

Abstract
In order to properly comprehend the epigenetic dysregulation that occurs during the course of disease, there is a need to characterize the epigenetic variability in healthy individuals that arises in response to aging and exposures, and to understand such variation within the biological context of the DNA sequence. We analyzed the methylation of 26,486 autosomal CpG loci in blood from 205 healthy subjects, using three complementary approaches to assess the association between methylation, age or exposures and local sequence features, such as CpG island status, repeat sequences, location within a polycomb target gene or proximity to a transcription factor binding site. We clustered CpGs (1) using unsupervised recursively partitioned mixture modeling (RPMM) and (2) bioinformatically-informed methods and (3) also employed a marginal model-based (non-clustering) approach. We observed associations between age and methylation and hair dye use and methylation, where the direction and magnitude was contingent on the local sequence features of the CpGs. Our results demonstrate that CpGs are differentially methylated dependent upon the genomic features of the sequence in which they are embedded, and that CpG methylation is associated with age and hair dye use in a CpG context-dependent manner in healthy individuals.
Keywords: methylation array, epigenetics, exposures, aging, Infinium, hair dye
Introduction
Epigenetics can be defined as stable and heritable changes that either alter or have the potential to alter gene expression without changing the DNA sequence.1 DNA methylation is the most commonly studied epigenetic modification in humans due to its stability and amenability to measurement. The covalent attachment of a methyl group to cytosine at the 5-carbon of the pyrimidine ring occurs primarily in the context of CpG dinucleotides.2 CpGs are disproportionately concentrated in enriched regions referred to as CpG islands (CGI), which tend to be differentially located in the promoter regions of genes. Methylation of CGIs in gene promoter regions is typically associated with transcriptional repression, although CGIs are generally not methylated in non-pathologic cells,2 but exceptions exist, as in the case of X-inactivation, imprinting3 or tissue differentiation.4–8 However, 70–90% of all CpGs in the human genome are not situated within CGIs and are typically methylated under normal conditions, helping to maintain genomic stability9,10 and suppress expression of transposable elements.11
Although the term epigenetics was originally coined in 1942,12 this discipline has burgeoned over the last three decades with the major advances initially found in cancer biology. However, in recent years, the study of the epigenetics of aging has emerged as a novel field, seeking to discern the epigenetic contribution to the highly complex process of aging in the context of the environment, broadly conceived.1 Alterations in DNA methylation have been associated with aging-related diseases, including insulin-resistant diabetes mellitus (type 2),13 Alzheimer disease,14 cardiovascular disease15,16 and cancer.3 Part of the key to understanding how altered DNA methylation patterns associate with aging is to determine whether they occur in response to endogenous or exogenous environmental exposures, are preprogrammed as a course of life, are primarily a stochastic process, or if the patterns of DNA methylation that develop over time in a tissue reflect an amalgamation of all of these inputs.
Gaining a better understanding of the potential for local sequence features and genomic context to influence the methylation state of CpGs will enhance our comprehension of how DNA methylation is regulated under normal conditions and becomes altered by aging and exposures and will provide important clues to the role of epigenetics in pathogenesis. While the methylome of the peripheral blood mononuclear cell has been described in reference 17, enhancing our ability to understand the normal state of blood cells, we have focused upon the role of aging and the environment in explaining inter-individual differences in DNA methylation. Considerable epidemiologic and basic research is currently being conducted investigating the patterns of DNA methylation in peripheral blood for a plethora of pathological conditions thought to be related to altered epigenetic states.18 Hence, it is imperative that we further define the intrinsic factors, such as sequence context, affecting DNA methylation in the non-pathologic state. We have previously demonstrated that CpG loci can be clustered in DNA extracted from the blood of healthy individuals according to their methylation patterns and that the extent and direction of correlation between CpG methylation and age is dependent upon CGI context.19 Here, we have extended this research to the evaluation of 26,486 autosomal CpG sites for methylation in blood DNA from 205 healthy subjects to investigate the relationships among patterns of DNA methylation and age, gender, environmental exposures and sequence features in healthy individuals. We demonstrate that intrinsic biological characteristics, such as local sequence features surrounding CpGs, may interact with aging and the environment to influence DNA methylation, and suggest an appropriate approach and methodology for assessing associations of methylation with aging and environmental exposures in healthy people.
Results
To assess the complex relationship of DNA methylation with age, gender and various exposures, we used DNA from blood samples of 205 healthy individuals (a description of the study population is provided in Sup. Table S1) and high-density methylation array technology to analyze CpG methylation via three complementary approaches. We first used a data-driven approach, clustering CpGs (as opposed to subjects) by relative methylation across all subjects using an unsupervised model-based hierarchical clustering algorithm. Our second approach applied classes derived externally from bioinformatic considerations, fitting the methylation data into these bioinformatic classes. Finally, to supplement the CpG cluster-based analyses, we developed marginal models to further assess the interactions between aging/exposures and local DNA sequence features with regard to methylation without needing to cluster the CpGs. An overview of our analytic strategy is presented in Figure 1.
Figure 1.

Overview of the analytic strategy for this study. Methylation data for 26,486 autosomal CpGs was assayed for each of 205 blood specimens by the Infinium HumanMethylation27k BeadArray. Three complementary approaches were used to assess the association between DNA methylation and age, sex and select environmental exposures, while taking into account variation in DNA sequence features of each CpG: (1) unsupervised clustering of CpGs by recursively partitioned mixture modeling (RPMM) into 32 CpG classes based on like methylation patterns, followed by evaluation of an association of mean methylation of the CpGs for each resultant class with age, gender or exposures; (2) clustering of CpGs into 41 classes based on bioinformatic attributes (CpG sequence features), again followed by evaluation of an association of mean methylation of the CpGs for each resultant class with age, gender or exposures; and (3) a marginal-model based analysis (no clustering), assessing interactions between DNA sequence features and age, gender and exposures, with respect to methylation.
Association of exposures with unsupervised CpG class methylation.
Previous work by our group identified a correlation of CpG methylation with age in peripheral blood DNA from healthy individuals, the magnitude and direction of which depended upon the CGI-status of the CpGs.19 We sought to expand on these findings with a larger pool of healthy study subjects using a denser methylation array, and to examine relationships of CpG methylation with several well-characterized exposures and potential cancer risk factors, while taking into account variability in propensity for methylation among CpGs. CpG loci were clustered by unsupervised recursively partitioned mixture model (RPMM),20 based on methylation (β) Z-scores, into 32 methylation classes. RPMM was chosen for its efficient and effective performance in clustering high-dimensional methylation array data (Sup. Analysis S1). The resultant CpG classes demonstrated interclass variability in their degree of methylation (Fig. 2; see Sup. Fig. 1 for additional detail).
Figure 2.

Adjusted association of exposures with unsupervised RPMM class methylation. The colored dots indicate the degree of average class methylation. The y-axis represents the t-statistic for the association of class methylation and the corresponding exposure from the multiple regression models, while the 32 CpG classes are depicted on the x-axis. The p value for the omnibus test (first difference test) of significance for the association of each exposure and average class methylation is found at the top left of the corresponding exposure plot. The red dotted lines represent the 95th percentile of the permutation distribution of the maximum absolute value (over 32 classes) of the regression coefficient t-statistics as a control for multiple comparisons. Note: age was adjusted for gender; gender was adjusted for age and hair dye use; all other models were adjusted for age and gender. *Restricted to ever-smokers. **Compared to non-drinkers (zero alcoholic drinks per week).
To evaluate the associations between methylation and aging/exposures, Spearman's rank correlation coefficient with each respective exposure was calculated for the average methylation within each class (32 such class-specific averages per subject). This approach was taken based on the notion that CpGs within classes should possess similarities that influence the direction and magnitude of methylation. Overall association of class methylation and aging/exposures was assessed using two separate omnibus tests (Table 1), described in detail in the Methods section. CpG class methylation was significantly correlated with age (P1st difference = 0.007; Psupremum < 0.001), cigarette pack-years restricted to ever-smokers (P1st difference = 0.004; Psupremum = 0.006) and lifetime ever-use of hair dye (P1st difference = 0.003; Psupremum = 0.01). After controlling for potential confounding factors using multiple linear regression (Table 1), there was still an overall association of CpG class methylation and age, adjusted for gender (P1st difference = 0.006; Psupremum < 0.001); and hair dye use after adjusting for age and gender (P1st difference = 0.006; Psupremum = 0.06); while the association between CpG methylation class and pack-years was borderline significant based on the “1st difference” test (P1st difference = 0.06), which is designed to account for structure among the class methylation-exposure relationships but was non-significant by the omnibus test based on maximum absolute value (Psupremum = 0.41), after adjusting for age and gender.
Table 1.
Omnibus tests of association for exposures and unsupervised RPMM CpG class methylation
| Omnibus p value | |||||
| Spearman correlation | Multivariable regression | ||||
| Exposure | Supremum | 1st Difference | Supremum | 1st Difference | |
| Age (years)a | <0.001 | 0.007 | <0.001 | 0.006 | |
| Female genderb | 0.04 | 0.20 | 0.23 | 0.64 | |
| Cigarette smoking | |||||
| Ever-smokingc | 0.06 | 0.07 | 0.29 | 0.28 | |
| Pack-yearsc,d | 0.006 | 0.004 | 0.41 | 0.06 | |
| Alcohol consumptionc,e | |||||
| Non-drinker (reference) | - | - | - | - | |
| ≤6.5 drinks/week | 0.29 | 0.66 | 0.49 | 0.60 | |
| ≥6.5 drinks/week | 0.34 | 0.42 | 0.47 | 0.60 | |
| Hair dye-usec | 0.01 | 0.003 | 0.06 | 0.006 | |
| Arsenic (µg/g)c,f | 0.31 | 0.26 | 0.17 | 0.06 | |
| Selenium (µg/g)c,f | 0.63 | 0.52 | 0.59 | 0.43 | |
| Ultraviolet radiation exposure | |||||
| Tanning lamp usec | 0.02 | 0.14 | 0.11 | 0.37 | |
| Lifetime painful sunburnsc | 0.89 | 0.88 | 0.67 | 0.65 | |
Note: Supremum test = test based on the maximum absolute value. 1st-difference test = test for structural relationships based on maximum absolute 1st difference.
The multiple regression model was adjusted for gender.
The multiple regression model was adjusted for age and hair dye use.
The multiple regression model was adjusted for age and gender.
Restricted to ever-smokers.
Median = 6.5 alcoholic drinks per week among drinkers.
Measured in toenail clippings.
When considering individual CpG classes (Fig. 2), after adjusting for potential confounding, classes with relatively greater extents of methylation (denoted by blue dots) were observed to have an inverse relationship between methylation and aging, although most were non-significant (controlling for multiple comparisons); while in the relatively unmethylated CpG classes (denoted by yellow dots), methylation tended to be positively associated with age, although none were individually significant (controlling for multiple comparisons). Hair dye use, arsenic and tanning lamp use also displayed similar patterns to that of age and methylation by class but no significant associations were observed for any individual class.
Cross-validation of unsupervised CpG class and methylation-exposure associations.
Methylation array data was available for peripheral blood from a second population of 92 healthy subjects (validation subjects). To assess the robustness of the unsupervised classes, CpG loci were again clustered into 32 classes by RPMM using the methylation data from the validation subjects, and class membership (CpG loci) was compared by cross-tabulation of the CpG classes derived from the primary study subjects and validation subjects. There was substantial concordance of CpG loci between the two sets of classes, indicating that the unsupervised clustering of CpGs by RPMM has a high-level of reproducibility in blood from healthy subjects (Sup. Fig. S2).
Next, we sought to validate the observed methylation-exposure associations. The two sets of CpG classes, one derived via RPMM from the primary study subjects (primary classes) and one from the validation subjects (validation classes) were applied (i.e., used to define class-specific methylation averages) to each study population, resulting in four comparisons: primary classes x primary subjects, primary classes x validation subjects, validation classes x primary subjects and validation classes x validation subjects. For each of the 32 classes in each comparison, Spearman's correlation and multiple regression were used to evaluate the association between methylation and exposures that were available in both data sets (age, gender, smoking and alcoholic drinks per week). Finally, omnibus tests of overall association of each exposure with class methylation were performed using a supremum test statistic, described in detail in the Methods section. Of the aforementioned exposures, only alcohol consumption (p = 0.001) and race/ethnicity (p < 0.001) differed by study population (Sup. Table S1) with the validation subjects less likely to be non-drinkers and more likely to consume >6.5 drinks per week and more likely to be of a racial/ethnic background other than Caucasian, although the vast majority identified as non-Hispanic Caucasians.
When applied to the same study population, each of the 2 sets of CpG classes, derived respectively from the primary study subjects and validation subjects, were similar with regard to correlation of class methylation and exposures, which was sustained after adjusting for potential confounders in the multiple regression models (Sup. Table S2). However a higher degree of variation was observed for each set of CpG classes when applied across populations, possibly indicative of inherent unaccounted differences between the populations.
Association of sequence features with unsupervised CpG class.
To investigate how the genomic context around specific CpG sites may impact the associations between exposures and methylation, we examined variability of the individual unsupervised (RPMM-derived) classes with regard to specific local sequence features. Interclass variability by sequence feature for the CpG loci was observed (Fig. 3). The classes with relatively high levels of methylation had higher proportions of CpGs within non-long terminal repeat (non-LTR) transposable elements, including LINE-1, LINE-2, Alu and mammalian wide-interspersed repeat (MIR) elements. Conversely, unmethylated CpG classes predominately contained loci residing within CGIs and had a higher proportion of CpGs located within 1,000 bases (1 kb) of at least one putative transcription factor binding site (TFBS). There was also variability among classes with respect to percent of CpGs located within a polycomb group (PcG) target gene,21–24 with the frequency of CpG loci associated with PcG targets within classes ranging from 4.0% to 32.8%; five classes had more than 20% of member loci that were associated with PcG targets.
Figure 3.

Frequency of sequence features associated with unsupervised RPMM class CpG loci. Classes are represented on each plot by the colored dots, which indicate the degree of average class methylation. The y-axis represents the frequency of CpG loci in each class associated with the sequence feature of interest, while the 32 CpG classes are depicted on the x-axis. Abbreviations: CGI = CpG island; PcG Target = located in a polycomb group protein target gene; TFBS, located within 1 kb of a transcription factor binding site; LINE, long interspersed nuclear element; MIR, mammalian wide-interspersed repeat element.
Bioinformatically-derived CpG class.
Motivated by the interclass variability by sequence features observed in the unsupervised RPMM-based clustering, we next utilized a bioinformatically-informed classification scheme, subdividing CpG sites by their sequence features to account for intricate interactions between them. Taking into consideration presence in a CGI, PcG target gene, LINE-1, LINE-2, Alu and MIR elements and proximity (≤1kb) to a TFBS, we obtained 41 classes containing at least one CpG based on various combinations of the aforementioned bioinformatic attributes. Classes are denoted by the applicable attributes separated by a “|” (e.g., a class of CpGs located in CGI and LINE-1 element would be symbolized as CGI|LINE1). The distribution of CpG loci by bioinformatically-derived class is presented in Supplemental Table S3. There was an overall significant relationship between age and bioinformatically-derived CpG class methylation (Table 2) by omnibus tests (supremum) of per class Spearman's rank correlation (p = 0.001) and multivariable regression (p = 0.001), adjusting for gender. Dye-use (p = 0.04) and female gender (p = 0.05) were also significantly correlated with class using an omnibus test of Spearman's coefficient but lost significance after adjusting for age and gender and age and dye-use, respectively.
Table 2.
Omnibus tests of association for exposures and bioinformatically-derived CpG class methylation
| Omnibus p value (Supremum) | |||
| Exposure | Spearman Correlation | Multiple Regression | |
| Age (years)a | 0.001 | 0.001 | |
| Female genderb | 0.05 | 0.56 | |
| Cigarette smoking | |||
| Ever-smokingc | 0.15 | 0.29 | |
| Pack-yearsc,d | 0.11 | 0.84 | |
| Alcohol consumptionc,e | |||
| Non-drinker (reference) | - | - | |
| ≤6.5 drinks/week | 0.14 | 0.21 | |
| >6.5 drinks/week | 0.17 | 0.20 | |
| Hair dye-usec | 0.04 | 0.38 | |
| Arsenic (µg/g)c,f | 0.25 | 0.09 | |
| Selenium (µg/g)c,f | 0.92 | 0.82 | |
| Ultraviolet radiation exposure | |||
| Tanning lamp usec | 0.15 | 0.47 | |
| Lifetime painful sunburnsc | 0.87 | 0.54 | |
The multiple regression model was adjusted for gender.
The multiple regression model was adjusted for age and hair dye use.
The multiple regression model was adjusted for age and gender.
Restricted to ever-smokers.
Median = 6.5 alcoholic drinks per week among drinkers.
Measured in toenail clippings.
There was a significant inverse association of age and methylation for several individual bioinformatically-derived classes (Fig. 4). These 8 classes (each class is shown in brackets) included [CGI|MIR], [PcG target|TFBS], [PcG target], [TFBS], [PcG target|MIR|TFBS], [MIR|TFBS], [CGI|PcG target|LINE2] and [LINE2|TFBS]; all of which had relatively higher degrees of average methylation. No other exposures were associated with methylation of individual bioinformatically-derived classes (after controlling for multiple comparisons).
Figure 4.

Adjusted association of exposures with bioinformatically-derived CpG class methylation. The colored dots indicate the degree of average class methylation. The y-axis represents the t-statistic for the association of class methylation and the corresponding exposure from the multiple regression models, while the 41 bioinformatically-derived Cp6 classes are listed numerically on the x-axis; the bioinformatic attributes corresponding to the numbers are provided in Supplemental Table S3. The p-value for the omnibus test (supremum) of significance for the association of each exposure and average class methylation is found at the top left the corresponding exposure plot. The red dotted lines represent the 95th percentile of the permutation distribution of the maximum absolute value (over 32 classes) of the regression coefficient t-statistics as a control for multiple comparisons. Note: age was adjusted for gender; gender was adjusted for age and hair dye use; all other models were adjusted for age and gender. There were 8 classes significantly associated with age: (12) CGI|MIR; (16) PcG|TFBS; (20) PcG; (23) TFBS; (24) PcG|MIR|TFBS; (27) MIR|TFBS; (29) CGI|PcG|LINE2; (31) LINE2|TFBS. Abbreviations: CGI = CpG island; PcG = located in a polycomb group protein target gene; TFBS = located within 1 kb of a transcription factor binding site; LINE = long interspersed nuclear element; MIR = mammalian wide-interspersed repeat element. *Restricted to ever-smokers. **Compared to non-drinkers (zero alcoholic drinks per week).
In response to recent literature suggesting a role in transcriptional control and differentiation,25,26 we conducted a subanalysis of CpG island shores (defined as sequences within 2 kb distance of CGI). However, we found no association between their methylation and exposures (Sup. Table S4) and thus have not further included them in our analyses.
Model-based analysis of exposure-sequence feature interactions.
Building on the bioinformatically-derived classes, a model-based approach (independent of CpG clustering) was employed to further assess the relationship between exposures, sequence features and methylation. To do this, we developed separate marginal models for each exposure of interest, adjusted for potential confounders and examined the main effect and interactions of each exposure and sequence feature with respect to DNA methylation. The models substantiate the inclusion of the sequence features used in the bioinformatically-derived classification of CpG loci, showing them to each be independently associated with methylation at p < 0.00001, with the exception of PcG targets, which is non-significant in all but one of the models (Sup. Table S5–S14).
A summary of the results from the marginal models for the association of exposures and methylation, overall and by sequence features of the CpG loci, are presented in Table 3 (the individual models are presented in entirety in Sup. Table S5–S14). Age was inversely associated with overall average methylation (p = 0.002). When considering CpG loci by sequence feature, there was no significant effect of age (adjusted for gender) on methylation of CpGs located in CGIs, although there was a significant inverse interaction (Pinteraction = 0.02; Sup Table S5); however methylation significantly decreased with age (adjusted for gender) for CpGs associated with all other sequence features, with significant interactions with LINE-2 (Pinteraction = 0.0003), MIR elements (Pinteraction < 0.0001) or PcG target genes (Pinteraction = 0.0003). There was no significant effect of any other exposures assessed, overall or by sequence feature, albeit there was an interaction between ever-use of hair dye (adjusted for age and gender) and methylation of CpGs in LINE-1 elements (Pinteraction = 0.04; Sup. Table S6) and an interaction between ever-use of tanning lamps (adjusted for age and gender) and methylation of CpG loci located within a PcG target gene (Pinteraction = 0.04; Sup. Table S13).
Table 3.
Summary of results for the marginal model-based assessment of the association of exposures and methylation, overall and by sequence feature
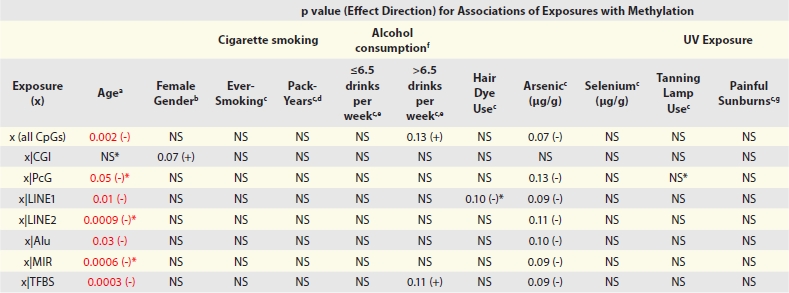 |
Abbreviations: CGI = CpG island; PcG, polycomb group protein target gene; TFBS, located within 1 kb of a transcription factor binding site; LINE, long interspersed nuclear element; MIR, mammalian wide-interspersed repeat element. Notes: x represents the exposure of interest for each respective model; The effect of exposures on methylation of CpG loci by sequence feature is denoted as: x|[feature]; p values >0.15 are denoted by NS. * Significant interaction term in the marginal model (Sup. Tables S5–S14) aPer decade; Adjusted for gender. bAdjusted for age and hair dye use. cAdjusted for age and gender. dRestricted to ever-smokers. eRelative to non-drinkers. fMedian = 6.5 alcoholic drinks per week among drinkers. gTotal lifetime number. This table summarizes the results from the marginal models for each of the 11 exposures assessed. Each column represents the model results for a specific exposure (x) and rows represent the overall or sequence feature-specific estimates of the association of each exposure with methylation (determined through linear combinations of the main effect and interaction term), presenting the p value and direction of the effect in parentheses.
Discussion
Epigenetic research in human subjects has been ongoing for decades but has primarily focused on alterations related to cancer. However, in order to properly understand aberrant epigenetic regulation that occurs during the course of disease, the normal methylome must be described. More specifically, there is a need to characterize the epigenetic state in non-pathologic tissues from healthy individuals to identify the variability in the overall profile of DNA methylation across individuals, and to clarify the relationship of that variability with aging or environmental exposures. Elucidation of the methylation patterns of CpG loci embedded in different genomic sequences or proximal to different features will critically inform our comprehension of alterations in epigenetic regulation that occur through pathologic processes and the mechanisms by which these alterations arise.
The study of the epigenetics of aging in healthy individuals is emerging as a novel discipline, seeking to discern the epigenomic changes that occur during the course of life. Early studies of this phenomenon examined candidate loci, such as individual gene promoters and “global” methylation markers, finding increased methylation of many of these specific gene promoters with aging,27–32 while methylation of the “global” markers (e.g., LINE-1, Alu, LUMA, CCGG, etc.,) decreased,33–36 giving rise to the notion that we lose global methylation with age, while we gain localized promoter methylation.37 In accordance with these earlier reports, we present here evidence, based on a genome-wide approach, of an association between aging and DNA methylation, the magnitude and direction of which is dependent upon the genomic context of the sequence in which the CpG is embedded. This is demonstrated by our marginal model results, which show no effect of age on CGI methylation but a decrease in methylation, overall and for all other sequence features considered, including several repeat sequences, with varying effects. This is additionally corroborated by our previous results in reference 19, which clustered 1,413 CpG loci into methylation classes using blood samples from 30 healthy adult subjects and finding the association of methylation with age to be CGI-context dependent. Furthermore, our present results indicate that inter- and intra-genomic differences in methylation acquired with age are more complex than just CGI vs. non-CGI, but rather vary according to biological differences in DNA sequence, as exemplified by the complex interactions observed in our bioinformatically-derived clustering approach.
We also undertook a more thorough examination of the relationship between methylation and environmental exposures experienced by the subjects studied. In doing so, we identified an association between hair dye use and methylation, where ever-use of hair dye was inversely associated with methylation among the more highly methylated unsupervised (RPMM-based) classes and positively associated with methylation in the classes with low methylation and higher CpG island contents. This finding was further supported by our marginal model estimates, which indicate an interaction between use of hair dye and methylation of CpGs in LINE-1 elements. However, using the bioinformatically-informed classification scheme, after adjusting for age and gender, we no longer observe a significant association between methylation and ever-use of hair dye by class. This may suggest that while the bioinformatically-derived classes are meaningful, they either do not fully explain the genomic context which accounts for differences in methylation between CpG loci or are over-parsing CpGs based on bioinformatic features, sacrificing statistical power for detection of associations. While several varieties of hair dyes exist, oxidative (permanent) dyes comprise 80% of the market share in the US.38 The main components of oxidative dyes include primary intermediates and couplers, composed of various forms of arylamines, oxidants and alkalinizing agents.39 A recent review concluded that there is no consistent evidence of genotoxicity from biomonitoring studies of hair dye exposure40 but there are some epidemiologic reports of increased risk of bladder41,42 and hematopoietic cancers43–45 among hair dye users, albeit the literature is conflicting.46 In light of our findings, further studies are indicated to examine the effect of hair dye use on epigenetic endpoints and the impact of these alterations on disease susceptibility.
We found no overall association of class methylation with ever-smoking but there was a borderline association among ever-smokers of pack-years with methylation using the RPMM-based approach, although the direction was contrary to what would be expected and thus further research is required to determine whether this effect is real or spurious. No association was observed for alcohol consumption, arsenic or selenium exposure (measured via toenail clippings). However, it is important to note that although the measured exposures may not be significantly associated with methylation in peripheral blood, they may be affecting methylation in other tissue types not measured in this study. Additionally, in response to evidence that ultraviolet (UV) exposure modifies the immune system,47,48 which could potentially result in altered methylation signatures in peripheral blood, we assessed measures of UV exposure, including ever-use of tanning lamps and lifetime number of painful sunburns. CpG class methylation was not associated with either UV measure, although we did observe an interaction between ever-use of tanning lamps and methylation of CpGs located within PcG target genes, the significance of which is unknown.
A key strength of this study is the employment of three complementary analytic strategies for evaluating the impact of aging and exposures on DNA methylation: (1) unsupervised clustering by recursively partitioned mixture modeling (RPMM), (2) a bioinformatically-informed clustering approach and (3) a marginal-model based analysis. Each of the 3 methodologies used bears its own set of strengths and weaknesses, with each making a positive contribution to the analysis and filling in for potential shortcomings of the others. The bioinformatically-derived clustering approach takes into account intricate interactions between DNA sequence features of the CpGs but is limited in scope to the sequence features that we considered and could potentially over-partition the data. Conversely, the unsupervised (RPMM-based) clustering approach has the capacity to capture variation in methylation due to unknown or poorly-understood features and interactions that would otherwise be unaccounted for since it clusters based on like-methylation patterns rather than specific DNA sequence attributes, although the source of variation may not be as easily interpreted. Additionally, the data-driven RPMM approach suffers from the weaknesses of all 2-stage latent variable approaches, i.e., “double-dipping” where the data are used twice (once to predict the latent variables and once again to assess their associations with other variables/phenotypes). In general, 2-stage approaches provide reasonably unbiased point estimates but can often underestimate standard errors.49 Finally, the addition of the marginal model-based (non-clustering) approach allows us to specifically analyze the interaction of each exposure of interest with each sequence feature. However, this assessment is limited to evaluation of 1st order interactions, whereas the cluster analyses may better capture more complex relationships between aging/exposures, variation in the DNA sequence and methylation.
Our results clearly demonstrate that the genomic context of CpGs is important when assessing associations of methylation with aging or exposures. They also indicate that simple consideration of CpG island status is not sufficient with respect to methylation, but rather that other variations in DNA sequence should be taken into account. Moreover, we have provided additional evidence that DNA methylation is associated with age and novel evidence for an association with hair dye use, each operating in a CpG context-dependent manner. Proper careful analysis of CpG loci with respect to methylation patterns in response to aging and exposures in healthy individuals, such as we have described here, will help us to gain insight into the mechanics of DNA methylation and epigenetic control. Ultimately, such conception of normal epigenetic variation will help to guide future research of aberrant methylation that occurs during the course of disease, enhancing our understanding of pathologic processes.
Methods
Study population.
The primary study population was composed of 205 healthy subjects with no prior history of cancer who served as shared controls for two case-control studies on bladder and skin cancer, and for whom peripheral blood (buffy coat) was available. Briefly, controls were population-based New Hampshire residents, ages 28–74 years.50 Upon enrollment, consenting subjects underwent personal interviews furnishing sociodemographic and exposure information, and provided a toenail sample used to assess the burden of arsenic and selenium in the body via inductively coupled plasma mass spectrometry.
A second study population of 92 healthy control subjects from a case-control study of head and neck squamous cell carcinoma (HNSCC) was used for cross-validation of the unsupervised CpG clustering (validation subjects), and has also been previously described in reference 51. Population-based control subjects were randomly selected from a larger pool recruited from the greater Boston area, ages 32–86 years. All subjects completed a self-administered questionnaire, providing sociodemographic and exposure information.
Institutional Review Board approval was obtained for sample collection and use of patient data for all subjects included in this study. All subjects provided written informed consent for participation in this study.
Methylation profiling.
DNA was extracted from peripheral blood buffy coats using the QIAmp DNA mini kit (Qiagen, Valencia, CA) according to the manufacturer's recommendation and was subsequently sodium bisulfite converted using the EZ DNA methylation kit (Zymo Research, Orange, CA). The bisulfite-converted DNA was analyzed using the Infinium HumanMethylation27 BeadChip array (Illumina, San Diego, CA) according to the manufacturer's recommendations at the Genomics Core Facility at the UCSF Institute for Human Genetics (San Francisco, CA). Analysis was conducted in 2 batches across 42 BeadChips. Outliers were detected using array control probes supplied by Illumina to diagnose problems such as poor bisulfite conversion, batch or BeadChip effect or color-specific problems. Specifically, Mahalanobis distances were determined based on fitted mean vector and variance-covariance matrix, and arrays with large distances (inconsistent with multivariate normality52) were discarded. The methylation status for each individual CpG locus was calculated as the ratio of fluorescent signals (β = Max(M,0)/[Max(M,0) + Max(U,0) + 100]), ranging from 0–1, using the average probe intensity for the methylated (M) and unmethylated (U) alleles. Beta (β) = 1 indicates complete methylation; β = 0 represents no methylation. Only the 26,486 autosomal CpGs were considered in the statistical analyses. We and others, have previously demonstrated that methylation of CpG loci detected through BeadArray platforms can be replicated using alternative detection techniques including pyrosequencing, Massarray analysis and quantitative methylation-specific PCR.53–58
Statistical analysis.
To capture relative, CpG-specific heterogeneity across specimens, methylation β values Bij were transformed to Z-scores (conferring robustness to biochemical range) by calculating mean 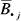 and standard deviation Sj for each individual CpG j and subsequently computing
and standard deviation Sj for each individual CpG j and subsequently computing 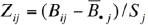 . CpG loci were clustered into methylation classes based on Z-scores using a recursively partitioned mixture model (RPMM) 20 adapted for Gaussian distributions. This likelihood-based hierarchical clustering algorithm has processing and memory requirements that are less burdensome than commonly used metric-based hierarchical clustering procedures, thereby granting computational feasibility to the clustering of 26,486 CpGs. In fact, by comparing the consistency of RPMM clustering to that of metric clustering (using Euclidean distance with Ward's linkage) by pairwise analysis of 100 resampling experiments, we have demonstrated that RPMM provides more consistent clustering than metric hierarchical clustering for this dataset (Sup. Analysis S1). In addition, its hierarchical presentation of classes confers robustness, compared with other mixture model algorithms, in the selection of the number of classes. The model was arbitrarily pruned after 5 splits (Q = 5), yielding 32 CpG methylation classes. For each of the 205 control subjects, 32 corresponding aggregate methylation values were obtained by averaging together average β values from all CpGs within the class. RPMM classes are labeled by 5-letter combinations of L (left) and R (right), denoting the direction of each of the 5-splits in the dendogram.
. CpG loci were clustered into methylation classes based on Z-scores using a recursively partitioned mixture model (RPMM) 20 adapted for Gaussian distributions. This likelihood-based hierarchical clustering algorithm has processing and memory requirements that are less burdensome than commonly used metric-based hierarchical clustering procedures, thereby granting computational feasibility to the clustering of 26,486 CpGs. In fact, by comparing the consistency of RPMM clustering to that of metric clustering (using Euclidean distance with Ward's linkage) by pairwise analysis of 100 resampling experiments, we have demonstrated that RPMM provides more consistent clustering than metric hierarchical clustering for this dataset (Sup. Analysis S1). In addition, its hierarchical presentation of classes confers robustness, compared with other mixture model algorithms, in the selection of the number of classes. The model was arbitrarily pruned after 5 splits (Q = 5), yielding 32 CpG methylation classes. For each of the 205 control subjects, 32 corresponding aggregate methylation values were obtained by averaging together average β values from all CpGs within the class. RPMM classes are labeled by 5-letter combinations of L (left) and R (right), denoting the direction of each of the 5-splits in the dendogram.
For each of these 32 aggregate measures, Spearman's rank correlation coefficient was used to measure the correlation between subject-specific exposure and subject- and class-specific aggregate methylation. Multiple linear regression models were used to assess the association of exposures and aggregate methylation, while adjusting for potential confounding variables. The model for the association of aggregate methylation and age (continuous, centered at the median) was adjusted for gender; the model for the association of aggregate methylation and gender was adjusted for age and hair dye use (ever/never); the model for the association with pack-years of smoking (continuous) was restricted to ever-smokers and was adjusted for age and gender; the respective models for ≤ and >6.5 alcoholic drinks per week (median) were compared to non-drinkers and adjusted for age and gender; and models for smoking (ever/never), hair dye use (ever/never), arsenic exposure (measured from toenail clippings as µg/g), selenium exposure (measured from toenail clippings as µg/g), tanning lamp use (ever/never) and number of lifetime painful sunburns (continuous) were all adjusted for age and gender.
Omnibus tests for overall association between exposure and aggregate CpG class methylation were obtained by permutation test. Two types of tests were used. The first type of test is a supremum test, analogous to a Kolmogorov-Smirnov test: specifically, for each hypothesized association, a test statistic was constructed as either the maximum absolute correlation or the maximum absolute t-statistic for the appropriate coefficient from the regression model, where the maximum was computed over the 32 individual correlations or regression models. The corresponding null distribution was obtained by randomly permuting the individual exposure or phenotype variable with respect to aggregate methylation values and potential confounders and computing the corresponding test statistic. 10,000 permutations were used and a hypothesized association was considered significant when p ≤ 0.05. Since this test is inefficient for detecting structural dependencies between classes that are adjacent with respect to a natural ordering (e.g., CpG classes ordered alphabetically by RPMM label or numerically by mean methylation) we employed a second type of test statistic, a “1st-difference” test: the sum of the squares of the first-order differences in smoothed correlation or t-statistic, where the smoothing was obtained by fitting a generalized additive model (GAM) to the statistics (with respect to the assumed order of the classes) and extracting the predicted smooth. GAMs were fit using the mgcv library in R. Since the classes must have a natural ordering in order for the “1st difference” test to be meaningful, this test was not applied to the bioinformatically-derived classes.
Additionally, an alternative clustering of CpGs was obtained by considering epigenetically relevant bioinformatic attributes of each CpG, including CpG island status,59 PcG target status of associated gene (i.e., gene was described as a PcG target in at least one of21–24), presence within 1 kb of at least one of 258 computationally predicted TFBS sequences obtained from the tfbsConsSites track of the UCSC Genomes Browser site (TFBS Z-score >2) and situation within each of the following classes of repetitive elements as defined by the Repeatmasker track of Genomes Browser: Alu, LINE-1, LINE-2 and MIR. This bioinformatic classification resulted in 41 distinct CpG classes containing at least one CpG, summarized in Supplemental Table S3.
Finally, to further analyze the interaction of each exposure of interest with each bioinformatic attribute with respect to CpG methylation, we fit attribute x exposure/confounder interaction regression models (marginal models). For each regression, we assumed the following data-generating model:
Where Yij = sin−1(Bij1/2) is the variance-stabilized methylation value obtained by arcsine transformation of average β value, Bij, for subject i and CpG j, xi is a vector of exposures/phenotypes and confounding variables, zj is a vector of CpG-specific attributes, ⊗ denotes Kronecker product, mj and aj are zero-mean CpG-specific effects, εij is a zero-mean error term, T symbolizes a transpose operation and the remaining coefficients are the focus of biological interest. Specifically, the vector α represents overall effect of exposure or phenotype on DNA methylation, γ represents the effects of individual CpG attributes on DNA methylation and δ represents the extent to which various CpG-specific attributes modify the effect of exposure or phenotype. Estimates were obtained in a two-stage approach by first computing individual regression coefficients 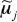 and
and 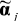 for the model
for the model 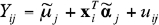 , then fitting the models
, then fitting the models 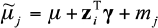 and
and 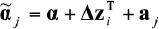 to obtain estimates
to obtain estimates 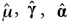 and the coefficient matrix estimate
and the coefficient matrix estimate 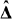 and finally vectorizing to obtain
and finally vectorizing to obtain 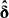 . This marginal-models approach is similar in spirit to the generalized estimating equation (GEE) popular in longitudinal data analysis. Statistical inference was obtained by bootstrap, i.e., obtaining 500 representatives of the sampling distribution by constructing 500 bootstrap data sets, each of which was obtained by sampling, with replacement, 205 data vectors consisting of methylation data concatenated with exposure/phenotype covariate data. We acknowledge that small biases in estimates will arise from the extent to which
. This marginal-models approach is similar in spirit to the generalized estimating equation (GEE) popular in longitudinal data analysis. Statistical inference was obtained by bootstrap, i.e., obtaining 500 representatives of the sampling distribution by constructing 500 bootstrap data sets, each of which was obtained by sampling, with replacement, 205 data vectors consisting of methylation data concatenated with exposure/phenotype covariate data. We acknowledge that small biases in estimates will arise from the extent to which 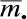 and
and 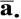 , computed over all the autosomal CpGs on the 27K array, would differ from the corresponding values obtained from all CpGs on the human genome, but conjecture that the bias is small for dense arrays, and that the resulting regression estimates will be representative of all human genome CpGs that conform to the selection criteria used by Illumina for inclusion on the 27K array.
, computed over all the autosomal CpGs on the 27K array, would differ from the corresponding values obtained from all CpGs on the human genome, but conjecture that the bias is small for dense arrays, and that the resulting regression estimates will be representative of all human genome CpGs that conform to the selection criteria used by Illumina for inclusion on the 27K array.
All statistical analyses were performed using the R statistical package (v. 2.11.1).
Acknowledgments
This work was supported by the Flight Attendant Medical Research Institute grant YCSA 052341 to C.M.; and the National Institutes of Health (R01CA121147 to K.K., R01CA100679 to K.K., R01CA078609 to K.K., R01CA126939 to K.K., R01CA057494 to M.K., P42ES007373 to M.K., R01CA082354 to H.N.).
Supplementary Material
References
- 1.Calvanese V, Lara E, Kahn A, Fraga MF. The role of epigenetics in aging and age-related diseases. Ageing Res Rev. 2009;8:268–276. doi: 10.1016/j.arr.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Tost J. DNA methylation: an introduction to the biology and the disease-associated changes of a promising biomarker. Mol Biotechnol. 2010;44:71–81. doi: 10.1007/s12033-009-9216-2. [DOI] [PubMed] [Google Scholar]
- 3.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 4.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Illingworth R, Kerr A, Desousa D, Jorgensen H, Ellis P, Stalker J, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rakyan VK, Down TA, Thorne NP, Flicek P, Kulesha E, Graf S, et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs) Genome Res. 2008;18:1518–1529. doi: 10.1101/gr.077479.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rakyan VK, Hildmann T, Novik KL, Lewin J, Tost J, Cox AV, et al. DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project. PLoS Biol. 2004;2:405. doi: 10.1371/journal.pbio.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schilling E, Rehli M. Global, comparative analysis of tissue-specific promoter CpG methylation. Genomics. 2007;90:314–323. doi: 10.1016/j.ygeno.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 9.Ehrlich M. DNA hypomethylation, cancer, the immunodeficiency, centromeric region instability, facial anomalies syndrome and chromosomal rearrangements. J Nutr. 2002;132:2424–2429. doi: 10.1093/jn/132.8.2424S. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann MJ, Schulz WA. Causes and consequences of DNA hypomethylation in human cancer. Biochem Cell Biol. 2005;83:296–321. doi: 10.1139/o05-036. [DOI] [PubMed] [Google Scholar]
- 11.Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775:138–162. doi: 10.1016/j.bbcan.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Waddington C. The epigenotype. Endeavor. 1:18–20. [Google Scholar]
- 13.Ronn T, Poulsen P, Hansson O, Holmkvist J, Almgren P, Nilsson P, et al. Age influences DNA methylation and gene expression of COX7A1 in human skeletal muscle. Diabetologia. 2008;51:1159–1168. doi: 10.1007/s00125-008-1018-8. [DOI] [PubMed] [Google Scholar]
- 14.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes and therapies. Lancet Neurol. 2009;8:1056–1072. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 15.Ordovas JM, Smith CE. Epigenetics and cardiovascular disease. Nat Rev Cardiol. 2010;7:510–519. doi: 10.1038/nrcardio.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turunen MP, Aavik E, Yla-Herttuala S. Epigenetics and atherosclerosis. Biochim Biophys Acta. 2009;1790:886–891. doi: 10.1016/j.bbagen.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010;8:1000533. doi: 10.1371/journal.pbio.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levenson VV. DNA methylation as a universal biomarker. Expert Rev Mol Diagn. 2010;10:481–488. doi: 10.1586/erm.10.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houseman EA, Christensen BC, Yeh RF, Marsit CJ, Karagas MR, Wrensch M, et al. Model-based clustering of DNA methylation array data: a recursive-partitioning algorithm for high-dimensional data arising as a mixture of beta distributions. BMC Bioinformatics. 2008;9:365. doi: 10.1186/1471-2105-9-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 24.Squazzo SL, O'Geen H, Komashko VM, Krig SR, Jin VX, Jang SW, et al. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res. 2006;16:890–900. doi: 10.1101/gr.5306606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi EK, Uyeno S, Nishida N, Okumoto T, Fujimura S, Aoki Y, et al. Alterations of c-fos gene methylation in the processes of aging and tumorigenesis in human liver. Mutat Res. 1996;354:123–128. doi: 10.1016/0027-5107(96)00056-5. [DOI] [PubMed] [Google Scholar]
- 28.Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 29.Issa JP, Vertino PM, Boehm CD, Newsham IF, Baylin SB. Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Proc Natl Acad Sci USA. 1996;93:11757–11762. doi: 10.1073/pnas.93.21.11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.So K, Tamura G, Honda T, Homma N, Waki T, Togawa N, et al. Multiple tumor suppressor genes are increasingly methylated with age in non-neoplastic gastric epithelia. Cancer Sci. 2006;97:1155–1158. doi: 10.1111/j.1349-7006.2006.00302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takatsu M, Uyeno S, Komura J, Watanabe M, Ono T. Age-dependent alterations in mRNA level and promoter methylation of collagen alpha1(I) gene in human periodontal ligament. Mech Ageing Dev. 1999;110:37–48. doi: 10.1016/s0047-6374(99)00041-x. [DOI] [PubMed] [Google Scholar]
- 32.Kwabi-Addo B, Chung W, Shen L, Ittmann M, Wheeler T, Jelinek J, et al. Age-related DNA methylation changes in normal human prostate tissues. Clin Cancer Res. 2007;13:3796–3802. doi: 10.1158/1078-0432.CCR-07-0085. [DOI] [PubMed] [Google Scholar]
- 33.Jintaridth P, Mutirangura A. Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol Genomics. 2010 doi: 10.1152/physiolgenomics.00146.2009. In press. [DOI] [PubMed] [Google Scholar]
- 34.Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drinkwater RD, Blake TJ, Morley AA, Turner DR. Human lymphocytes aged in vivo have reduced levels of methylation in transcriptionally active and inactive DNA. Mutat Res. 1989;219:29–37. doi: 10.1016/0921-8734(89)90038-6. [DOI] [PubMed] [Google Scholar]
- 36.Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, et al. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. 2009;130:234–239. doi: 10.1016/j.mad.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Issa JP. Aging, DNA methylation and cancer. Crit Rev Oncol Hematol. 1999;32:31–43. doi: 10.1016/s1040-8428(99)00019-0. [DOI] [PubMed] [Google Scholar]
- 38.Corbett JE, Sharma RK, Dressler WE. Cosmetic Toxicology. In: Marquardt H, Schafer SG, McClellan RO, Welsch F, editors. Toxicology. San Diego, CA: Academic Press; 1999. pp. 899–918. [Google Scholar]
- 39.Nohynek GJ, Antignac E, Re T, Toutain H. Safety assessment of personal care products/cosmetics and their ingredients. Toxicol Appl Pharmacol. 2010;243:239–259. doi: 10.1016/j.taap.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 40.Preston RJ, Skare JA, Aardema MJ. A review of biomonitoring studies measuring genotoxicity in humans exposed to hair dyes. Mutagenesis. 2010;25:17–23. doi: 10.1093/mutage/gep044. [DOI] [PubMed] [Google Scholar]
- 41.Andrew AS, Schned AR, Heaney JA, Karagas MR. Bladder cancer risk and personal hair dye use. Int J Cancer. 2004;109:581–586. doi: 10.1002/ijc.11729. [DOI] [PubMed] [Google Scholar]
- 42.Gago-Dominguez M, Bell DA, Watson MA, Yuan JM, Castelao JE, Hein DW, et al. Permanent hair dyes and bladder cancer: risk modification by cytochrome P4501A2 and N-acetyltransferases 1 and 2. Carcinogenesis. 2003;24:483–489. doi: 10.1093/carcin/24.3.483. [DOI] [PubMed] [Google Scholar]
- 43.Holly EA, Lele C, Bracci PM. Hair-color products and risk for non-Hodgkin's lymphoma: a population-based study in the San Francisco bay area. Am J Public Health. 1998;88:1767–1773. doi: 10.2105/ajph.88.12.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miligi L, Seniori Costantini A, Crosignani P, Fontana A, Masala G, Nanni O, et al. Occupational, environmental and life-style factors associated with the risk of hematolymphopoietic malignancies in women. Am J Ind Med. 1999;36:60–69. doi: 10.1002/(sici)1097-0274(199907)36:1<60::aid-ajim9>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 45.Nagata C, Shimizu H, Hirashima K, Kakishita E, Fujimura K, Niho Y, et al. Hair dye use and occupational exposure to organic solvents as risk factors for myelodysplastic syndrome. Leuk Res. 1999;23:57–62. doi: 10.1016/s0145-2126(98)00135-0. [DOI] [PubMed] [Google Scholar]
- 46.Rollison DE, Helzlsouer KJ, Pinney SM. Personal hair dye use and cancer: a systematic literature review and evaluation of exposure assessment in studies published since 1992. J Toxicol Environ Health B Crit Rev. 2006;9:413–439. doi: 10.1080/10937400600681455. [DOI] [PubMed] [Google Scholar]
- 47.Welsh MM, Karagas MR, Applebaum KM, Spencer SK, Perry AE, Nelson HH. A role for ultraviolet radiation immunosuppression in non-melanoma skin cancer as evidenced by gene-environment interactions. Carcinogenesis. 2008;29:1950–1954. doi: 10.1093/carcin/bgn160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Welsh MM, Applebaum KM, Spencer SK, Perry AE, Karagas MR, Nelson HH. CTLA4 variants, UV-induced tolerance and risk of non-melanoma skin cancer. Cancer Res. 2009;69:6158–6163. doi: 10.1158/0008-5472.CAN-09-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanchez B, Budtz-Jorgensen E, Ryan L. An estimating equations approach to fitting latent exposure models with longitudinal health outcomes. Ann Appl Stat. 2009;3:830–856. [Google Scholar]
- 50.Wilhelm CS, Kelsey KT, Butler R, Plaza S, Gagne L, Zens MS, et al. Implications of LINE1 methylation for bladder cancer risk in women. Clin Cancer Res. 2010;16:1682–1689. doi: 10.1158/1078-0432.CCR-09-2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Applebaum KM, McClean MD, Nelson HH, Marsit CJ, Christensen BC, Kelsey KT. Smoking modifies the relationship between XRCC1 haplotypes and HPV16-negative head and neck squamous cell carcinoma. Int J Cancer. 2009;124:2690–2696. doi: 10.1002/ijc.24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Houseman E, Coull B, Ryan L. A functional-based distribution diagnostic for a linear model with correlated outcomes. Biometrika. 2006;93:911–926. [Google Scholar]
- 53.Poage GM, Christensen BC, Houseman EA, McClean MD, Wiencke JK, Posner MR, et al. Genetic and epigenetic somatic alterations in head and neck squamous cell carcinomas are globally coordinated but not locally targeted. PLoS One. 2010;5:9651. doi: 10.1371/journal.pone.0009651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Christensen BC, Kelsey KT, Zheng S, Houseman EA, Marsit CJ, Wrensch MR, et al. Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 2010;6:1001043. doi: 10.1371/journal.pgen.1001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marsit CJ, Houseman EA, Christensen BC, Gagne L, Wrensch MR, Nelson HH, et al. Identification of methylated genes associated with aggressive bladder cancer. PLoS One. 2010;5:12334. doi: 10.1371/journal.pone.0012334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolff EM, Chihara Y, Pan F, Weisenberger DJ, Siegmund KD, Sugano K, et al. Unique DNA methylation patterns distinguish noninvasive and invasive urothelial cancers and establish an epigenetic field defect in premalignant tissue. Cancer Res. 2010;70:8169–8178. doi: 10.1158/0008-5472.CAN-10-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin Z, Hegarty J, Cappel J, Yu W, Chen X, Faber P, et al. Identification of disease-associated DNA methylation in intestinal tissues from patients with inflammatory bowel disease. Clin Genet. 2010 doi: 10.1111/j.1399-0004.2010.01546.x. [DOI] [PubMed] [Google Scholar]
- 58.Ang PW, Loh M, Liem N, Lim PL, Grieu F, Vaithilingam A, et al. Comprehensive profiling of DNA methylation in colorectal cancer reveals subgroups with distinct clinicopathological and molecular features. BMC Cancer. 2010;10:227. doi: 10.1186/1471-2407-10-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci USA. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.