

Abstract
Understanding the interactions of nanomaterials with biosystems is a critical goal in both biomedicine and environmental science. Engineered nanoparticles provide excellent tools for probing this interface. In this Feature Article we will summarize one of the themes presented in our recent Langmuir Lecture, discussing the use of monolayer design to understand and control the interactions of nanoparticles with biomolecules and cells.
Introduction
Nanoparticles provide promising platforms for a wide variety of biomedical applications, including biosensing,1 imaging2 and delivery.3 Clearly, understanding how these synthetic materials interact with biomolecules4, cells5, tissues6, and (ultimately) patients7 is central to applications of these systems in biomedicine; conversely, however, the use of nanoparticles in industrial applications creates growing environmental concerns regarding distribution and toxicity.8
The goal of understanding the interactions of nanoparticles with biosystems is truly multidimensional: nanoparticles come in a myriad of shapes and sizes, and can be decorated with an almost infinite variety of functionality.9 While there are a considerable number of studies that provide data on specific systems, there have also been those that have picked specific axes including particle size and shape and explored them in a comprehensive fashion.10 In our research, we have chosen to focus on how chemical functionality at the surface of the nanoparticle affects the interactions of nanoparticles with biosystems.11 In the course of our studies, we have adopted a standardized platform for our study that provides stability and controlled display of functional groups (Figure 1). Since surface functionality dictates the interactions of nanoparticles with the outside world, our approach provides information that can be generalized to nanoparticles with a variety of core materials exhibiting an array of physical properties.
Figure 1.

Nanoparticle monolayer design featuring stability and controlled presentation of functionality.
Nanoparticle-Protein Interactions
Finding the tabula rasa
The key to effective structure-property correlation is the proper choice of control. For studying the interactions of nanoparticles with biosystems, the ideal starting point would be a fully non-interacting particle (tabula rasa, i.e. “blank slate”) that could then be functionalized with specific headgroups, allowing us to ascertain the interactions of these functional groups with biosystems in a quantitative fashion. In our initial studies of nanoparticle-protein interactions, we investigated the interactions of the protease chymotrypsin (ChT) with mercaptoundecanoic acid-functionalized nanoparticles (NP-MUA) featuring a 2 nm diameter core gold core (Figure 2a).12 In this system the anionic NP-MUA particles were complementary to the positively charged patch on ChT, resulting in inhibition of activity. A two-step inhibition process was observed, however, with a rapid reversible inhibition step followed by a slower irreversible process; our hypothesis for this behavior was that the initial inhibition was driven by electrostatics forces, whereas the slower denaturation process involved interaction of hydrophobic residues of ChT with the nonpolar interior of the NP-MUA monolayer. As access to the interior would be headgroup dependent, simple alkyl ligands clearly do not provide the blank slate we desired.
Figure 2.

Effect of monolayer on nanoparticle-protein (ChT) interactions. a) Simple alkanethiol-based monolayer results in protein denaturation. b) TEG-functionalized particles are non-interacting. c) Termination of the TEG layer with carboxylate groups results in reversible binding to ChT that stabilizes the protein towards denaturation.
Oligo(ethylene glycol) (OEG) functionality provides one of the best materials for making non-interacting surfaces.13 Our thought was that appending a short OEG segment to the exterior of a hydrophobic particle would retain the micelle-like stabilization of the hydrophobic ligand shell interior, while providing a non-interacting exterior. Building upon our prior studies of ChT, we found that neutral tetra(ethylene glycol)-functionalized CdSe quantum dots (3.2 nm core diameter) showed no appreciable interaction with ChT (Figure 2b).14 In contrast, anionic particles (Figure 2c) strongly inhibited ChT, albeit fully reversibly. These observations are in agreement with the desired limitation of interaction to the charged headgroup, allowing us to isolate this electrostatic interaction from other complicating behaviors. Significantly, four is the magic number of ethylene glycol repeats required, as later studies indicate substantially increased rates of denaturation with shorter tri(ethylene glycol) ligand analogs.15
Controlling enzyme activity using nanoparticles
Given that core materials can be interchanged, we shifted our research from CdSe back to gold nanoparticles, observing a number of noteworthy properties for the analogous 2 nm diameter gold core Au-TCOOH particles (Figure 3). First, Au-TCOOH particles actually stabilize ChT against denaturation at interfaces, an important issue in biotechnology. Two mechanisms were identified for stabilization, namely binding of ChT in the active conformation coupled with the nanoparticle going selectively to the interface, and shielding the ChT from interfacial denaturation (Figure 3a). 16
Figure 3.

a) Schematic representation of ChT and ChT/Au-TCOOH complex at air-water interface. b) Rates of ChT (0.8 μM) activity decay of ChT alone and ChT with varying concentrations of Au-TCOOH.
A second unexpected result of ChT/Au-TCOOH binding was a dramatic alteration of the substrate selectivity of ChT: free ChT is a promiscuous enzyme, readily hydrolyzing appropriate anionic, cationic and neutral substrates, but upon binding to Au-TCOOH, the hydrolysis of cationic substrates is accelerated and that of anionic substrates retarded (Figure 4).17 Further kinetic studies established that this behavior resulted from interactions of the anionic monolayer surface of the nanoparticle with charged substrates and reaction products.18
Figure 4.

a) Chemical structure of the TCOOH ligand and schematic depiction of substrate-monolayer interaction induced enzyme selectivity. b) Structures of the modified SPNA substrates 1–3 and c) the initial rates of ChT hydrolysis of these modified substrates. Inset: normalized activity of Au-TCOOH bound ChT towards substrates 1–3.
Having explored the effects of charge, we next looked at the effects of hydrophobicity on nanoparticle-ChT affinity and the stability of the protein in the resulting complexes. For these studies we used nanoparticles featuring amino acid termini (Figure 5), allowing us to readily tailor the nanoparticle surface.19 The binding affinity followed an expected trend, with increasing nanoparticle surface hydrophobicity resulting in increased binding (Figure 5b). Protein stability, surprisingly, was also increased with increasing hydrophobicity (Figure 5c). This result is interesting given the general belief that hydrophobic surfaces are detrimental to protein stability.20
Figure 5.
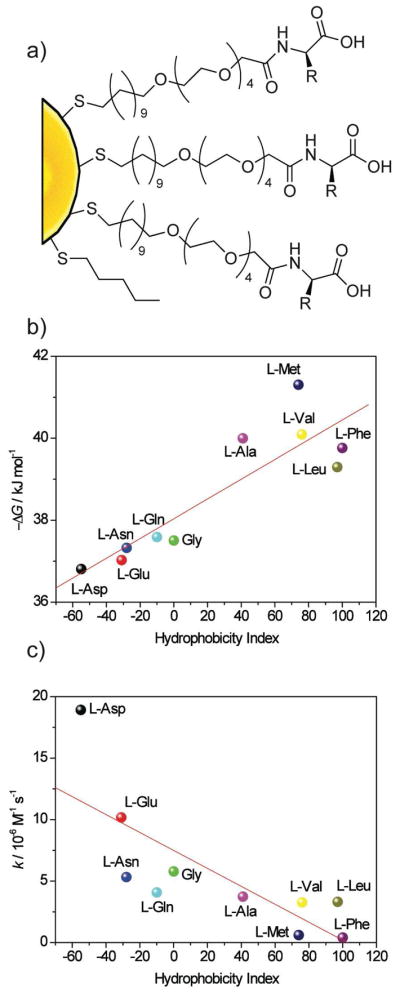
a) Amino acid decorated gold nanoparticles. b) Correlation between Gibbs free energy of NP-ChT interaction and hydrophobicity index of amino acid side chains. c) Correlation between the denaturation rate constants (k) of ChT and the hydrophobicity index of amino acid side chains in nanoparticles.
Nanoparticles as artificial proteins
The ability to tailor the nanoparticle surface in order to bind proteins makes these materials excellent candidates for mimicking protein surfaces. As an added benefit, the overall diameter of 2 nm core gold nanoparticles with our ligands is 7–8 nm,21 consistent in scale to a mid-sized globular protein. We have used isothermal titration calorimetry (ITC) to probe the thermodynamics of nanoparticle-protein interactions (Figure 6).22 These studies revealed that with amino acid-terminated ligands, protein-nanoparticle interactions strongly resembled protein-protein interactions (particularly in regard to entropy-enthalpy properties), with nanoparticles replicating protein-protein interactions better than any other known synthetic system (Figure 6c).23
Figure 6.
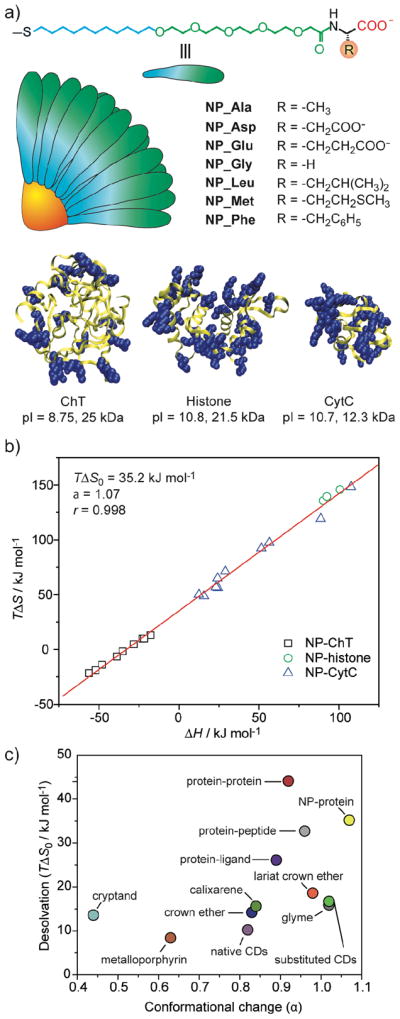
a) Structural features and relative sizes of amino acid-functionalized gold nanoparticles and proteins. The blue overlapping spheres in the proteins represent the positively charged residues on their surface. b) Plots of entropy (TΔS) versus enthalpy (ΔH) for NP-protein (number of data set n = 23) interactions. c) Slope (α) and intercept (TΔS0) values for various host-guest systems. Protein-ligand interactions have been divided into protein-peptide and protein-other (protein-ligand) interactions.
Beyond having similar behavior to proteins, appropriately charged nanoparticles can function as protein mimics. In one example, nanoparticles were used to disrupt protein-protein interactions between cytochrome c (cyt c) and cytochrome c peroxidase. 24 In these studies, the nanoparticle binds the target protein, preventing protein-protein interactions. This binding is quite biomimetic, as further studies using H/D exchange showed that facial selectivity of the particles to cyt c is dictated by particle functionality (Figure 7).25
Figure 7.

a) Chemical structures of functionalized gold nanoparticles, b) schematic depiction of surface interactions with Cyt c, and c) solvent accessibility (high, red; medium, green; low, blue; not observed, grey) for Cyt c in complex with Au-TAsp or Au-TPhe.
Concurrent with our protein binding studies, we explored functional DNA recognition by nanoparticles. Histones are highly cationic proteins that bind and condense DNA into nucleosomes, regulating gene transcription. Our hypothesis was that nanoparticles could be engineered to replicate the ability of histones to regulate DNA transcription. To test this hypothesis, we synthesized cationic nanoparticles and bound them to DNA a 37-mer primer for T7 RNA polymerase (Figure 8).26 Binding of the particles was verified using centrifugation, and transcription assays using T7 demonstrated that the particle bound DNA with high affinity, completely inhibiting translation (Figure 8c,d).
Figure 8.

a) Schematic representation of cationic nanoparticle, showing the interaction between the trimethylammonium sidechains and the anionic DNA. b) Relative sizes of the 37-mer DNA duplex (extended conformation) and particle. c) Representative acrylamide gel electrophoresis of the RNA products. Lanes are numbered with colloid (nanoparticle) equivalents used in each assay. d) The amount of RNA detected relative to levels produced in the absence of colloids (100% transcription).
Nanoparticles and cells
Nanoparticles can interact with living systems in a variety of fashions. This diversity makes nanoparticles promising players in the area of biomedicine.27 In our research we have employed nanoparticles as both delivery vehicles28 and as potential therapeutics in their own right.29 These studies have used our ability to control the surface properties of the particles through ligand design, and are generally (but not always) based on the tabula rasa structures presented previously.
Nanoparticles for DNA and protein delivery
Our initial delivery research focused on DNA transfection. These studies were built upon our previous studies of DNA binding (vide supra), and were motivated by the thought that binding DNA to small histone-sized particles should efficiently condense DNA plasmids, facilitating delivery. In our initial studies, we found that 2 nm core gold nanoparticles provided quite efficient gene delivery vehicles, with an observed transfection efficiency 7-fold that of PEI.30
While our initial cationic particles of study were efficient transfection agents, they were somewhat more toxic than one would desire. Our thought was that the quaternary ammonium headgroup was perhaps to blame. To circumvent this concern, we synthesized a set of more biocompatible 2 nm core diameter gold particles featuring amino acid headgroups (Figure 9).31 Complexation of these particles revealed that the dendritic lysinebased headgroup generated substantially smaller nanoplexes than the other particles (Figure 9a). While gene delivery is a very complex multi-step process, the initial step of cellular uptake would be expected to be facilitated by smaller-sized assemblies; this prediction is borne out in the observed transfection efficiencies, where the lysine dendron-based particle was found to be a much more effective delivery vehicle (Figure 9c). Significantly, no toxicity was observed at the concentrations used for transfection for any of the amino acidterminated nanoparticle systems (Figure 9d).
Figure 9.

a) Schematic illustration of the monolayer protected gold nanoparticles used as transfection vectors. b) Chemical structures of headgroups presented on the surface of the nanoparticles, with nanoparticle-plasmid DNA nanoplex diameters. c) Effective transfection using NP-LysG1 and NP-Lys relative to positive controls, NP-TMA and polylysine (pLys). No appreciable enzyme activity in absence of vectors. Inset showing solution color during β-Gal activity assay performed after transfection. Color change: yellow (substrate) to red (product). d) Cell viability determined by Alamar blue assay at the end of transfection showing low toxicity for amino acid-terminated ligands.
Given that our 2 nm cationic gold particles could transport highly anionic DNA across the cell membrane, we thought that anionic proteins could perhaps be delivered in similar fashion. Protein-based therapeutics have the potential to revolutionize medicine, but only if they can be delivered intact and active. For our studies we chose β-galactosidase (β-Gal) as a model payload for intracellular delivery. β-Gal presents a particularly challenging protein to transport, owing to its large size (465 kD) and negative charge (pI 4.6). Our initial attempts to deliver β-Gal using simple cationic nanoparticles based on our prior β-Gal binding studies32 were unsuccessful. To provide a more sophisticated receptor, we turned to peptide-capped particles, using a TEG linker to prevent protein denaturation (Figure 10).33 The exterior 4-mer peptide headgroup of the ligand was comprised of both strongly and weakly basic amino acid residues (Arg, Lys and His), and serves multiple roles. The cationic peptide headgroup provides protein surface recognition through favorable electrostatic interaction as well as plasma membrane association, additionally, the proton-sponge imidazole group of histidine provides “endosomal buffering” and potential escape for the complexes; in practice, these particles were quite effective for delivering β-Gal into cells. Our initial studies using FITC-tagged β-Gal demonstrated intracellular delivery, and further studies using FM 4–64 (an endosomal marker) showed very little co-localization, indicating that the β-Gal was not trapped in endosomes. Most significantly, the uptaken protein retained activity, as demonstrated through X-gal staining of the cells (Figure 10c-e).
Figure 10.

a) Schematic representation of intracellular delivery of functional protein using gold nanoparticles. b) Structure of the HKRK nanoparticle. (c-e) X-gal staining after transfection. (c) HeLa with protein only. Transfected (d) HeLa and (e) C2C12 cells with NP_Pep/β-gal (100 nM/50 nM). (f) The percent of transfection with NP_Pep/β-gal (100 nM/50 nM) in different cell lines. (g) Dose-dependent protein delivery into HeLa cells at 2:1 NP_Pep/β-gal.
Sensing Proteins and Cells using Nanoparticle “Noses”
As shown above, nanoparticles provide excellent scaffolds for binding biomacromolecules, presenting a size that is large enough to effectively interact with target biomolecules with high affinity. These systems are also tunable in terms of charge, hydrophobicity and surface topology. As such, monolayer-functionalized nanoparticles are ideal for applications where selective binding of biological systems is required. One such application is “chemical nose/tongue” sensing, an approach modeled after olfaction that relies on pattern recognition of sensor arrays. In our studies, we have applied this strategy to both protein and cell sensing using the tunability of the nanoparticle-surface to provide the required selectivity.34 An additional advantage of our nanoparticles is that they bind proteins reversibly. This property renders sensors, as opposed to the dosimeters that would arise from irreversible binding.
Protein Sensing
In our initial studies, we explored the use of nanoparticle-polymer sensors to identify proteins. In this case we used a displacement strategy where polymer fluorescence is quenched by gold nanoparticles. This polymer is displaced from the particle surface by addition of protein analytes, generating a change in fluorescence (Figure 11a-b). For this approach to be effective, the polymer used to transduce particle-protein interaction needs to be both highly fluorescent and bind the particle with affinities commensurate with those of the analyte proteins, i.e. pico- to nanomolar. The poly(phenylene ethynylene) (PPE) polymers studied extensively by Bunz provided ideal properties for our assays.35
Figure 11.

a) Displacement of quenched fluorescent polymer by protein analyte with concomitant restoration of fluorescence. b) Pattern generation through differential release of fluorescent polymers from gold nanoparticles. c) Chemical structure of cationic gold nanoparticles (NP1-NP6) and anionic fluorescent polymer PPE-CO2 (n ~ 12). d) Fluorescence response (ΔI) patterns of the NP-PPE sensor array (NP1 – NP6) against various proteins (CC: cytochrome c, β-Gal: β-galactosidase, PhosA: acid phosphatase, PhosB: alkaline phosphatase, SubA: subtilisin. e) Canonical score plot for the first two factors of simplified fluorescence response patterns obtained with NP-PPE assembly arrays against 5 μM proteins. The canonical scores were calculated by LDA for the identification of seven proteins. The 95% confidence ellipses for the individual proteins are also shown.
Our first sensing efforts were focused on sensing of individual proteins in solution. Using a set of six nanoparticles, we were readily able to sense seven different analyte proteins, including four featuring similar size and charge (Figure 11c-e).36 From the fluorescence profiles, it is clear that there is a distinct signature for each analyte protein. These signatures were analyzed using Linear Discriminant Analysis (LDA), allowing us to differentiate between the analyte proteins. An important issue with nose-type sensors is that the concentration response can be rather complex; to provide our sensor with the ability to identify proteins at arbitrary concentrations, we used an optical density-based approach, diluting protein concentrations to an absorbance of 0.005 at 280 nm. Using this approach we were able to identify the proteins at concentrations of 4–215 nM depending on the proteins extinction coefficient. Once the protein was identified, we could then calculate the initial protein concentration from the absorbance using its extinction coefficient.
Having effectively sensed proteins in “clean” solutions, we next addressed the substantially more difficult of protein sensing in serum; human serum is a complex fluid consisting of >20,000 different proteins, with an overall concentration of ~1 mM. The predominant constituent of human serum is human serum albumin (HSA, ~700 mM), making sensing of proteins in serum an exercise much akin to finding needles in a haystack. Our initial goal in designing a sensor for serum was to be able to detect small changes in protein levels. To this end, we used human serum and “spiked” it with analyte proteins while maintaining constant protein concentration. Our initial efforts using the same polymer-particle system were unsuccessful using serum spiked with either 5 mM or 500 nM protein.
The lack of responsiveness of our sensor system in serum was puzzling. After considerable study, we determined that polymer aggregation was the culprit, as self-quenching counteracted the fluorogenesis arising from displacement of the probe polymer from the particle surface. To avoid this aggregation, we explored the use of green fluorescent protein (GFP) as an alternative protein37 due to the biocompatible nature of this fluorophore. This polymer/protein substitution was facilitated by the fact that our initial polymer and GFP (pI 5.92) are both anionic, allowing us to use the same family of nanoparticles. After screening our nanoparticle library, we identified five nanoparticles that provided effective identification of five different serum proteins (including HSA) spiked into undiluted human serum at 500 nM. This sensor system is highly discriminating, as a 500 nM change in HSA concentration corresponds to 0.065% change in the concentration of this species (Figure 12).
Figure 12.
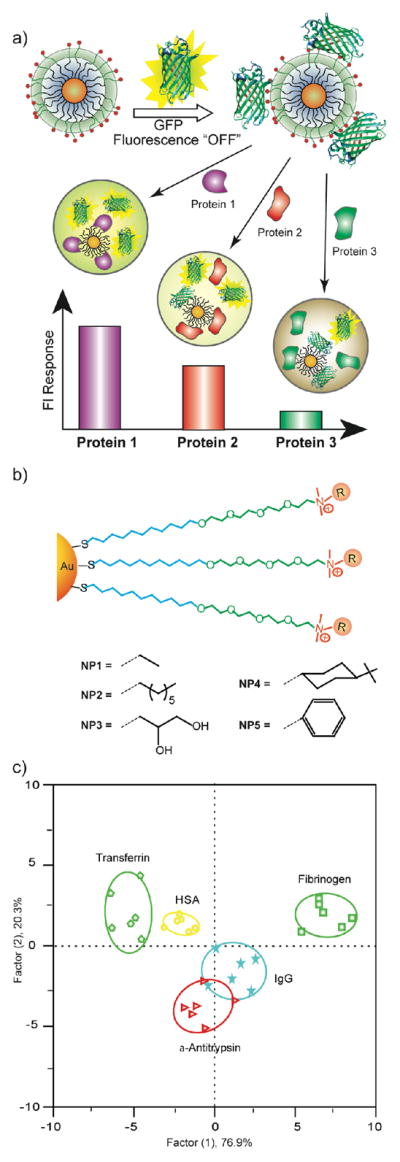
a) Schematic illustration of the competitive binding between protein and quenched nanoparticle-GFP complexes and protein aggregation leading to the fluorescence light-up or further quenching. b) Chemical structure of cationic gold nanoparticles. c) Canonical score plot for the fluorescence patterns as obtained from LDA against five protein analytes at fixed concentration (500 nM) with 95% confidence ellipses.
Bacteria sensing
Having shown that we can differentiate small changes in protein analyte levels in complex solutions, it would seem to make sense that one can differentiate the complex mixtures of biomolecules found on cell surfaces, e.g. bacteria. Clearly, rapid sensing and identification would be an important tool for biomedical, environmental, and security applications. As with our serum sensing, initial studies using the original polymer were unsuccessful, in this case due to aggregation on the bacteria surface. To overcome this issue we used a synthetic “swallow-tail” polymer designed to be non-aggregating for the sensing process (Figure 13a). Using this polymer and just three particles, we were able to identify twelve different bacteria (105 cells/mL), including both Gram-positive and – negative.38 Most importantly, we were able to effectively discriminate between three different strains of E. coli., a key aspect in distinguishing between pathogenic and relatively benign bacteria (Figure 13c).
Figure 13.

a) Receptor and transducer components of the bacteria sensors. b) Fluorescence response patterns of nanoparticle-polymer constructs in the presence of various bacteria (OD600 = 0.05). c) Canonical score plot for the fluorescence response patterns as determined with LDA.
Sensing of mammalian cells: detection and identification of cancer cells
Given our success with bacteria, we felt that our array-based sensing strategy should be applicable to mammalian cells as well, providing a diagnostic tool for cancer detection. For these studies we began by differentiating between cell types: using three particles and (interestingly) our original polymer, we were able to readily differentiate between human liver, cervix, breast and testis cells.39 This result is not surprising, as these cells have different functions that would be expected to generate cell surface differences. We next set our sights on a more challenging target, namely differentiation of cell state. Using the same particles, we were able to distinguish between three different human breast cell lines (normal, cancerous, and metastatic)40; while these studies were successful, the fact that these three cell lines came from three different individuals raised a concern that we were sensing individual-to–individual variations as opposed to cell state. To address this concern, we chose three isogenic cell lines (once again normal, cancerous, and metastatic) derived from BALB/c mice (Figure 14). As before, these cells could be readily distinguished, demonstrating the promise of our method for both cancer detection and identification.41
Figure 14.

a) Molecular structures of nanoparticles and polymers. b) Schematic of fluorophore displacement cell detection array. c) Change in fluorescence intensities for three cell lines of same genotype CDBgo, TD cell and V14 using nanoparticle-polymer supramolecular complexes. d) Canonical score plot for the first two factors of simplified fluorescence response patterns obtained with NP–PPE assembly arrays against different mammalian cell types.
Summary and Outlook
Nanoparticles provide highly promising systems for fundamental and applied biomedical research. In our studies, we have developed nanoparticles that are functional mimics of proteins, replicating the surface properties and hence interactions of these biomolecules. Building upon these properties, we have developed new delivery and sensing systems, exploiting the tunability through engineering of the particle monolayer. Beyond these applications, nanoparticles are emerging as therapeutics in their own right, with the capability of modulating cellular processes based on their surface functionality. Taken together, it is clear that there is much still to be learned in fundamental terms regarding the complex interactions of nanomaterials with biosystems. Simultaneously, the properties already demonstrated are ripe for translation into biomedicine, providing new tools for the diagnosis and treatment of disease.
Acknowledgments
V.R. acknowledges N.I.H. (EB012246-01, EB012246-01, GM07717 and the NSF (CHE-0808945, VR), MRSEC facilities, and the Center for Hierarchical Manufacturing (DMI-0531171).
References
- 1.(a) Miranda OR, Chen HT, You CC, Mortenson DE, Yang XC, Bunz UHF, Rotello VM. J Am Chem Soc. 2010;132:5285–5289. doi: 10.1021/ja1006756. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Peng G, Hakim M, Broza YY, Brillan S, Abdah-Bortnyak R, Kuten A, Tisch U, Haick H. Brit J Cancer. 2010;103:542–551. doi: 10.1038/sj.bjc.6605810. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Miranda OR, Creran B, Rotello VM. Curr Opin Chem Biol. 2010;14:728–736. doi: 10.1016/j.cbpa.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Sharrna P, Brown S, Walter G, Santra S, Moudgil B. Adv Colloid Interface Sci. 2006;123:471–485. doi: 10.1016/j.cis.2006.05.026. [DOI] [PubMed] [Google Scholar]; (b) Popović Z, Liu W, Chauhan VP, Lee J, Wong C, Greytak AB, Insin N, Nocera DG, Fukumura D, Jain RK, Bawendi MG. Angew Chem Int Edit. 2010;49:8649–8652. doi: 10.1002/anie.201003142. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R. Nat Biotechnol. 2000;18:410–414. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- 3.(a) Duncan B, Kim C, Rotello VM. J Controlled Release. 2010;148:122–127. doi: 10.1016/j.jconrel.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Han G, Ghosh P, De M, Rotello VM. Nanobiotechnology. 2007;3:40–45. [Google Scholar]; (c) Han G, Ghosh P, Rotello VM. Nanomedicine. 2007;2:113–123. doi: 10.2217/17435889.2.1.113. [DOI] [PubMed] [Google Scholar]
- 4.Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AKR, Han MS, Mirkin CA. Science. 2006;312:1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- 5.Verma A, Uzun O, Hu Y, Hu Y, Han HS, Watson N, Chen S, Irvine DJ, Stellacci F. Nat Mater. 2008;7:588–595. doi: 10.1038/nmat2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu Z-J, Carboni R, Quercio MJ, Jr, Yan Bo, Miranda OR, Anderton DL, Arcaro KF, Rotello VM, Vachet RW. Small. 2010;6:2261–2265. doi: 10.1002/smll.201000989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis ME. Mol Pharmaceutics. 2009;6:659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 8.(a) Peralta-Videa JR, Zhao L, Lopez-Moreno ML, De la Rosa G, Hong J, Gardea-Torresdey JL. J Hazard Mater. 2010;186:1–15. doi: 10.1016/j.jhazmat.2010.11.020. [DOI] [PubMed] [Google Scholar]; (b) Kumar P, Robins A, Vardoulakis S, Britter R. Atmos Environ. 2010;44:5035–5052. [Google Scholar]
- 9.(a) De M, Miranda OR, Rana S, Rotello VM. Chem Commun. 2009;16:2157–2159. doi: 10.1039/b900552h. [DOI] [PubMed] [Google Scholar]; (b) De M, Ghosh P, Rotello VM. Adv Mat. 2008;20:4225–4241. [Google Scholar]; (c) Ghosh P, Han G, De M, Kim CK, Rotello VM. Adv Drug Del Rev. 2008;60:1307–1315. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 10.(a) Cedervall T, Lynch I, Foy M, Berggard T, Donnelly SC, Cagney G, Linse S, Dawson KA. Angew Chem Int Edit. 2007;46:5754–5756. doi: 10.1002/anie.200700465. [DOI] [PubMed] [Google Scholar]; (b) Lym YS, Haynes CL. J Am Chem Soc. 2010;132:4834–4832. doi: 10.1021/ja910846q. [DOI] [PubMed] [Google Scholar]; (c) Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE. Adv Drug Del Rev. 2009;61:428–437. doi: 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) He Q, Zhang Z, Gao F, Li Y, Shi J. Small. 2011;7:271–280. doi: 10.1002/smll.201001459. [DOI] [PubMed] [Google Scholar]
- 11.You CC, Verma A, Rotello VM. Soft Matter. 2006;2:190–204. doi: 10.1039/b517354j.You C-C, De M, Rotello VM. Curr Opin Chem Biol. 2005;9:639–646. doi: 10.1016/j.cbpa.2005.09.012.Phillips RL, Miranda OR, Mortenson DE, Subramani C, Rotello VM, Bunz UHF. Soft Matter. 2009;5:607–612.Carver AM, De M, Bayraktar H, Rana S, Rotello VM, Knapp MJ. J Am Chem Soc. 2009;131:3798–3799. doi: 10.1021/ja806064t.De M, Rana S, Rotello VM. Macromol Biosci. 2009;9:174–178. doi: 10.1002/mabi.200800289.Bayraktar H, Srivastava S, You CC, Rotello VM, Knapp MJ. Soft Matter. 2008;4:751–756. doi: 10.1039/b716386j.(g)
- 12.(a) Fischer NO, McIntosh CM, Simard JM, Rotello VM. Proc Natl Acad Sci U S A. 2002;99:5018–5023. doi: 10.1073/pnas.082644099. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fischer NO, Verma A, Goodman CM, Simard JM, Rotello VM. J Am Chem Soc. 2003;125:13387–13391. doi: 10.1021/ja0352505. [DOI] [PubMed] [Google Scholar]; (c) Verma A, Simard JM, Rotello VM. Langmuir. 2004;20:4178–4181. doi: 10.1021/la036183v. [DOI] [PubMed] [Google Scholar]; (d) De M, Rotello VM. Chem Commun. 2008;30:3504–3506. doi: 10.1039/b805242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herrwerth S, Eck W, Reinhardt S, Grunze M. J Am Chem Soc. 2003;125:9359–9366. doi: 10.1021/ja034820y. [DOI] [PubMed] [Google Scholar]
- 14.Hong R, Fischer NO, Verma A, Goodman CM, Emrick TS, Rotello VM. J Am Chem Soc. 2004;126:739–743. doi: 10.1021/ja037470o.For studies on TEG-terminated gold nanoparticles see: Jordan BJ, Hong R, Han G, Rana S, Rotello VM. Nanotechnology. 2009;20:43004. doi: 10.1088/0957-4484/20/43/434004.
- 15.You CC, De M, Rotello VM. Org Lett. 2005;7:5685–5687. doi: 10.1021/ol052367k. [DOI] [PubMed] [Google Scholar]
- 16.Jordan BJ, Hong R, Gider B, Hill J, Emrick T, Rotello V. Soft Matter. 2006:558–560. doi: 10.1039/b603980d. [DOI] [PubMed] [Google Scholar]
- 17.Hong R, Emrick T, Rotello VM. J Am Chem Soc. 2004;126:13572–13573. doi: 10.1021/ja0461163. [DOI] [PubMed] [Google Scholar]
- 18.You CC, Agasti S, De M, Knapp MJ, Rotello VM. J Am Chem Soc. 2006;128:14612–14618. doi: 10.1021/ja064433z. [DOI] [PubMed] [Google Scholar]
- 19.You C-C, De M, Rotello VM. J Am Chem Soc. 2005;127:12873–12881. doi: 10.1021/ja0512881. [DOI] [PubMed] [Google Scholar]; (b) You C-C, Agasti SS, Rotello VM. Chem Eur J. 2008;14:143–150. doi: 10.1002/chem.200701234. [DOI] [PubMed] [Google Scholar]
- 20.(a) Sethuraman A, Belfort G. Biophys J. 2005;88:1322–1333. doi: 10.1529/biophysj.104.051797. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Strickler SS, Gribenko AV, Gribenko AV, Keiffer TR, Tomlinson J, Reihle T, Loladze VV, Makhatadze GI. Biochemistry. 2006;45:2761–2766. doi: 10.1021/bi0600143. [DOI] [PubMed] [Google Scholar]; (c) Dong H, Mukaiyama A, Tadokoro T, Koga Y, Takano K. J Mol Biol. 2008;378:264–272. doi: 10.1016/j.jmb.2008.02.039. [DOI] [PubMed] [Google Scholar]
- 21.(a) Agasti SS, You CC, Arumugan P, Rotello VM. J Mater Chem. 2008;18:70–73. [Google Scholar]; (b) Cooke G, Garety JF, Hewage SG, Rabani G, Rotello VM, Woisel P. Chem Commun. 2006;39:4119–4121. doi: 10.1039/b608543a. [DOI] [PubMed] [Google Scholar]
- 22.De M, You CC, Srivastava S, Rotello VM. J Am Chem Soc. 2007;129:10747–10753. doi: 10.1021/ja071642q. [DOI] [PubMed] [Google Scholar]
- 23.You CC, Chompoosor A, Rotello VM. Nanotoday. 2007;2:34–43. [Google Scholar]
- 24.Bayraktar H, Ghosh P, Rotello VM, Knapp MJ. Chem Commun. 2006;13:1390–1392. doi: 10.1039/b516096k. [DOI] [PubMed] [Google Scholar]
- 25.Bayraktar H, You C-C, Rotello VM, Knapp MJ. J Am Chem Soc. 2007;129:2732–2733. doi: 10.1021/ja067497i. [DOI] [PubMed] [Google Scholar]
- 26.(a) McIntosh CM, Esposito EA, Boal AK, Simard JM, Martin CT, Rotello VM. J Am Chem Soc. 2001;123:7626–7629. doi: 10.1021/ja015556g. [DOI] [PubMed] [Google Scholar]; (b) Han G, Martin CM, Rotello VM. Chem Biol Drug Des. 2006;67:78–82. doi: 10.1111/j.1747-0285.2005.00324.x. [DOI] [PubMed] [Google Scholar]; (c) Han G, Chari NS, Verma A, Hong R, Martin CT, Rotello VM. Bioconjugate Chem. 2005;16:1356–1359. doi: 10.1021/bc050173j. [DOI] [PubMed] [Google Scholar]
- 27.(a) Agasti SS, Rana S, Park MH, Kim CK, You CC, Rotello VM. Adv Drug Deliv Rev. 2010;162:316–328. doi: 10.1016/j.addr.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) LeDuc PR, Wong MS, Ferreira PM, Groff RE, Haslingerm K, Koonce MP, Lee WY, Love JC, McCammon JA, Monteiro-Riviere NA, Rotello VM, Rubloff GW, Westervelt R, Yoda M. Nat Nanotechnol. 2007;2:3–7. doi: 10.1038/nnano.2006.180. [DOI] [PubMed] [Google Scholar]
- 28.(a) Kim B, Han G, Toley BJ, Kim CK, Rotello VM, Forbes NS. Nat Nanotechnol. 2010;5:465–472. doi: 10.1038/nnano.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim C-K, Ghosh P, Rotello VM. Nanoscale. 2009;1:61–67. doi: 10.1039/b9nr00112c. [DOI] [PubMed] [Google Scholar]; (c) Agasti S, Chompoosor A, You C-C, Ghosh P, Kim C-K, Rotello Vincent M. J Am Chem Soc. 2009;131:5728–5729. doi: 10.1021/ja900591t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kim CK, Ghosh P, Pagliuca C, Zhu Z-J, Menichetti S, Rotello VM. J Am Chem Soc. 2009;131:1360–1361. doi: 10.1021/ja808137c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hong R, Han G, Kim B, Forbes NS, Rotello VM. J Am Chem Soc. 2006;128:1078–1079. doi: 10.1021/ja056726i. [DOI] [PubMed] [Google Scholar]
- 29.(a) Kim CK, Agasti SS, Zhu ZJ, Isaacs L, Rotello VM. Nature Chem. 2010;2:962–966. doi: 10.1038/nchem.858. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chompoosor A, Saha K, Ghosh PS, Macarthy DJ, Miranda OR, Zhu ZJ, Arcaro KF, Rotello VM. Small. 2010;6:2246–2249. doi: 10.1002/smll.201000463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sandhu KK, McIntosh CM, Simard JM, Smith SW, Rotello VM. Bioconjugate Chem. 2002;13:3–6. doi: 10.1021/bc015545c. [DOI] [PubMed] [Google Scholar]
- 31.(a) Ghosh PS, Kim CK, Han G, Forbes NS, Rotello VM. ACS Nano. 2008;2:2213–2218. doi: 10.1021/nn800507t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh PS, Han G, Erdogan B, Rosado O, Krovi SA, Rotello VM. Chem Biol Drug Des. 2007;70:13–18. doi: 10.1111/j.1747-0285.2007.00534.x. [DOI] [PubMed] [Google Scholar]; (c) Ghosh PS, Han G, Erdogan B, Rosado O, Rotello VM. J Pept Sci. 2008;14:134–138. doi: 10.1002/psc.947. [DOI] [PubMed] [Google Scholar]
- 32.Verma A, Simard JM, Worrall JWE, Rotello VM. J Am Chem Soc. 2004;126:13987–13991. doi: 10.1021/ja046572r. [DOI] [PubMed] [Google Scholar]
- 33.Ghosh P, Yang X, Arvizo R, Zhu ZJ, Mo Z, Rotello VM. J Am Chem Soc. 2010;132:2642–2645. doi: 10.1021/ja907887z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bunz UHF, Rotello VM. Angew Chem Int Edit. 2010;49:3268–3279. doi: 10.1002/anie.200906928. [DOI] [PubMed] [Google Scholar]
- 35.(a) Kim IB, Dunkhorst A, Gilbert J, Bunz UHF. Macromolecules. 2005;38:4560–4562. [Google Scholar]; (b) Kim IB, Phillips R, Bunz UHF. Macromolecules. 2007;40:5290–5293. [Google Scholar]
- 36.(a) You CC, Miranda OR, Gider B, Ghosh PS, Kim I-B, Erdogan B, Krovi SA, Bunz UHF, Rotello VM. Nature Nanotech. 2007;2:318–323. doi: 10.1038/nnano.2007.99. [DOI] [PubMed] [Google Scholar]; (b) Miranda OR, You CC, Phillips R, Kim IK, Ghosh PS, Bunz UHF, Rotello VM. J Am Chem Soc. 2007;129:9856–9857. doi: 10.1021/ja0737927. [DOI] [PubMed] [Google Scholar]
- 37.De M, Rana S, Akpinar H, Miranda OR, Arvizo RR, Bunz UHF, Rotello VM. Nature Chem. 2009;1:461–465. doi: 10.1038/nchem.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phillips RL, Miranda OR, You CC, Rotello VM, Bunz UHF. Angew Chem Int Edit. 2008;47:2590–2594. doi: 10.1002/anie.200703369. [DOI] [PubMed] [Google Scholar]
- 39.Bajaj A, Miranda OR, Kim IK, Phillips RL, Jerry DJ, Bunz UHF, Rotello VM. Proc Natl Acad Sci U S A. 2009;106:10912–10916. doi: 10.1073/pnas.0900975106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bajaj A, Rana S, Miranda OR, Yawe JC, Jerry DJ, Bunz UHF, Rotello VM. Chem Sci. 2010;1:134–138. [Google Scholar]
- 41.Bajaj A, Miranda OR, Phillips R, Kim IB, Jerry DJ, Bunz UHF, Rotello VM. J Am Chem Soc. 2010;132:1018–1022. doi: 10.1021/ja9061272. [DOI] [PMC free article] [PubMed] [Google Scholar]