

Abstract
Many microRNAs (miRNAs) are encoded within the introns of RNA Pol II transcripts, often as polycistronic precursors. Here we demonstrate the optimization of an intron encoding three endogenous miRNAs for the ectopic expression of heterologous anti-HIV-1 siRNAs processed from a single RNA polymerase II primary miRNA. Our expression system, designated as MCM7, is engineered from the intron embedded, tri-cistronic mir106b cluster that endogenously expresses miR-106b, miR-93 and miR-25. Manipulation of the mir106b cluster demonstrated a strict requirement for maintenance of the native flanking pri-miRNA sequences and key structural features of the native miRNAs for efficient siRNA processing. As a model for testing the efficacy of this approach, we have replaced the three endogenous miRNAs with siRNAs targeting the tat and rev transcripts of HIV-1. This study has enabled us to establish guidelines for optimal processing of the engineered miRNA mimics into functional siRNAs. In addition, we demonstrate that the incorporation of a small nucleolar RNA TAR chimeric decoy (snoRNA) inserted within the MCM7 intron resulted ina substantial enhancement of HIV suppression in long term acute infectious HIV-1 challenges.
Keywords: RNAi, HIV-1, intron, MCM7, MicroRNAs, siRNAs, polycistron, TAR, nucleolar
INTRODUCTION
Since its discovery in the worm C. elegans1, and subsequent demonstration in essentially all higher organisms, including humans2, RNA interference (RNAi) has become the method of choice for studying gene function in mammalian cells and is also attracting increasing attention as a therapeutic modality3–5. RNAi-mediated knockdown or post transcriptional gene silencing (PTGS) of the gene of interest is achieved either by transfection of synthetic small interfering RNAs (siRNAs)2, by expression of short hairpin RNAs (shRNA)6, 7 or by mimics8 of their naturally occurring counterparts, the miRNAs9. A wide range of viral diseases have been successfully targeted in cell culture and animal models by either siRNA or shRNA vector-based approaches4, 10, 11. Stable expression systems can be efficiently delivered to mammalian cells by viral vectors in cell culture and in vivo, resulting in long term silencing of the target gene12–14.
Traditional small-molecule based drug treatments of viral diseases are burdened with two related serious limitations: the great genetic diversity of the various strains and the frequent evolution of drug-resistant strains under the selective pressure of treatment4, 15. This problem was quickly recognized as a limitation that was also shared by RNAi for treating RNA viruses with high mutation rates. In one of the first published papers on the use of RNAi against a viral target in cell culture, Gitlin et al10 demonstrated that while effective inhibition of poliovirus replication could be achieved, escape mutants in the target sequence of the siRNA effector emerged upon extended culturing of the infected cells. Similar observations were subsequently reported by various other researchers for other viruses including HIV16–20. Some researchers have attempted to bypass the problem of escape by using a combination of anti-viral inhibitory agents 4, 15. The feasibility of this strategy was first demonstrated by Song et al21, who observed more pronounced and longer-lasting siRNA-mediated protection against HIV infection upon simultaneous targeting of both CCR5 and p24 than either of the individual targets alone. Similar effects were reported for targeting of HCV by two chemically synthesized siRNAs19. Successful incorporation of multiple effector molecules (including non-RNAi effectors) within the same expression vector have subsequently been reported by various researchers for targeting both HIV and HCV22–27. In a comprehensive study of siRNAs targeting HIV, ter Brake et al screened 86 different shRNA constructs targeting conserved sequences within 8 different regions of the HIV genome, before combining their two and three best candidates as separate Pol III expression cassettes26. This resulted in markedly improved inhibition of virus replication and substantially delayed emergence of escape mutants. A computational model of the propagation of HIV infection in cells expressing RNAi suggested that simultaneous expression of 4 optimally designed RNAi effectors might be sufficient to achieve a complete protection against the emergence of escape mutants27, 28 although it remains to be seen whether this prediction bears out experimentally.
The ability to express multiple RNAi effector units from a single Pol II polycistronic transcript is potentially very important for avoidance of toxicities observed when shRNAs are ectopically expressed from strong Pol III promoters29, 30. This problem of Pol III expressed shRNA toxicity can be circumvented by taking advantage of endogenous RNAi transcripts, miRNAs31–33, for expression of transgenes. MiRNAs are structurally and functionally very similar to siRNAs, and are incorporated into RISC34. They are primarily transcribed by RNA Pol II as parts of larger primary transcripts35, 36 that undergo initial processing in the nucleus by the microprocessor complex, consisting of the nuclease Drosha and its specificity binding partner, DGCR8/Pasha37–41. The resulting imperfect hairpin of approximately 65 nt, the pre-miRNA, is then exported to the cytoplasm through exportin-542–44, where it is further processed by Dicer. For the majority of miRNAs, one strand is asymmetrically loaded into RISC to function as the guide strand32, 45–47. Endogenous miRNAs are often expressed as intronic clusters from a single polycistronic transcript and the miRNAs simultaneously downregulate the expression of multiple mRNAs48–52.
Cullen and co-workers pioneered the use of Pol II-based miRNA expression cassettes through their demonstration that the region of the primary transcript of miR-30 corresponding to the mature miRNA may be replaced with a heterologous stem without compromising activity8, enabling the generation of a miRNA-based expression cassette with generalizable targeting properties. MiR-30 based expression constructs have been incorporated into lentiviral vectors and used to generate whole libraries of validated targeting vectors53–55, allowing temporal control of target gene silencing for functional studies56. The known localization of many miRNA genes within introns has prompted researchers to attempt expression of miRNAs from polycistronic transcripts. Zhou et al expressed an siRNA from a miR-30 background within an intron of an EGFP expression cassette, thus directly coupling miRNA and marker gene expression57. MiR-155 has been used by others in a similar fashion58, 59, including the simultaneous expression of two active miRNAs from the embedded intron, allowing dual targeting58. Results from various studies based on different miRNA backgrounds are somewhat conflicting, but an emerging consensus appears to be that proper processing of the chimeric transcript requires maintaining the secondary structure of at least one helical turn immediately preceding the Drosha processing site, with possible contributions also from the terminal loop60, 61.
In this work we have developed a platform for the incorporation of multiple heterologous miRNAs which allows for the simultaneous expression of up to three individual siRNAs as a means of multiplexing siRNA production. This platform is based upon a naturally occurring miRNA cluster, mir106b-mir93-mir2548, encoded entirely within intron 13 of the putative protein-encoding gene MCM7 on chromosome 7. Within the intron, the three micro units are closely spaced, with only 124 and 145 nt separating the three pre-miRNA hairpins. This spacing makes this system useful for incorporation into viral vectors that have spatial limitations for gene insertions. We have cloned the native cluster, including parts of upstream and downstream exons, verified the targeting efficacy and strand selectivity of the original miRNAs, and modified the original cluster to facilitate independent replacement of each unit with heterologous, trans-targeting siRNAs. As a model system for testing this platform, we have used HIV-1 as a target and inserted three different siRNAs targeting different sites within the HIV-1 genome. When the three siRNAs were expressed in T-lymphocyte cell lines and challenged with two different strains of HIV, the siRNAs effectively suppressed replication of HIV LAI, but were less effective in suppressing the more pathogenic lab strain HIV IIIB. In an effort to further suppress HIV replication, we incorporated a small nucleolar localizing TAR decoy(U16TAR12, 62) in three different positions of the MCM7 intron. In each case a mature sized chimeric construct was produced. In acute challenges with HIV LAI and IIIB the inclusion of the TAR decoy within the intron resulted in a marked enhancement of anti-HIV function. Our studies provide both a platform unit for multiplexing siRNAs and other small RNAs as well as a novel therapeutic construct for the treatment of HIV infection.
RESULTS
Generation of a mir106b-based expression system
With the aim of generating a siRNA multiplexing expression system within a single transcript, we sought to use a naturally occurring polycistronic miRNA as a scaffold. We based our work on the tricistronic mir106b cluster located in intron 13 of the protein encoding MCM7 gene on chromosome 748. This cluster, which consists of miR106b, miR-93 and miR-25, is relatively compact, and the entire intron is less than 800 bp long. We initially cloned the mir106b cluster into a CMV-driven expression plasmid, pcDNA3.1, by PCR amplification of human genomic DNA. Two versions were cloned, harboring the intron sequence only (MCM7-I), or incorporating an additional ~100 nt of flanking exon sequences on either side (MCM7-E) (Fig. 1). Functional processing of mature miRNAs and their predicted passenger strands (miR*) from our expression plasmid was then tested by co-transfecting various expression plasmids with dual luciferase psiCHECK reporter constructs that harbored targets specific for each of the two strands of the miRNAs. Each psiCHECK 2.2-based reporter (derived from psiCHECK 2 by extending the polylinker for cloning) contains a short stretch of target sequence with perfect complementarity to a specific miR (or its miR* partner) inserted into the 3′UTR of Renilla luciferase. Normalized reporter expression is a measure of productive RISC loading and reporter mRNA cleavage by miR (or miR*). As shown in Fig. 2A, co-transfections with the two versions of the MCM7 plasmid resulted in equivalent levels of silencing for all three mature miRNAs and their passengers strands, suggesting that inclusion of exonic regions did not substantially enhance activity. The greatest level of silencing, 65%, was observed for miR-25, while slightly lower levels were observed for miR-93 (60%) and miR-106B (50%). This order of efficacy was reversed for the passenger strand specific reporters, with negligible knockdown observed for miR-25*, 25% for miR-93* and 30% for miR-106b* (Fig. 2A). The inverse relationship between the levels of silencing mediated by the two strands of the miRNAs is consistent with the asymmetric nature of strand selection, in which one strand is preferentially utilized by RISC at the expense of the other63, 64. Our observation that silencing by the designated mature miRNA strand was in all cases superior to that of the designated passenger strand, is consistent with assigned asymmetries of strand utilization for the three miRNAs of this cluster, which is based on cloning of recoverable short RNAs.
FIG. 1. Overview of mir-106b polycistron based expression plasmids.
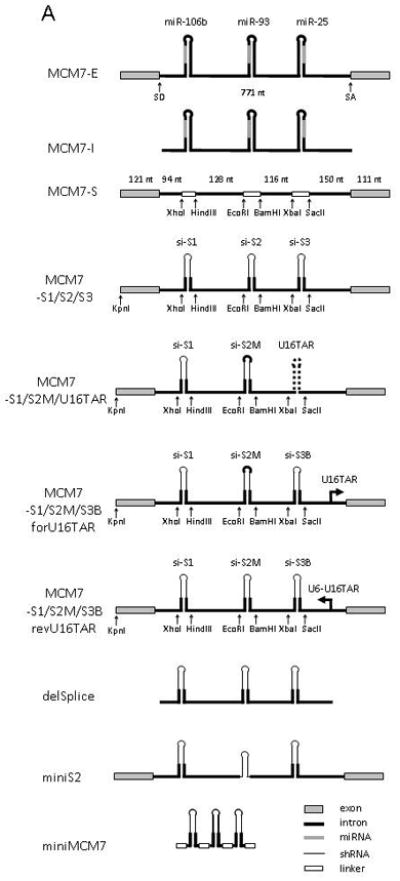

(A) Schematic diagram of the mir-106b polycistronic miRNA expression plasmids and the derived siRNA expression plasmid used in this study. The “MCM7” name was used to indicate that the mir-106b cluster is located in intron 13 of the protein encoding gene MCM7 on chromosome 7q22.1. MCM7-E, -I and –S, refers to exon, intron and scaffold, respectively. MCM7-S1/S2/S3 refers to targeting of three independent targets sites, S1-S3, in the viral HIV-1 genome. U16TAR decoy refers to the nucleolar RNA TAR decoy, with forU16TAR being U16TAR expressed in the forward direction and revU16TAR in the opposite orientation. The constructs delSplice, miniS2 and miniMCM7, represent deletion variants of MCM7-S1/S2/S3. (B) Example of replacing the native miR stem-loop with an HIV-1 targeting shRNA sequence using the introduced restriction sites in MCM7-S, here illustrated with miR-106b and S1 using XhoI and HindIII. Mature miRNA/guide strand in green, predicted passenger strand (miR*) in red, heterologous sequences in uppercase, restriction sites in bold.
FIG. 2. Functional expression of miRNAs from the MCM7 plasmid.
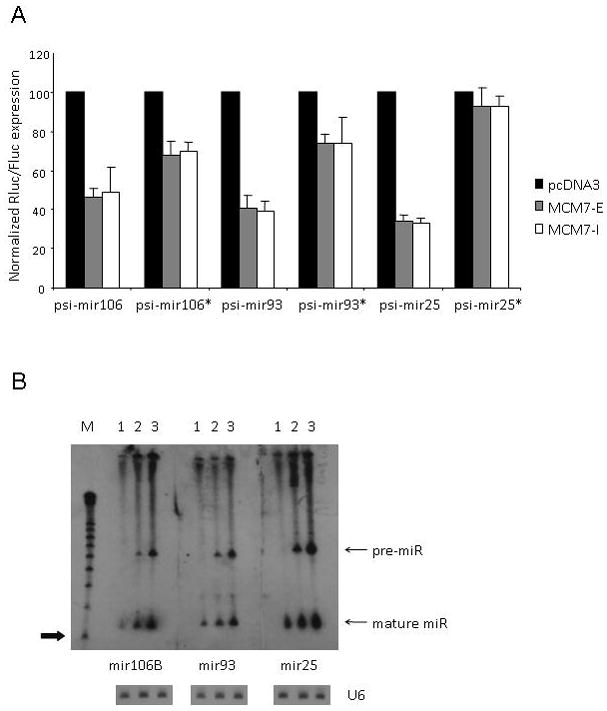
(A) Knockdown of miR- and miR*-sensitive Renilla Luciferase reporters (psi-mir) after transient co-transfection of HCT116 cells with MCM7 plasmids. Rluc activity was normalized to the internal Firefly Luciferase control and values expressed relative to levels for control-transfected cells (± SD). (B) Northern analysis of endogenous expression levels of miR-106b, miR-93 and miR-25 in HCT116 cells (lane 1) and after transient transfection of MCM7-I (lane 2) and –E (lane 3). Lower and upper bands correspond to the mature miRNA and the pre-miRNA precursor, respectively. Arrow indicates mobility of a 20-nt RNA size marker. U6 snRNA was probed as an internal RNA loading control.
Mature miRNA expression from our plasmids was also evaluated using Northern blot analyses of transfected human HCT116 cells. All three mature miRNAs and their precursors were easily detectable by 5′-radiolabeled complementary oligonucleotide probes (Fig. 2B). Variable background levels of mature miRNAs were detected in control-transfected cells, indicative of endogenous miRNA expression in HCT116 cells. Our target inhibition data suggested that the exon-deficient version of the miRNA cluster was as effective as the exon-inclusive version, but Northern blot analyses indicated that the exon-inclusive construct consistently produced higher levels of mature miRNA. Based on the Northern analyses, we decided to proceed with the exon-inclusive version of our expression construct as a scaffold for heterologous miRNA expression.
Expression of heterologous siRNAs from the mir106b scaffold
We next sought to modify the MCM7-E plasmid in a manner that would allow us to replace the individual pre-miRNA stem-loop structures with any given sequence or hairpin structure. To accomplish this we introduced unique restriction sites on both sides and as close as possible to the three pre-miRNA sequences, using a minimal number of nucleotide substitutions. Importantly, secondary structure predictions did not indicate that the substitutions would disrupt the endogenous miRNA stem loop structure (data not shown). A miRNA depleted scaffold of the MCM7 intron was generated by PCR-amplification of each of the flanking and intervening pri-miR “spacer” sequences with restriction site-tagged primers followed by re-assembly of the individual units in a suitable vector in the native sequence order. We next subcloned this MCM7 scaffold into a pcDNA3.1-derived plasmid to generate MCM7-S (Fig. 1A). In MCM7-S the mir106b stem-loops (and a few flanking nucleotides) are essentially replaced with small linkers that serve as stuffers for insertions of any sequence using the introduced restriction sites. This platform itself should be useful to investigators wishing to insert their own miRNA clusters. We next introduced stem-loop structures, S1-S3, targeting three conserved sites in the HIV-1 genome called sites 1, 2 and 3. S1 targets the common exon of early HIV-1 tat and rev transcripts while S2 and S3 target rev and tat, respectively.
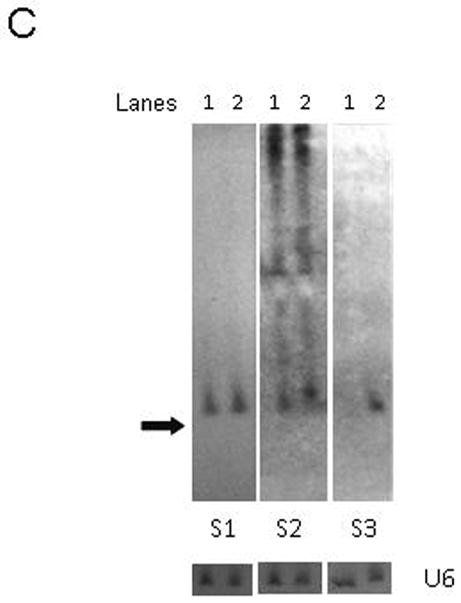
The heterologous hairpins were cloned as cassettes generated by PCR extension of overlapping oligonucletides followed by restriction endonuclease digestion. We designed our HIV-1 targeting hairpins by maintaining the natural pri-miR sequences at the base of the stems up to the final closing base pair proximal to the mature miRNA and then simply replaced the upper part of the structure with an artificial, fully paired 23 bp stem and an artificial 5-base loop (Fig. 1B). The upper part of this hybrid structure closely resembles a classical Pol III-promoted shRNA construct. The final construct was designated MCM7-S1/S2/S3. We tested this construct for processing of each of the siRNA species by co-transfection assays with individual S1, S2 and S3 target site specific dual luciferase reporters (psi-Sx). As shown in Fig. 3A, the two first units S1 and S2 worked efficiently and reduced luciferase reporter activity by approximately 75%.. The third unit, S3, exhibited reduced knockdown ability (less than 50%). Importantly, the intrinsic activity of the S3 target, in the context of a shRNA construct does not differ significantly from S1 or S2 (data not shown). Hence, the diminished activity suggested that si-S3 was processed less efficiently than si-S1 and si-S2. Consistent with this, a Northern blot analysis demonstrated lower levels of mature si-S3 than si-S1 or si-S2 (Fig. 3B). Note that for the Northern analysis the MCM7-S1/S2/S3 was transcribed by the Pol-II U1 snRNA promoter to avoid interference with the CMV-driven GFP marker used as a transfection control. Our data suggests that the restriction site modified mir106b expression system can be used to direct Pol-II promoted expression of three different siRNAs and that at least two of these siRNAs had comparable target knockdown efficacy relative to the native miRNAs.
FIG. 3. Functional expression of siRNAs from the MCM7-S1/S2/S3 plasmid.
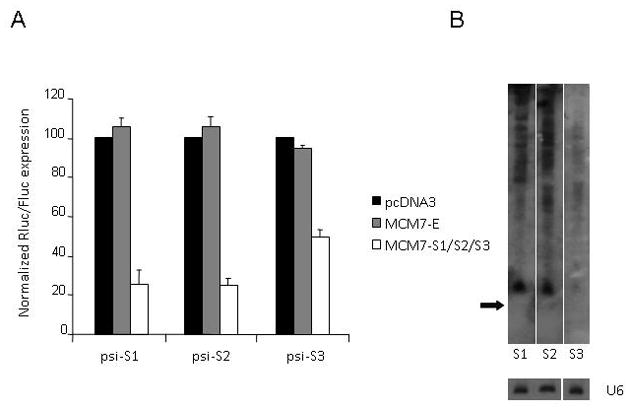
(A) Knockdown of HIV-1 siRNA guide strand sensitive RLuc reporters (psi-Sx) after transient co-transfection of HEK293 cells with MCM7 plasmids. Rluc activity was normalized to the internal Firefly Luciferase control and values expressed relative to levels for control-transfected cells (± SD). (B) Northern analysis of siRNA guide strand expression levels after transient transfection of HCT116 cells with the MCM7-S1/S2/S3 construct. Arrow indicates mobility of a 20-nt RNA size marker. Note that for the Northern blot analysis, MCM7-S1/S2/S3 expression was driven from a Pol-II U1-promoter plasmid to avoid interference from the CMV-driven GFP marker which was used as a transfection control. U6 snRNA was probed as an internal RNA loading control.
Removal of flanking pri-miRNA sequences abolishes siRNA functionality
Using the established knockdown efficiency of MCM7-S1/S2/S3 as measured by dual luciferase reporters, we set out to determine whether the flanking pri-miRNA sequences are crucial for functional processing of siRNAs, or if we could configure the MCM7 expression platform in a simpler manner. First we individually replaced the longer mi/shRNA hybrid stem-loops with a simple 23 nt-stem shRNA. We did this by direct cloning of oligo cassettes that encompassed the restriction sites and shRNA sequences only, thereby deleting the pre-miR leader sequences as well as a few flanking nucleotides. Reporter co-transfections with these minimal constructs, designated miniSx’s (see Fig. 1A for a diagram of miniS2), demonstrated that removal of pre-miR leader sequences completely abolished activity of the siRNA (Fig. 4A). Notably, removal of the leader sequence of one pre-miRNA unit had little effect on the functional release of siRNAs from the two remaining units (Fig. 4A), which suggests that each unit is processed independently. Next, we generated a pcDNA3-derived expression construct, miniMCM7 (Fig. 1A), in which all sequences flanking each of the three mi/shRNA units were removed and replaced by short, unstructured 6 nt linkers. This construct exhibited only marginal silencing activity (Fig. 4B and data not shown). Thus, structural features within the mir106b scaffold are critical for efficient siRNA processing. Our results are consistent with recently published data indicating that effective Drosha/DGCR8 mediated nuclear processing of the primary transcript depends on a base-paired stem of about one full helical turn flanked by unstructured regions of the sequence outside of the pre-miRNA51.
FIG. 4. Limited efficacy of siRNAs expressed from MCM7-S1/S2/S3 deletion mutants.
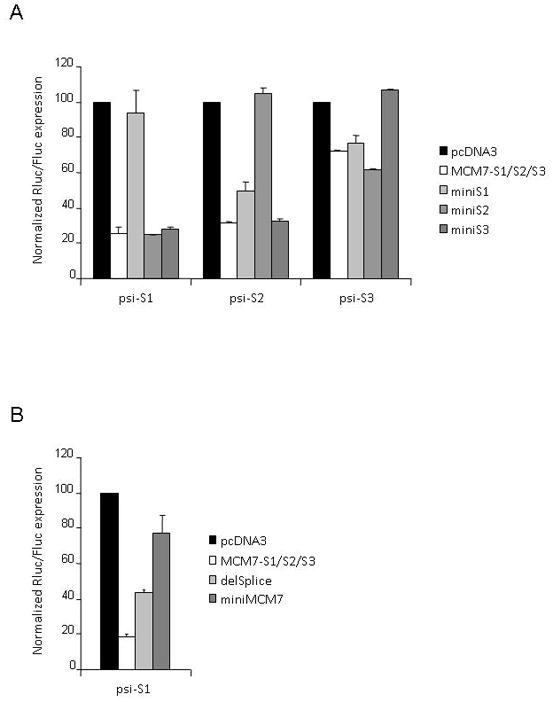
(A) Knockdown of siRNA-sensitive RLuc reporters after transient co-transfection of HEK293 cells with MCM7-S1/S2/S3 plasmids lacking the lower part of the miRNA stem-loop structure (miniSx). (B) Knockdown of siRNA-sensitive Renilla Luciferase reporters in MCM7- S1/S2/S3 plasmids lacking the flanking exon and the splice sites (delSplice) or missing the pri-miR-derived sequences between the stem-loop structures (miniMCM7). See Fig. 1A for construct diagrams. Rluc activity was normalized to the internal Firefly Luciferase control and values expressed relative to levels for control-transfected cells (± SD).
Splicing enhances siRNA functionality in the mir106b system
Among the various features missing in miniMCM7 is the exon-intron-exon structure. In order to test whether splicing affected siRNA functionality, we cloned a shorter version of MCM7-S1/S2/S3, delSplice, in which flanking MCM7-derived exons and all splicing signals within the introns were removed (Fig. 1A). This construct exhibited a 2-fold reduction in silencing of psi-S1 in co-transfection assays (Fig. 4B). Reduced silencing potential was also observed for the other two siRNAs expressed from the tri-cistronic construct (data not shown). These data are similar to our observation that inclusion of flanking exons enhances the expression of the native miRNAs from this cluster. Clearly, however, removal of splicing potential does not in of itself abolish the ability of intron-embedded polycistronic miRNAs to be processed by Drosha/DGCR8. This is consistent with recent observations by others in which deletion of splice sites only marginally affected pre-miRNA generation from an intron51.
Strand selectivity of the S2 unit is improved by a full miR-93 mimic
When expressed as standard shRNAs from U6 promoters, all three RNAi effectors display favorable asymmetry in strand utilization (data not shown). We decided to investigate whether or not asymmetry was retained in the polycistronic miRNA expression background. Co-transfection experiments with strand-specific reporters demonstrated highly variable strand selectivity for the various units. The reporters for the presumptive guide and passenger strands of si-S2 were silenced almost equally by MCM7-S1/S2/S3, indicating little discrimination between strands in RISC incorporation (Fig. 5A). In contrast, si-S1 and si-S3 demonstrated negligible and marginal passenger strand activities, respectively (data not shown). The poor strand selectivity of S2 is clearly undesirable as this increases the likelihood of passenger strand mediated off-target effects. The poor strand selectivity of si-S2 compared to its parent miR-93 in the context of the polycistronic transcript may result from skewed processing of the hybrid transcript due to changes in secondary structure. In an attempt to control for such an effect, we changed our “simple” S2 mi/shRNA hybrid to a version, designated S2M, in which the passenger strand was selectively mutated in order to closely mimic the secondary structure of miR-93 while retaining the desired S2 guide strand (Fig. 5B). This miR-93 mimic exhibited no passenger strand activity (in co-transfection experiments with a new reporter specific for the mutated passenger strand, psi-S2M-AS) while still silencing the psi-S2 reporter more than 70% (Fig. 5A). Importantly, the MCM7-S1/S2M/S3 construct maintained S1 and S3 targeting activities (data not shown). These observations suggest that in order to achieve the desired combination of efficacy and specificity, polycistronic miRNA-based expression cassettes should maintain the general secondary structures of the pre-miRNA leader sequences and heterologous stems.
FIG. 5. Improved asymmetry of S2 siRNA RISC loading by a miR-93 structural mimic.
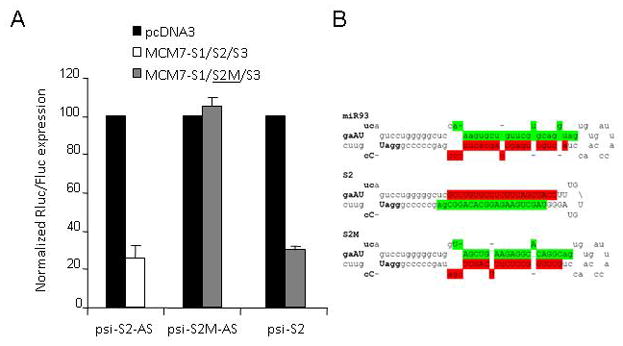
(A) Knockdown of passenger strand-sensitive RLuc reporters (psi-S2-AS or psi-S2M-AS) after transient co-transfection of HEK293 cells with MCM7-S1/S2/S3 plasmid or a miR-93 mimic (S2M). Rluc activity was normalized to the internal Firefly Luciferase control and values expressed relative to levels for control-transfected cells (± SD). (B) Predicted stem-loop structure of miR-93, the fully base paired S2 hairpin and its miR-93 mimic, S2M. Mature miRNA/guide strand in green, predicted star/passenger strand in red, heterologous sequences in uppercase, restriction sites (EcoRI and BamHI) in bold.
Increased expression of si-S3 from a miR-93 mimic but not a miR106 nor a miR-25 mimic
Having achieved efficient knockdown and good strand selectivity for the S1 and S2 units, we next addressed the issue of poor functionality from the S3 unit. To improve S3, we decided to investigate whether the secondary structures of the first two units might be transferable to the third unit in a heterologous tri-cistron. The S3 siRNA was therefore incorporated into the third position as either a S1/miR-106b, a S2M/miR-93 or a miR25 structural mimic, generating the hybrids S3A, S3B, and S3C, respectively. While the S3A hairpin resulted in only a modest improvement of psi-S3 knockdown and S3C resulted in no improvement (data not shown), the S3B variant improved silencing to 80% (Fig. 6A). Concomitant with this improvement in functionality, Northern blot analyses demonstrated a marked increase in the levels of mature si-S3 from S3B as compared to S3 (Fig. 6C). Notably, MCM7-S1/S2M/S3B maintained knockdown efficacy of the S1 and S2 units (Fig. 6A), while exhibiting good strand selectivity for S3 (Fig. 6B). We are thus confident that this final configuration of the MCM7 intron is appropriate for use in other applications besides HIV-1 infection.
FIG. 6. Optimized S3B containing cluster maintains the knockdown efficiencies of S1 and S2M, but has greatly improved S3 efficacy.
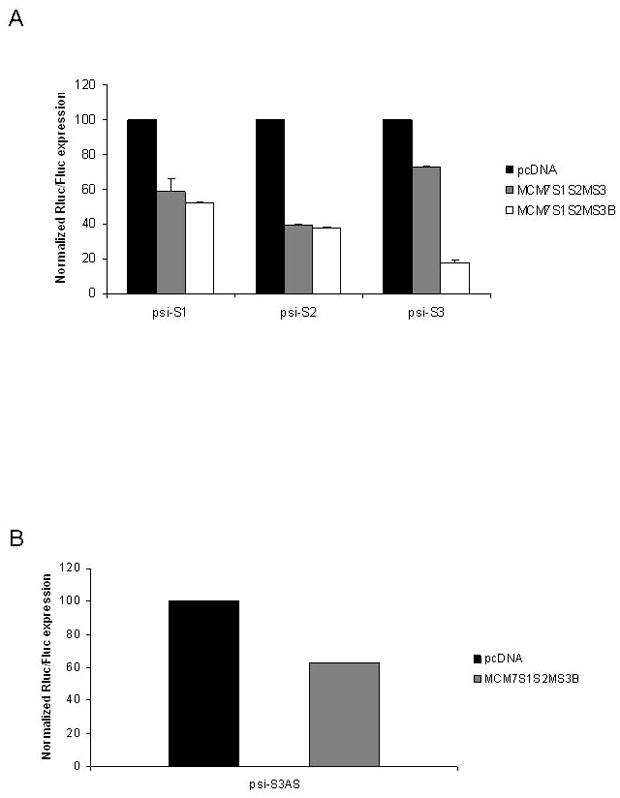
Plasmids containing the three anti-HIV miRNA mimics were co-transfected with psi-CHECK vectors harboring the targets for each of the three siRNAs targeting S1, S2 and S3. The S3 unit was tested in the context of S1 and S2M in its original configuration (MCM7-S1/S2M/S3) or in the optimized configuration (MCM7-S1/S2M/S3B). The MCM7-S1/S2M/S3B cluster provides comparable levels of knockdown for sites S1 and S2, but demonstrated enhanced knockdown of the S3 target (A). Good strand selectivity is maintained for MCM7-S1/S2M/S3B when the target is inserted in the anti-sense orientation (B). The data are presented as the means of triplicate measurements. (C) Northern analysis of siRNA guide strand expression levels after transient transfection of HCT116 cells. RNA from MCM7-S1/S2/S3 (lane 1) and MCM7-S1/S2M/S3B (lane 2) transfected cells, respectively. Arrow indicates mobility of a 20-nt RNA size marker. U6 snRNA was probed as an internal RNA loading control.
Incorporation of U16 snoRNA-TAR decoy hybrid RNA to increase the functionality of the MCM7 system
In targeting HIV-1 it is important to use a combinatorial inhibitory approach and it seems wise not to depend upon a single inhibitory modality, such as RNAi. Prior results from our lab demonstrated a nucleolar localizing RNA TAR decoy (U16TAR) served as a good inhibitory agent of HIV-1 replication62. We therefore decided to create combinations of the anti-HIV siRNAs with the U16TAR decoy. In the first construct S3B was replaced with the U16TAR element. Two additional constructs incorporating U16TAR were also made. One of these incorporated a U6 Pol III promoter driven U16TAR decoy downstream of the siRNAs and transcribed in the opposite orientation with respect to the siRNAs (revU16TAR). We also inserted the U16TAR decoy downstream of the siRNAs in an intron position shown to be favorable to snoRNA processing65, 66 and designated this as forU16TAR. The forU16TAR is co-transcribed with the siRNAs from the upstream Pol II promoter (Fig. 1). Expression analyses of the siRNAs as well as the U16TAR decoy by Northern blotting revealed good expression of the siRNAs and the U16TAR RNA, with the most robust U16TAR expression deriving from the revTARd (Fig. 7).
FIG. 7. Expression of the RNAs in the optimized MCM7 constructs.
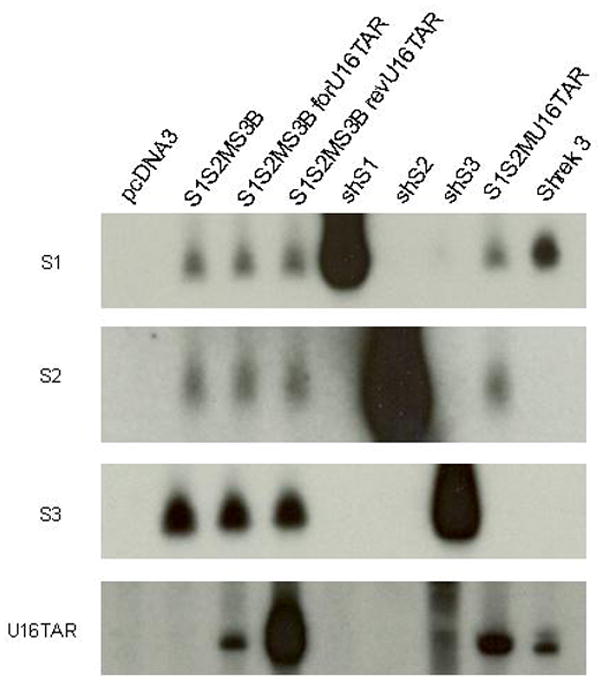
HEK293 cells were transiently transfected with each of the optimized MCM7 constructs and controls. Forty-eight hours following transfection, total RNA was collected, electrophoresed in an 8% polyacrylamide gel with 8M urea, blotted onto a nylon membrane, and hybridized with the corresponding 32P-labled probes. Hybridization to U6-driven short hairpin siRNAs S1, S2, S3, and the U16TAR decoy expressed from Shrek 368 were used as positive controls. RNA prepared from an empty vector (pcDNA) transfected cell line was used as a negative control. S1, S2, and S3 siRNAs are approximately 21 nucleotides. The U16TAR decoy is 132 nucleotides.
We reasoned that the forU16TAR should be processed from the intron in a fashion similar to endogenous snoRNA processing65 while the U16TAR inserted in place of a miRNA unit could potentially be processed by the miRNA processing enzyme, Drosha. To address this possibility, we took advantage of a Tet-inducible anti-Drosha shRNA construct67. Following addition of doxycycline to induce the anti-Drosha shRNA we monitored processing of the siRNAs as well as the U16TAR by Northern analyses. While the processing of the S1 and S2 siRNAs and endogenous miR21 RNA were inhibited by Drosha knockdown, there was no inhibition of the U16TARdecoy (Supplementary Figure 1). Interestingly, the steady state level of the U6 driven revU16TAR was somewhat elevated following Drosha knockdown. We conclude from these results that the processing of the forU16TAR and U16TAR takes place via the snoRNA processing mechanism.
Optimized multicistronic expression units suppresses HIV-1 replication
To test the functionality of the MCM7-S1/S2M/S3B/U16TAR constructs, we transiently co-transfected HEK293 cells with the various vectors and HIV pNL4-3proviral DNA. On the days indicated, supernatants were collected and assayed for HIV replication using a branched DNA (b-DNA) assay which measures viral RNA (Fig. 8). All of the constructs provided two or more logs of inhibition of HIV RNA production, but the revU16TAR and forU16TAR containing MCM7 constructs provided the most potent knockdown over the course of the assay and were at least twice as inhibitory as the MCM7-S1/S2M/S3B construct.
FIG. 8. Anti-HIV-1 activity of the optimized MCM7 constructs.
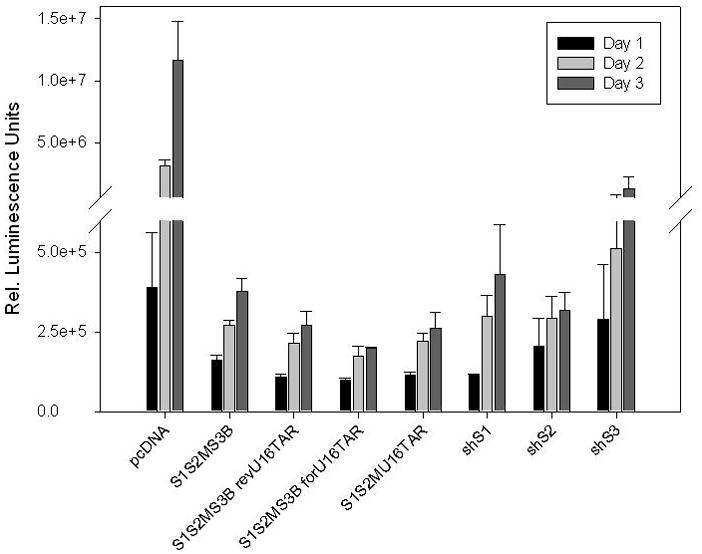
HEK293 cells were co-transfected with the indicated vectors and HIV-1 NL4-3 at a ratio of 4:1. The empty pcDNA vector served as the negative control. To demonstrate a higher combinatorial knockdown efficacy, the MCM7 constructs were compared to individually expressed Pol III hairpins: shS1, shS2, and shS3. Culture supernatants were collected at 24, 48, and 72 hour time points, analyzed by the bDNA assay (Panomics), and values are depicted as means ± standard deviation. As compared to the MCM7-S1/S2M/S3B construct, the addition of the TAR decoy in both the reverse and forward orientations provided a two fold enhancement in the inhibition of HIV-1 replication and was also more potent than the individual Pol III shRNA expression units.
The U16TAR decoy enhances viral inhibition of the MCM7- S1/S2M/S3B cluster
The most robust U16TAR decoy expression was obtained from the revU16TAR insertion which did not compromise expression of the siRNAs. Therefore, we incorporated this configuration into the pHIV7 lentiviral vector which was used to transduce human T-cell lines for testing in HIV acute challenge assays. We also replaced the CMV promoter with the human U1 snRNA promoter and SV40 poly-A sequence. The entire unit was inserted into pHIV7 in an orientation opposite to that of the CMV packaging promoter to prevent splicing of the MCM7 intron during packaging. CEM and SupT1 cells were stably transduced with the control pHIV7 vector, MCM7-S1/S2M/S3B or MCM7-S1/S2M/S3B revU16TAR. The transduced cells were sorted for EGFP expression and subsequently challenged with HIV-1. For the CEM cells, the challenge virus was the HIV IIIB while the SupT1 cells were challenged with HIV-LAI. Supernatants were collected at various time points and levels of viral p24 were measured by ELISA (Fig. 9). Although the MCI7-S1/S2M/S3B construct provided good inhibition with HIV LAI, it was less protective when challenged with HIV IIIB. Inclusion of the revU16TAR resulted in substantially better inhibition of both strains of HIV-1.
FIG. 9. Addition of the U16TAR decoy confers greater anti-HIV-1 activity.
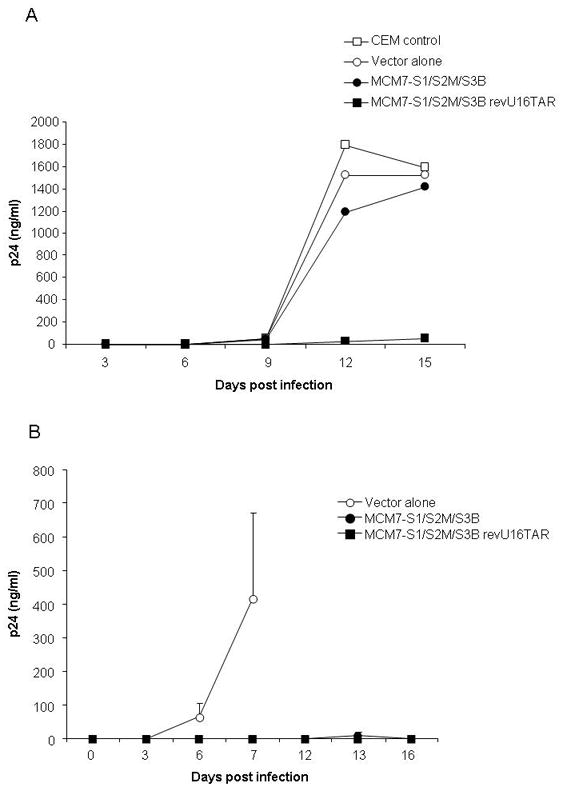
Stably transduced CEM-T cells (A) and SupT1 cells (B) expressing the indicated vectors were challenged with HIV-IIIB and HIV-LAI, respectively. Culture supernatants were collected at various time points and analyzed by a p24 ELISA assay. Data points are reported as means ± standard deviation. Standard deviation calculations for each data point in panel (A) were <1.3. The addition of the U16TAR decoy in the MCM7-S1/S2M/S3B revU16TAR (closed square) confers long-term viral inhibition as compared to the MCM7-S1/S2M/S3B construct (closed circle).
DISCUSSION
We have engineered a system for functional co-expression of three different heterologous siRNAs from a polycistronic, intronic, miRNA platform. This system is based on the mir106b cluster that is located in a 0.8 kb intron in the host gene MCM7. Accordingly, we have called the system MCM7 (Fig. 1). The native cluster was initially cloned into a CMV-promoted expression plasmid vector and each of the encoded miRNAs; miR-106b, miR-93 and miR-25 were shown to be functionally expressed from this backbone (Fig. 2). By introducing a small number of substitutions we created suitable restriction sites flanking each of the three miRNA stem loops that allowed for the insertion of any given stem loop at each of the three positions. As a test of this platform we inserted three different anti-HIV sequences and tested each unit for function. The HIV-1 targeting sequences were initially introduced as fully base paired shRNA-like structures that essentially replaced the pre-miRNA parts of the primary transcript, above the Drosha cleavage site51, 68. Knockdown efficiencies of the cognate targets of the heterologous siRNAs were comparable to those observed for the parental miRNAs of the wildtype polycistron (Fig. 3). Of importance, we noticed a strict requirement for the flanking pri-miRNA sequences for functionality. Knockdown efficacy was abolished by the removal of leader sequences below the Drosha cleavage site of the extended stem structure, or when the long intervening sequences between the different miRNA units were replaced with short unstructured linkers (Fig. 4). Moreover, siRNA expression levels and functionality were also reduced when we removed the flanking exon structure and splice sites, suggesting that splicing may enhance processing of polycistronic miRNAs.
Despite the efficient processing of at least two siRNAs from our miR/shRNA chimera (MCM7-S1/S2/S3), we observed two undesirable properties of the initial MCM7 tri-cistronic construct. First, the third unit, S3, was expressed at lower levels than the first and second units and concomitantly less active than the other two. Second, S2 displayed equivalent RISC incorporation of both strands. This may be a desirable property for some applications where both strands are intentionally designed to be functional, in our case it indicated that our design had poor strand asymmetry which increases the potential for passenger strand mediated off-target effects. We thus sought to correct both deficiencies without altering the HIV-1 targeting properties. Our approach was to introduce targeted mutations in the passenger strand in such a way as to convert the original miR/shRNA hybrids to structures that better resembled the native miRNAs of this cluster. Such a structural mimic of miR-93, S2M, improved the strand selectivity of the S2 siRNA to a level at which functional passenger strand activity was negligible, while still maintaining guide strand mediated functionality (Fig. 5). We suspect that the initial poor strand selectivity for this unit might stem from skewed Drosha processing of the primary transcript. Algorithms that predict Drosha processing sites based on conserved primary and secondary structure motifs69 in fact predicted the intended cleavage site for S1, while suggesting multiple cleavages sites for S2 (data not shown). If these predictions are accurate, the resulting siRNAs would most likely differ in strand loading based upon differences in thermodynamic end stabilities64, 70.
Once our MCM7 construct was optimized for efficient processing of the siRNAs, we sought to increase the construct’s multi-functionality by inserting a nucleolar localizing TAR decoy within the intron. This U16TAR RNA mimic binds Tat and subsequently sequesters it from the virus and inhibits HIV-1 replication62. Here, we demonstrate the processing and expression of the U16TAR decoy in three different configurations within the modified MCM7 intron (revU16TAR, for U16TAR, U16TAR) (Fig. 7). When these various combinations of the MCM7-S1/S2M/S3B and U16TAR decoy were tested for anti-viral activity, we observed better inhibition of HIV-1 as opposed to the parental MCM7-S1/S2M/S3B construct (Fig. 8). Since more robust expression of the U16TAR was observed from the MCM7-S1/S2M/S3B revU16TAR than the forU16TARd MCM7 cluster (Fig. 7), we decided to utilize the revU16TAR configuration for insertion into the pHIV7 lentiviral vector. From this assay, the MCM7-S1/S2M/S3B revU16TAR construct provided a marked reduction in HIV p24 antigen levels compared to the MCM7 cluster without the U16TAR decoy (Fig. 9). The HIV LAI isolate was inhibited by the MCM7-S1/S2M/S3 construct alone, but addition of the U16TAR decoy enhanced the inhibition (Fig. 9B) in similarity to the HIV IIIB challenges. However, the lack of significant inhibition by the MCM7-S1/S2M/S3B when challenged with HIV IIIB (Fig. 9A) was somewhat surprising and contrasted with our HIV pNL4-3 co-transfection results. In our hands, HIV IIIB is one of the more difficult lab isolates of HIV-1 to inhibit in cell culture challenges. The relatively low expression levels of the siRNAs from the integrated lentiviral vector construct, although important for minimizing toxicities, may allow HIV to overwhelm the RNAi effectors. We have previously observed that shRNAs can be overwhelmed by HIV in acute challenges, but when they are combined with other RNA based inhibitors, such as the TAR decoy and a ribozyme, virtually complete inhibition can be obtained12. It is important to note that on its own the U16TAR decoy is not a potent inhibitor of HIV replication, but in combination with one or more other anti-HIV RNAs there is an apparent synergy12, 71. Binding of the U16TAR to Tat may subsequently slow the progression of viral transcription to reach a critically low level that allows each of the S1, S2M, and S3B hairpins to function more effectively in downregulating the tat/rev transcripts.
Of note, our U16TAR decoy is a chimeric snoRNA that has been shown to exclusively localize within the nucleoli of human cells62. When encoded in an intron, the assembly of snoRNA particles is dependent upon the position within the intron or the stability of a stem structure flanking the snoRNA65. Here, we demonstrate expression of the U16TAR decoy configured in three different ways. In one configuration it is expressed from its own U6 promoter in an orientation opposite of the MCM7-S1/S2M/S3B cluster (revU16TAR). In the second configuration it is co-expressed with the MCM7-S1/S2M/S3B miRNAs via its insertion approximately 70 bases upstream of the 3′ splice signal (forU16TAR). Finally, we have inserted the U16TAR decoy unit in place of S3B within a structured stem (U16TAR). Although all three variants are expressed and processed, the strongest expression was obtained from the revU16TAR (Fig. 7). Drosha knockdown experiments did not diminish the levels of mature U16TAR in any of the constructs, despite the reduction in mature S1 and miR21 levels (Supplemental Figure 1).
In nature, multi-cistronic miRNA clusters are common, and the miRNAs produced from these often regulate the expression of many different transcripts simultaneously. The use of a miRNA cluster for concurrent expression of multiple siRNAs has several advantages. First, targeting of viral pathogens at multiple positions should, in theory, reduce the likelihood of viral escape. For HIV-1, it is well established that the potency of RNAi rapidly selects for escape mutants10, 16–20, and expression of multiple siRNAs by various means has been shown to improve both initial functionality and delay the emergence of resistance19, 21, 26, 27. This strategy is analogous to the use of multiple drugs in the current HAART treatments. This new expression system also has the advantage that the choice of a promoter is quite flexible, and constitutive, tissue specific or even HIV Tat inducible promoters72 can be used. Thus, levels of expression can be controlled to avert potential toxicities caused by high levels of expression of the miRNAs and resultant siRNAs30. Since the processing signals for the miRNAs are encoded within the miRNAs themselves as well as sequences immediately flanking the hairpins and there are no specific structural requirements for the 5′ and 3′ ends of the primary transcripts, this system should prove to be generally useful for expression of multiple hairpins from a variety of promoters.
In summary, we have engineered a novel mir106b-cluster based expression system, MCM7, for the delivery of multiple siRNAs from a single RNA Pol II promoted transcript. We have optimized the designs of the miRNA mimics for efficient functional release of all three of the ectopic siRNAs. In addition, we have included within the intron a nucleolar U16TAR decoy which is co-expressed with the siRNAs. The combined siRNAs and U16TAR decoy demonstrate potent inhibition of HIV-1 replication. Our miRNA-based siRNA expression system may also be useful as a general tool for silencing of multiple cellular targets and could find applications in both basic experimentation and in therapeutic applications where multiple siRNA or miRNA expression is required.
METHODS
Generation of polycistronic expression constructs
We cloned the entire mir-106b cluster downstream of a CMV promoter in the expression plasmid pcDNA3.1 by PCR of human genomic DNA using primers E-F1 and E-R4 (see details on all oligos in supplementary Table S1). The resulting PCR product was initially cloned into the TA vector pCR2.1 (Invitrogen) and then subcloned as a HindIII/XbaI fragment to pcDNA3.1 to generate pcDNA3-MCM7-E. pcDNA3-MCM7-E contains the mir-106b bearing intron from the MCM7 gene as well as parts of the upstream and downstream exon sequences. A mir-106b expression plasmid without the flanking exon structure, pcDNA3-MCM7-I, was cloned directly into pcDNA3.1 by PCR from genomic DNA using HindIII- and XbaI-tagged primers (I-F and I-R, respectively). Next, we built a mir-106b-based scaffold, pcDNA3-MCM7-S, that could be used for insertion of heterologous sequences at the positions of miR-106b, miR-93 and miR25. This was accomplished by PCR amplification of each of the four regions flanking miR-106b, miR-93 and miR25, and inserting those one by one into pBSIISK (region 1 used primers E-F1 and E-R1 and so forth). Each primer was tagged with restriction sites corresponding to the order of the multiple cloning site (MCS) in pBSIISK (see Fig. 1A) in a manner that introduced minimal changes in the native mir-106b cluster sequence. After cloning all four regions into pBSIISK, the entire scaffold was cloned directionally as a KpnI/BssHII fragment into KpnI/AscI sites of the pcDNA3.1 modified plasmid pcDNA3-PL4 (in which the polylinker between HindIII and XbaI in pcDNA3.1 was replaced with a shorter linker containing AgeI, KpnI, XhoI, BamHI, AscI and HpaI sites, destroying the original HindIII and XbaI sites in the cloning process) to generate pcDNA3-MCM7-S.
Using pcDNA3-MCM7-S, heterologous hairpin structures were inserted into the original positions of miR-106b, miR-93 and miR25 as XhoI/HindIII, EcoRI/BamHI or XbaI/SacII fragments, respectively. Inserts were generated by PCR extension of partially annealed overlapping ~60 nt oligonucleotides, followed by digestion with the appropriate restriction enzymes. The U16TAR decoy structure was PCR amplified with the appropriate restriction enzymes from the U6-TAR RNA decoy harboring Shrek3 plasmid71 and cloned into S3B. Restriction sites ClaI and SwaI were introduced 70 nt upstream of the miRNA cluster’s 3′ splice site for insertion of the forward and reverse U16TAR decoy. The revU16TAR was cloned with a Pol III U6 promoter for expression of the decoy in the reverse orientation.
A splice-deficient version of pcDNA3-MCM7-S1/S2/S3 (called delSplice) with deleted splice signals was generated by PCR amplification from the full-length construct with KpnI-tagged delS-F and BssHII-tagged delS-R primers. The resulting KpnI+BssHII restriction product was cloned into KpnI+AscI sites of pcDNA3-PL4. A minimal hairpin cassette, named miniMCM7, was generated by cloning the S1, S2 and S3 pre-miRNA units into the MCS of pBSIISK, followed by subcloning the KpnI/BssHII fragment into KpnI/AscI sites of pcDNA3-PL4.
We created promoter derivatives of pcDNA3-MCM7-S1/S2M/S3B in which the original CMV promoter was replaced with the non-CMV-interfering U1 promoter. The U1 promoter version was generated similarly by replacing the CMV MluI-KpnI promoter fragment with a SpeI-KpnI fragment from a U1 plasmid (pKS-U1, our unpublished data). All constructs were verified by restriction analysis and sequencing.
To generate lentiviral vectors, the U1-MCM7-S1/S2M/S3B and U1-MCM7-S1/S2M/S3B revU16TAR fragments were excised from the pcDNA3 plasmid by Mlu I and Kpn I digestion and inserted into separate pHIV-7-GFP lentiviral vectors.
Cell culture
HEK293, HCT116, 293T and HT180 cells were purchased from ATCC (Manassas, VA). The cells were maintained in high glucose (4.5 g/l) DMEM supplemented with 2 mM glutamine, 10% FBS, and 2 mM Penicillin/Streptomycin. The human T-cell line CEM was cultured in RPMI 1640 medium supplemented with 10% FBS. SupT1 cells were cultured in advanced RPMI with 2 mM glutamine, 1% FBS, and 2 mM Penicillin/Streptomycin.
Construction of psiCHECK reporters
Dual luciferase (psiCHECK2, Promega) reporters were generated for the guide and passenger strands for each of the miRNA and anti-HIV-1 siRNA expressed from the various MCM7 constructs. The reporters contained the minimal target site of the respective putative RISC-loaded mi/siRNA strand plus 3 additional target derived sequences at each end, to account for slight variations in processing of miRNAs. Target sequences were cloned into XhoI+NotI sites in the 3′ UTR of the Renilla Luciferase gene of the psiCHECK2.2 (a version of the commercial psiCHECK2 with an extended MCS). Oligonucleotide cassettes bearing XhoI and NotI compatible 4-nt overhangs at each end were generated by annealing two in vitro phosphorylated partially overlapping oligonucleotides. Cleavage of the target site results in degradation of reporter mRNA, with a concomitant decrease in translated product, which can be detected by a luminescence based assay system. Firefly Luciferase expressed from the same vector serves as an internal normalization control.
Reporter co-transfection experiments
HEK293 or HCT116 cells, seeded one day before, were transfected in duplicate in 24-well plates at 80% confluency with a Lipofectamine 2000 (Invitrogen)-complexed mixture of 25 ng psiCHECK reporter plasmid and 250 ng MCM7 effector plasmid DNA per well. Twenty-four hours post-transfection, the cells were lysed with 120 μl Passive Lysis Buffer (Promega) and Luciferase levels were analyzed from 10 μl lysate using the Dual Luciferase reporter assay (50 μl of each substrate reagents, Promega) on a Veritas Microplate Luminometer (Turner Biosystems). Changes in expression of Renilla Luciferase (target) were calculated relative to firefly Luciferase (internal control) and normalized to levels in cells transfected with “empty” pcDNA3 control plasmid.
Northern blot analysis
HCT116 or HEK293 cells were transfected in 6-well plates at 80% confluency with a Lipofectamine2000 (Invitrogen)-complexed mixture of 2 μg MCM7 effector plasmid DNA and/or 400 ng eGFP control plasmid DNA per well. Total RNA was isolated forty hours post transfection using STAT-60 (Tel-Test Inc) and resuspended in nuclease-free water.
For detection of siRNA or miRNA expression, 10–20 μg total RNA were electrophoresed in 10% denaturing PAGE gels in 1X TBE buffer for 1–2h at 300V together with an RNA size marker (Decade Marker, Ambion, Austin, TX). The RNA was transferred to a positively charged nitrocellulose membrane, Hybond-N+ (Amersham Biosciences, Piscataway, NJ) by electroblotting. UV-fixed membranes were hybridized over night in PerfectHyb solution (Sigma-Aldrich, St. Louis, MA) at 37 °C with purified 5′-32P-labeled oligos complementary to the predicted mature miRNA or siRNA strand. The oligo probes (2 pmol) were end-labeled with PNK (10 units, NEB) and γ-32P-ATP (3 pmol) in 20 μl reactions for 30 min, and purified with G-25 Sephadex spin-columns (GE Healthcare). Hybridized membranes were washed once with 6X SSC/0.1% SDS, then twice with 2X SCC/0.1% SDS, before exposure to film (Biomax MS, Kodak, New Haven, CT) or PhosphorImager analysis. miR-106b probe 5′-ATCTGCACTGTCAGCACTTTA-3′, miR-93 probe 5′-CTACCTGCACGAACAGCACTTT-3′, miR-25 probe 5′-TCAGACCGAGACAAGTGCAATG-3′, S1 probe 5′-GCGGAGACAGCGACGAAGAGC -3′, S2 probe 5′-GCCTGTGCCTCTTCAGCTACC-3′, S3 probe, 5′-CATCTCCTATGGCAGGAAGAA-3′, TAR probe 5′-CCAGAGAGCTCCCAGGCTCAG-3′, miR-21 probe 5′-ATCGAATAGTCTGACTACAACT, U6 probe 5′-TATGGAACGCTTCTCGAATT-3′.
Lentiviral vector production and transduction of target cells
Lentiviral vector production, transduction, and generation of stable cell lines were generated as previously described26.
HIV-1 challenge assays
HEK 293 cells were co-transfected in triplicate with the various vectors of interest and HIV-1 NL4-3 at a ratio of 4:1. Culture supernatants were collected at 24, 48, and 72 hour time points, and analyzed by a branched DNA (bDNA) assay using the QuantiGene Reagent System (Panomics). The bDNA assay values for relative luciferase light units (RLU) were divided by the viable cells for each time point to give RLU/cell.
One millionCEM-T cells were infected with wild-type HIV-1 strain IIIB at an MOI of 0.001 in duplicate. After overnight incubation, cells were washed three times with Hank’s balanced salts solution and cultured in RPMI 1640 with 10% FBS. The SupT1-T cells (2 × 105 cells in 1 ml of medium) were challenged with 0.006 ng of measured CA-p24 of HIV-1 strain LAI in triplicate. At designated time points, culture supernatants were collected and analyzed for HIV replication by a p24 ELISA assay.
Supplementary Material
{kind=link}
Acknowledgments
This work was funded by a fellowship to LAA from the Alfred Benzon Foundation, a post-doctoral fellowship to MA from the Norwegian Research Council and NIH AI42552.AI29329 and HL07470 grants to JJR. KE is supported by ZonMW through a VICI grant. We thank A. Ehsani for his suggestions about the positions for inserting the Tar decoy and members of the Rossi lab for their support.
Footnotes
The authors declare no competing financial interest.
References
- 1.Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 3.Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431:371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 4.Dykxhoorn DM, Lieberman J. Silencing viral infection. PLoS Med. 2006;3:e242. doi: 10.1371/journal.pmed.0030242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim DH, Rossi JJ. Strategies for silencing human disease using RNA interference. Nat Rev Genet. 2007;8:173–184. doi: 10.1038/nrg2006. [DOI] [PubMed] [Google Scholar]
- 6.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 7.Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng Y, Wagner EJ, Cullen BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell. 2002;9:1327–1333. doi: 10.1016/s1097-2765(02)00541-5. [DOI] [PubMed] [Google Scholar]
- 9.Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- 10.Gitlin L, Karelsky S, Andino R. Short interfering RNA confers intracellular antiviral immunity in human cells. Nature. 2002;418:430–434. doi: 10.1038/nature00873. [DOI] [PubMed] [Google Scholar]
- 11.Lee NS, et al. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat Biotechnol. 2002;20:500–505. doi: 10.1038/nbt0502-500. [DOI] [PubMed] [Google Scholar]
- 12.Li MJ, et al. Inhibition of HIV-1 infection by lentiviral vectors expressing Pol III-promoted anti-HIV RNAs. Mol Ther. 2003;8:196–206. doi: 10.1016/s1525-0016(03)00165-5. [DOI] [PubMed] [Google Scholar]
- 13.Rubinson DA, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 14.Snove O, Jr, Rossi JJ. Expressing short hairpin RNAs in vivo. Nat Methods. 2006;3:689–695. doi: 10.1038/nmeth927. [DOI] [PubMed] [Google Scholar]
- 15.Grimm D, Kay MA. Combinatorial RNAi: A Winning Strategy for the Race Against Evolving Targets? Mol Ther. 2007 doi: 10.1038/sj.mt.6300116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boden D, Pusch O, Lee F, Tucker L, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J Virol. 2003;77:11531–11535. doi: 10.1128/JVI.77.21.11531-11535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das AT, et al. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol. 2004;78:2601–2605. doi: 10.1128/JVI.78.5.2601-2605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westerhout EM, Ooms M, Vink M, Das AT, Berkhout B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005;33:796–804. doi: 10.1093/nar/gki220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson JA, Richardson CD. Hepatitis C virus replicons escape RNA interference induced by a short interfering RNA directed against the NS5b coding region. J Virol. 2005;79:7050–7058. doi: 10.1128/JVI.79.11.7050-7058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Eije KJ, ter Brake O, Berkhout B. Human immunodeficiency virus type 1 escapeis restricted when conserved genome sequences are targeted by RNA interference. J Virol. 2008;82:2895–2903. doi: 10.1128/JVI.02035-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song E, et al. Sustained small interfering RNA-mediated human immunodeficiency virus type 1 inhibition in primary macrophages. J Virol. 2003;77:7174–7181. doi: 10.1128/JVI.77.13.7174-7181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersson MG, et al. Suppression of RNA interference by adenovirus virus-associated RNA. J Virol. 2005;79:9556–9565. doi: 10.1128/JVI.79.15.9556-9565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang LJ, Liu X, He J. Lentiviral siRNAs targeting multiple highly conserved RNA sequences of human immunodeficiency virus type 1. Gene Ther. 2005;12:1133–1144. doi: 10.1038/sj.gt.3302509. [DOI] [PubMed] [Google Scholar]
- 24.Henry SD, et al. Simultaneous targeting of HCV replication and viral binding with a single lentiviral vector containing multiple RNA interference expression cassettes. Mol Ther. 2006;14:485–493. doi: 10.1016/j.ymthe.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 25.Li M, Rossi JJ. Lentiviral vector delivery of siRNA and shRNA encoding genes into cultured and primary hematopoietic cells. Methods Mol Biol. 2005;309:261–272. doi: 10.1385/1-59259-935-4:261. [DOI] [PubMed] [Google Scholar]
- 26.ter Brake O, Konstantinova P, Ceylan M, Berkhout B. Silencing of HIV-1 with RNA interference: a multiple shRNA approach. Mol Ther. 2006;14:883–892. doi: 10.1016/j.ymthe.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 27.ter Brake O, et al. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Mol Ther. 2008;16:557–564. doi: 10.1038/sj.mt.6300382. [DOI] [PubMed] [Google Scholar]
- 28.Leonard JN, Schaffer DV. Computational design of antiviral RNA interference strategies that resist human immunodeficiency virus escape. J Virol. 2005;79:1645–1654. doi: 10.1128/JVI.79.3.1645-1654.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.An DS, et al. Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol Ther. 2006;14:494–504. doi: 10.1016/j.ymthe.2006.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimm D, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 31.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 32.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 33.Kim VN. Small RNAs: classification, biogenesis, and function. Mol Cells. 2005;19:1–15. [PubMed] [Google Scholar]
- 34.Mourelatos Z, et al. miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 2002;16:720–728. doi: 10.1101/gad.974702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. Rna. 2004;10:1957–1966. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee Y, et al. MicroRNA genes are transcribed by RNA polymerase II. Embo J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- 38.Gregory RI, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 39.Han J, et al. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landthaler M, Yalcin A, Tuschl T. The human DiGeorge syndrome critical region gene 8 and Its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol. 2004;14:2162–2167. doi: 10.1016/j.cub.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 41.Lee Y, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 42.Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 43.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng Y, Cullen BR. Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res. 2004;32:4776–4785. doi: 10.1093/nar/gkh824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chendrimada TP, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 47.Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. Embo J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altuvia Y, et al. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005;33:2697–2706. doi: 10.1093/nar/gki567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He L, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanzer A, Stadler PF. Molecular evolution of a microRNA cluster. J Mol Biol. 2004;339:327–335. doi: 10.1016/j.jmb.2004.03.065. [DOI] [PubMed] [Google Scholar]
- 51.Kim YK, Kim VN. Processing of intronic microRNAs. Embo J. 2007;26:775–783. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang K, Elledge SJ, Hannon GJ. Lessons from Nature: microRNA-based shRNA libraries. Nat Methods. 2006;3:707–714. doi: 10.1038/nmeth923. [DOI] [PubMed] [Google Scholar]
- 54.Silva JM, et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet. 2005;37:1281–1288. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- 55.Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2005;102:13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dickins RA, et al. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet. 2005;37:1289–1295. doi: 10.1038/ng1651. [DOI] [PubMed] [Google Scholar]
- 57.Zhou H, Xia XG, Xu Z. An RNA polymerase II construct synthesizes short-hairpin RNA with a quantitative indicator and mediates highly efficient RNAi. Nucleic Acids Res. 2005;33:e62. doi: 10.1093/nar/gni061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chung KH, et al. Polycistronic RNA polymerase II expression vectors for RNA interference based on BIC/miR-155. Nucleic Acids Res. 2006;34:e53. doi: 10.1093/nar/gkl143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Du G, Yonekubo J, Zeng Y, Osisami M, Frohman MA. Design of expression vectors for RNA interference based on miRNAs and RNA splicing. Febs J. 2006;273:5421–5427. doi: 10.1111/j.1742-4658.2006.05534.x. [DOI] [PubMed] [Google Scholar]
- 60.Han J, et al. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 61.Zeng Y, Yi R, Cullen BR. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. Embo J. 2005;24:138–148. doi: 10.1038/sj.emboj.7600491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Michienzi A, Li S, Zaia JA, Rossi JJ. A nucleolar TAR decoy inhibitor of HIV-1 replication. Proc Natl Acad Sci U S A. 2002;99:14047–14052. doi: 10.1073/pnas.212229599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tomari Y, Matranga C, Haley B, Martinez N, Zamore PD. A protein sensor for siRNA asymmetry. Science. 2004;306:1377–1380. doi: 10.1126/science.1102755. [DOI] [PubMed] [Google Scholar]
- 64.Schwarz DS, et al. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 65.Hirose T, Shu MD, Steitz JA. Splicing-dependent and -independent modes of assembly for intron-encoded box C/D snoRNPs in mammalian cells. Mol Cell. 2003;12:113–123. doi: 10.1016/s1097-2765(03)00267-3. [DOI] [PubMed] [Google Scholar]
- 66.Hirose T, Steitz JA. Position within the host intron is critical for efficient processing of box C/D snoRNAs in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:12914–12919. doi: 10.1073/pnas.231490998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aagaard L, et al. A facile lentiviral vector system for expression of doxycycline-inducible shRNAs: knockdown of the pre-miRNA processing enzyme Drosha. Mol Ther. 2007;15:938–945. doi: 10.1038/sj.mt.6300118. [DOI] [PubMed] [Google Scholar]
- 68.Pumfery A, et al. Potential use of pharmacological cyclin-dependent kinase inhibitors as anti-HIV therapeutics. Curr Pharm Des. 2006;12:1949–1961. doi: 10.2174/138161206777442083. [DOI] [PubMed] [Google Scholar]
- 69.Helvik SA, Snove O, Jr, Saetrom P. Reliable prediction of Drosha processing sites improves microRNA gene prediction. Bioinformatics. 2007;23:142–149. doi: 10.1093/bioinformatics/btl570. [DOI] [PubMed] [Google Scholar]
- 70.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 71.Li MJ, et al. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIVshRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol Ther. 2005;12:900–909. doi: 10.1016/j.ymthe.2005.07.524. [DOI] [PubMed] [Google Scholar]
- 72.Unwalla HJ, et al. Negative feedback inhibition of HIV-1 by TAT-inducible expression of siRNA. Nature biotechnology. 2004;22:1573–1578. doi: 10.1038/nbt1040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.