277. doi: 10.1038/cdd.2008.151. [DOI] [PubMed] [Google Scholar]
- 52.Yan W, Chen X. GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. The Journal of Biological Chemistry. 2006;281(12):7856–7862. doi: 10.1074/jbc.M512655200. [DOI] [PubMed] [Google Scholar]
- 53.Ellisen LW, Ramsayer KD, Johannessen CM, et al. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Molecular Cell. 2002;10(5):995–1005. doi: 10.1016/s1097-2765(02)00706-2. [DOI] [PubMed] [Google Scholar]
- 54.Liu G, Chen X. The ferredoxin reductase gene is regulated by the p53 family and sensitizes cells to oxidative stress-induced apoptosis. Oncogene. 2002;21(47):7195–7204. doi: 10.1038/sj.onc.1205862. [DOI] [PubMed] [Google Scholar]
- 55.Zdanov S, Bernard D, Debacq-Chainiaux F, et al. Normal or stress-induced fibroblast senescence involves COX-2 activity. Experimental Cell Research. 2007;313(14):3046–3056. doi: 10.1016/j.yexcr.2007.04.033. [DOI] [PubMed] [Google Scholar]
- 56.Klimova TA, Bell EL, Shroff EH, et al. Hyperoxia-induced premature senescence requires p53 and pRb, but not mitochondrial matrix ROS. FASEB Journal. 2009;23(3):783–794. doi: 10.1096/fj.08-114256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park IJ, Hwang JT, Young MK, Ha J, Ock JP. Differential modulation of AMPK signaling pathways by low or high levels of exogenous reactive oxygen species in colon cancer cells. Annals of the New York Academy of Sciences. 2006;1091:102–109. doi: 10.1196/annals.1378.059. [DOI] [PubMed] [Google Scholar]
- 58.Lee J, Kosaras B, Aleyasin H, et al. Role of cyclooxygenase-2 induction by transcription factor Sp1 and Sp3 in neuronal oxidative and DNA damage response. The FASEB Journal. 2006;20(13):2375–2377. doi: 10.1096/fj.06-5957fje. [DOI] [PubMed] [Google Scholar]
- 59.Andoh T, Chiueh CC, Chock PB. Cyclic GMP-dependent protein kinase regulates the expression of thioredoxin and thioredoxin peroxidase-1 during hormesis in response to oxidative stress-induced apoptosis. The Journal of Biological Chemistry. 2003;278(2):885–890. doi: 10.1074/jbc.M209914200. [DOI] [PubMed] [Google Scholar]
- 60.Chung HY, Cesari M, Anton S, et al. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Research Reviews. 2009;8(1):18–30. doi: 10.1016/j.arr.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bazan NG, Palacios-Pelaez R, Lukiw WJ. Hypoxia signaling to genes: significance in Alzheimer’s disease. Molecular Neurobiology. 2002;26(2-3):283–298. doi: 10.1385/MN:26:2-3:283. [DOI] [PubMed] [Google Scholar]