

Abstract
A new scheme, sketch-map, for obtaining a low-dimensional representation of the region of phase space explored during an enhanced dynamics simulation is proposed. We show evidence, from an examination of the distribution of pairwise distances between frames, that some features of the free-energy surface are inherently high-dimensional. This makes dimensionality reduction problematic because the data does not satisfy the assumptions made in conventional manifold learning algorithms We therefore propose that when dimensionality reduction is performed on trajectory data one should think of the resultant embedding as a quickly sketched set of directions rather than a road map. In other words, the embedding tells one about the connectivity between states but does not provide the vectors that correspond to the slow degrees of freedom. This realization informs the development of sketch-map, which endeavors to reproduce the proximity information from the high-dimensionality description in a space of lower dimensionality even when a faithful embedding is not possible.
Keywords: nonlinear dimensionality reduction, proteins, molecular dynamics
The dynamics of many of the molecules that appear in biology, materials science, and chemistry are highly complex. These molecules can undergo transitions involving large numbers of atoms between an enormous number of different configurations (1), which makes it difficult to comprehend these motions using only chemical intuition. Nevertheless, within this data there is a lot of correlation, and there is a strong body of evidence that the energetically accessible regions of phase space lie on a structure that has a low dimensionality (2–6). Therefore, low-dimensionality representations of the free-energy surface can give meaningful insight into phenomena and can provide collective variables (CVs) that can be used to accelerate the dynamics and to reconstruct the free-energy landscape. Methods exist for extracting this low-dimensionality structure by postprocessing the results of long unbiased molecular dynamics trajectories in which the entirety of the landscape is explored (3, 6–8). Unfortunately however, for many systems—in particular for atomistic simulations—obtaining information on interesting, long-time-scale motions using unbiased simulations requires heroic amounts of computational time (9). Therefore, for these types of problems one would ideally like to use dimensionality reduction in tandem with accelerated sampling. This has to work both ways—the method must be able to analyze data from accelerated sampling simulations on very rough free-energy surfaces. Furthermore, it should produce a mapping of phase space that can serve as an optimized, bespoke set of CVs for calculations that extract quantitative free energies.
Experiments have shown that the low-free-energy part of phase space has a complex structure with a nonuniform dimensionality (8), that it is nonlinear (2, 4), that it is nonuniformly sampled (8, 10), and that it is possibly fractal (4, 11). It therefore seems likely that three, four, or even more vectors would be required to faithfully describe these complex topologies using the currently available dimensionality-reduction technologies. In fact, even for relatively simple systems, which can be sampled using unbiased dynamics, a very careful analysis is required to obtain a satisfactory three-dimensional description (7). This is problematic when it comes to using these methods to educate accelerated sampling algorithms because these methods work best with very low numbers of CVs—ideally one or two (12). Furthermore, it is of paramount importance that these CVs map all the basins in the free-energy surface to different parts of the xy plane as barriers to motion in transverse degrees of freedom can hinder the convergence of the free energy. Hence, in this paper we introduce an algorithm, sketch-map, that endeavors to reconcile these two conflicting aims. In doing this we first present an analysis of an enhanced-sampling trajectory, which explores the energetically accessible configurations for a simple polypeptide. This analysis demonstrates that there is a characteristic length scale at which the most valuable topological information about the free-energy landscape is encoded. Therefore, the design of sketch-map is predicated on the assumption that it is not necessary to produce an isometric embedding of the high-dimensionality manifold. Rather, one must preserve the proximity information and ensure that points closer than this characteristic distance are mapped close together, while simultaneously ensuring that the farther apart points are well separated in the projection.
Background
The only dimensionality-reduction algorithm that has been widely adopted within the simulation community is principal component analysis (PCA) (2–5). In this method one runs a simulation trajectory and calculates the means and variances for a large number of collective coordinates. By diagonalizing the resulting covariance matrix one can obtain the directions in which there are the largest structural fluctuations—the directions that are assumed to span the essential substance of the dynamics. However, the assumption that low-energy regions lie in a linear subspace of the full dimensionality space renders PCA appropriate in local regions but results in a poor characterization of the global, nonlinear features (6).
These deficiencies of PCA have led researchers to investigate other, nonlinear manifold learning algorithms and in particular locally linear embedding (LLE) (13), Isomap (6, 14, 15), and diffusion maps (7, 8, 10, 16). The first of these, LLE, is a nonlinear approach, which seeks to combine a set of locally linear descriptions in the vicinity of each trajectory frame into a single, unified embedding (13). It is common knowledge that algorithms like this one are very sensitive to noise (17). This forces one to question how effective this algorithm can be for molecular trajectories, which are typically very noisy (8). The alternative then are global approaches, which seek to reproduce all the pairwise distances between the D-dimensional frames by distributing their embeddings in a lower, d-dimensional space. The grandfather of these methods is multidimensional scaling (MDS) (18), which can be solved as an eigenvector problem or by minimization of a stress function. When Euclidean distances are used the eigenvector solution is equivalent to PCA, so approaches involving stress function minimization are often preferred because they are more flexible. By using a different metric to calculate distances, one can use MDS to fit nonlinear manifolds (19, 20). For instance, assuming the manifold is isometric with a linear space, one can use the geodesic distance (the distance along the manifold). This idea is the basis of the Isomap algorithm in which geodesic distances are obtained by calculating the length of the shortest path through a fully connected graph that is created by joining the points that are closest together (19). Calculating geodesics in this way assumes that the high-dimensionality points lie in a convex subset of RD—i.e., it assumes that the low-dimensional manifold is uniformly sampled and there are no “holes.” Donoho and Grimes (21, 22), in the context of image articulation, have demonstrated that for relatively simple cases this approximation is not valid and that in these cases Isomap fails to find the correct parameter space up to a linear mapping.
Currently the most promising approach for trajectory dimensionality reduction is diffusion maps (23–25), which can be formulated in a way that makes it resilient to noisy and nonuniformly distributed data (8). In this approach one defines a weighted graph on the simulation data and then uses the first few eigenvalues of the Laplacian of the manifold as the embedding coordinates. This approach is exciting because for the systems examined the vectors spanning the low-dimensionality manifold are those in which large barriers to motion make diffusion slow (8, 16). That said, the method has thus far only been applied to relatively simple systems and not to systems that require one to use accelerated sampling to explore phase space.
To demonstrate our method we use in this paper the folding of polyalanine-12, modeled with a distance-dependent dielectric (ϵij = rij in Angstroms) that mimics some of the solvent effects. This system has been extensively studied (26) and has been shown to have a complex, funnel-shaped energy landscape with an alpha-helical global minimum that does not form during long MD simulations started from a random configuration (27). To accelerate the dynamics we therefore use the recently developed reconnaissance metadynamics method (see Materials and Methods) because with this method one can use a large number of CVs to characterize configurations and still obtain a qualitatively correct mapping of the free-energy surface (27). Furthermore, unlike in other papers on dimensionality reduction, we take advantage of the fact that changes in bond lengths, bond angles, and rigid peptide bond dihedrals along with the rotations of methyl groups are uninteresting. We therefore use only the 24 backbone dihedral angles (Fig. 1A) to characterize the various configurations visited during the trajectories.
Fig. 1.

Information on the distribution of torsional angles found in our reconnaissance metadynamics simulations. A shows the ala12 system examined and the backbone dihedral angles that were used as CVs. B shows a 2D projection of the distribution of angles found during reconnaissance. Here we show the distribution as a function of ψ in the third residue and of ϕ in the sixth residue, although the distribution of any pair of angles shows the same qualitative features. C shows (in red) a histogram for the distribution of distances between pairs of frames. Also shown in this figure is the distribution expected for a 24-dimensional, isotropic Gaussian with a standard deviation equal to 0.5 (black) and the distribution of distances expected for a set of points distributed uniformly across the 24-dimensional space (gray).
The Free-Energy Landscape of a Polypeptide
Before introducing our dimensionality-reduction algorithm it is perhaps useful to step back for a moment and to examine some qualitative features of the protein’s free-energy landscapes in detail. Therefore in Fig. 1B we project the set of configurations obtained from our reconnaissance metadynamics simulations onto two dihedrals. We find that, even for a trajectory in which relatively high energy states are sampled and regardless of the pair of dihedrals selected, the resulting distribution of angles is very similar to the Ramachandran plot. Hence, angles are not uniformly distributed across the available space and there are instead regions of high and low probability. This behavior was also seen by Sims et al. (28) when they examined the distribution of torsional angles for short peptide chains in higher dimensional spaces.
High-dimensionality spaces can often display very nonintuitive properties, which challenge our understanding of distance and proximity (29). We therefore cannot possibly expect to understand what structures are present simply by visualizing 2D projections. One quantity that can give us some feel as to whether or not it is feasible to represent the data in the lower dimensionality state is the histogram of pairwise distances, which is shown in Fig. 1C. Remarkably, the long range part of this distribution resembles that obtained from a uniform distribution of points in the full, 24-dimensional space.* In fact, only when r is less than eight is there a marked deviation from the uniform distribution—a slower decay toward zero. For values of r of about one this decay resembles that of a Gaussian distribution in the full, 24-dimensional space in agreement with what one would expect for the fluctuations within a harmonic basin. We therefore postulate that the most interesting distances are those between about two and eight because only here does the histogram resemble neither the Gaussian or uniform distribution.
Fig. 1C suggests that fitting protein free energy surfaces using dimensionality-reduction methods based on pure distance matching is impossible. The plain fact is that certain features of the distribution of distances are characteristic of points distributed in the full dimensionality space. This histogram can thus not be reproduced by projecting points in a space of lower dimensionality. In addition, it would appear that the free-energy surface has a complex topology. This appears in our analysis because we use torsional angles that are inherently non-Euclidean to characterize configurations. However, there is evidence from the literature that protein potential energy surfaces have fractal dimensionalities (4, 11) or an otherwise intrinsically non-Euclidean topology.
The theory of energy landscapes suggests that energetically accessible configurations take up only a tiny fraction of phase space because these configurations are clustered together in basins, in which fluctuations take place in a high-dimensionality space, that are themselves connected by a spider’s web of transition pathways (1). This picture is far more consistent with the information coming from our analysis of Fig. 1C and the structure of the Ramachandran plot than any picture in which all the low-energy regions of phase space lie on a low-dimensional, Euclidean manifold. Therefore, to test whether this is a realistic model for the energy landscape of ala12 we generated a set of points from a model potential that exhibits these features by importance sampling at a sufficiently large temperature for both basins and low-lying transition states to be sampled. The resulting collection of points thus resembles what could have been obtained from enhanced-sampling calculations and can be compared with the histogram of distances obtained for ala12 (Fig. 2). Similarly to what was observed for the protein (Fig. 1C) the distribution of pairwise distances only deviates from the histogram for a uniform distribution in the full-dimensionality, periodic space at short r, and at the shortest r the decay resembles that observed for the distribution of distances in a multivariate Gaussian in the full-dimensionality space. In fact the main qualitative difference for the two systems is that the deviation here is less pronounced, which is simply a consequence of the lower dimensionality of this potential. The similarities thus give us confidence in our conceptual picture for the shape of the protein free energy landscape in the high-dimensionality space.
Fig. 2.

Information on a model potential (V(θ,ϕ,ψ) = exp[3(3 - sin4(θ) - sin4(ϕ) - sin4(ψ))] - 1), which exhibits many of the features that we believe characterize complex free-energy landscapes. In A the isosurfaces that enclose 50, 80 and 90% of the probability density for a particle diffusing about this potential at a temperature of kBT = e3 - 1 are shown. In B the distribution of points extracted from this potential through importance sampling are shown and the 500 landmark points selected using a farthest point sampling strategy are highlighted. In this panel the size of the landmarks is related to their weights and their colors depict the value of one of the angles. A key for the coloring is shown in C, and for the remainder of this paper, wherever points are colored according to the value of an angle, we ask the reader to refer to this scale. Finally, in D we show a histogram of the distances between pairs of generated points (red). This is again compared with the distribution expected for a 3D, isotropic Gaussian (black) and the distribution for a set of points distributed uniformly across the 3D space (gray).
Dimensionality Reduction Algorithm
One simple way to introduce nonlinearity in manifold learning algorithms is to perform distance matching but with the distances transformed (31) or weighted (32) so as to enhance the importance of certain connections—often the short distances (33). The analysis of the previous sections suggests that, if we could make the algorithm focus on reproducing distances from the interesting part of the histogram (the part where the distribution does not correspond to a high-dimensionality uniform or Gaussian distribution), this would be a useful approach for trajectory data. Furthermore, we can justify this approach based on our picture for the structure of the free-energy landscape by noting that by doing this we are focusing on reproducing the relations and connections between nearby basins and are discarding all the high-dimensionality, unfittable data on the internal structure of basins and the relative positions of distant basins. Our method, sketch-map, then is essentially multidimensional scaling, in which the distances in both the high- and low-dimensional spaces are transformed by a sigmoid function, which maps monotonically 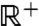 to [0,1). Hence, one produces the mapping by minimizing (for details see Materials and Methods) the following stress function:
to [0,1). Hence, one produces the mapping by minimizing (for details see Materials and Methods) the following stress function:
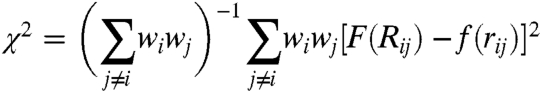 |
[1] |
where wi is the weight of point i and Rij = |Xi - Xj|(D) and rij = |xi - xj|(d) are the distances between points i and j in the high- and low-dimensionality spaces, respectively.† F and f are then both general sigmoid functions of the form:
| [2] |
where sσ,a,b(σ) = 1/2 and the exponents a and b determine the rate at which the function approaches 0 and 1, respectively. The same value of σ is used in both F and f as using different values simply corresponds to a scaling of coordinates. However, we distinguish between the values of a and b in the two functions by using aD and bD for F and ad and bd for f.
When selecting parameters for the high-dimensionality space sigmoid function F, one is essentially selecting the length scales over which the connectivity data in the high-dimensionality space is interesting. The analysis presented in the previous section would therefore suggest that we should tune σ, aD, and bD so that for small values of Rij, where the histogram resembles that of a full-dimensional multivariate Gaussian, F(Rij) ≈ 0.0, while for large values of Rij, where the histogram of distances resembles that of a set of points uniformly distributed in the full-dimensional space, F(Rij) ≈ 1.0. This ensures that, once minimized, points that are close together in the D-dimensional space are mapped close together in the d-dimensional space and vice versa. Furthermore, because the error in the reproduction the distance Rij contributes an amount to χ2 that is proportional to F′(R), a function that is peaked in the vicinity of σ, only a cursory attempt is made to reproduce the precise distribution of near and far neighbors around any given point. Meanwhile, the major focus during optimization is the reproduction of distances close to the value of the method’s critical parameter, σ, which selects the interesting length scale for the problem. The values of aD and bD are far less important and, much like when similar functions are used to calculate continuous versions of coordination numbers, the performance of the method only depends weakly on their values.
When the same parameters are used in the two sigmoid functions of Eq. 1 sketch-map, like MDS, will reproduce all pairwise distances if the configurations lie in a linear subspace of dimension d. However, given that we know the points are not distributed in this way, this choice is not appropriate and is in fact detrimental because, as shown in Fig. 1C, at short-range uninteresting fluctuations occur in the full D-dimensional space. Hence, distance matching involves the impossible task of mapping a manifold, which has parts where the radial density grows as rD-1, into a space where radial density can grow only as rd-1. In sketch-map we therefore use different a and b parameters for the two sigmoid functions to bypass this intractable problem. We note that for any distribution where the radial density around points grows as rD-1 the corresponding histogram of distances, transformed by sσ,a,b(r), is approximately equal to sD/a-1 for small s. Therefore, for small s, the histograms of (differently) transformed distances for two distributions with radial densities that grow as rD-1 and rd-1 will be similar if ad/d ≈ aD/D.
The minimization of Eq. 1 scales quadratically with the number of data points so when fitting a trajectory using sketch-map the first step is to select a small number of landmark frames (34), which, as detailed elsewhere, can be done either by selecting points at random or by using a farthest point sampling strategy (FPS) (35, 36). One can then assign weights to the landmarks based either on an estimate of the free energy, if available, or by computing the number of trajectory frames within each landmark’s Voronoi polyhedron to ensure that reproduction of the structure in the low-energy parts of the landscape is weighted more in the fitting. Finally, once the minimization is completed, one can calculate the projection, x, of any high-dimensionality point X by minimizing:
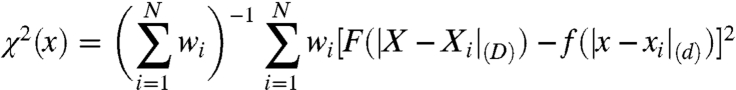 |
[3] |
where Xi is one of the landmark points and xi is its low-dimensional projection. A global minimum for this quantity can be obtained by calculating the value of χ2(x) on a grid and then using the lowest-lying point as a start point for a conjugate gradient minimization. The code for performing sketch-map is available online at sketchmap.berlios.de.
Results
Dimensionality Reduction Example.
Before fitting the reconnaissance metadynamics data we first fit the data from the model potential shown in Fig. 2. Five hundred landmarks points were selected using a FPS strategy from the 5,000 frames generated by importance sampling. Their weights were then set equal to the number of frames within each landmark’s Voronoi polyhedron. In the sketch-map result shown in Fig. 3 all eight basins are well separated and the majority of the connections are reproduced. This is an impressive result as this distribution is periodic in three dimensions and is thus not isometric with a linear, two-dimensional space. Nevertheless, unlike the other manifold learning algorithms we tested (see SI Text), sketch-map is able to circumvent this issue by breaking four of the connections between basins. The resulting embedding thus unrolls the box and gives the net shown in Fig. 3 rather than simply squashing the box onto the plane. This clear picture for the shape of phase space that emerges from our sketch-map projection is very appealing from the point of view of our eventual aim of using this method in tandem with biased MD.
Fig. 3.

A 2D, sketch-map projection of the landmark points selected from the dataset depicted in Fig. 2. This model has eight minima in the free-energy surface, which appear at (± π/2, ± π/2, ± π/2). Projections of these points are indicated on this figure using labeled circles, while the various transition pathways are shown as dashed lines. Parameters for the sigmoid functions were chosen based on the histogram of distances (Fig. 2D) as σ = 2, aD = 3, bD = 9, ad = 2, and bd = 2. Projected points are colored, using the key shown in Fig. 2C, in accordance with the value of one of the three underlying variables.
Polyalanine-12.
For the considerably more complex ala12 landscape we selected 1,000 landmark points from our reconnaissance metadynamics trajectories and again set their weights equal to the number of the remaining frames within each landmark’s Voronoi polyhedron. Sketch-map parameters (given in Fig. 4) were then selected based on the shape of the histogram shown in Fig. 1C. After fitting we projected the nonlandmarks points, using (Eq. 3). The final result is shown in Fig. 4, where points are colored in accordance with the number of residues that were identified as being part of an alpha helix or beta sheet by the STRIDE algorithm (37). Fig. 4 shows that embedded points are clustered in basins much like what is observed in full-dimensional description. Furthermore, there is a clear-cut separation between the regions of the plane that correspond to helix-like and sheet-like secondary structures. In the areas around each of these quintessential protein configurations STRIDE identifies the structures as being mostly composed of coils and turns.
Fig. 4.

Results for a projection of the frames obtained from the reconnaissance metadynamics simulations of ala12. The parameters of the sigmoid functions were chosen to be σ = 6, aD = bD = 12, ad = 1, and bd = 2. Points not included in the landmark set were embedded with the out-of-sample extension described in the text. The 2D projection is shown, with the embedded points colored in accordance with the number of residues that, according to STRIDE, are part of an alpha helix or beta sheet. A key for the color scheme is also shown, together with snapshots of a few selected configurations.
Fig. 5 gives more detailed diagnostic information and also compares the results of sketch-map with those from pure distance matching.‡ The panels that show the embedded points colored in accordance with one of the backbone dihedrals demonstrate that sketch-map is better at clustering together points with similar values for a particular dihedral. More revealing, though, is the analysis of the joint probability distribution of low and high-dimensional pairwise distances between frames. Obviously, if the embedding is exact all density should be concentrated along the diagonal. However, as discussed above, this goal cannot be achieved, because of the intrinsically high-dimensional nature of the distribution of configurations at both short and large distances. Fig. 5 shows that, for distance matching, there is a sizable density in the region of the histogram corresponding to projection of points close together when they are in actuality fact far apart. This is disastrous in terms of using these coordinates to provide a coarse-grained description of configuration space as it means that structures that are very different from each other cannot be distinguished. In contrast, the histogram for the sketch-map result (Fig. 5D) result demonstrates that this algorithm only projects points that lie closer than σ in the high-dimensionality space close together.
Fig. 5.

A and B show diagnostic information on the embedding of the ala12 trajectory frames obtained by pure distance matching, while C and D refer to the results of sketch-map. Similar figures for other dimensionality reduction algorithms are provided in SI Text. In A an C the low-dimensional embedding of the frames is shown, colored according to two of the backbone dihedrals. B and D depict the joint probability distribution of the distances between two frames in high dimension (Rij) and the distance between the corresponding low-dimensional projections, (rij).
Conclusions
For proteins and other chemical systems the manifold on which the energetically accessible region of phase space lies has a small volume but a very complex structure. It consists of small, high-dimensionality basins that are connected by a spider’s web of transition pathways and its structure can be thought of in terms of a hierarchy of different length scales. On the smallest of these scales harmonic fluctuations in the full-dimensionality space take place. Changes in secondary and tertiary structure, meanwhile, take place over longer scales. Evidence presented here and elsewhere has demonstrated that one can recognize these different length scales by examining the distribution of distances between trajectory frames and that estimates of the dimensionality of the manifold depend on the length scale at which one examines the problem. Therefore, we contend that, when creating a low-dimensionality projection of a trajectory, one should first examine the distribution of distances and thereby identify the interesting length scale. Then, when projecting the data, an algorithm like sketch-map can be used so that the fitting effort is directed toward reproducing distances in the range that has been identified as interesting. Using these ideas we were able to produce a 2D mapping from a description of a set of protein configurations based on backbone dihedral angles. This mapping is able to reproduce the qualitative features of the free-energy landscape and clearly separates configurations with different protein secondary structures. Furthermore, in SI Text we show that sketch-map produces a qualitatively similar mapping when the set of distances between the Cα atoms is used to describe configurations. It may well be that for larger systems analysis of the distribution of distances will provide evidence of multiple interesting length scales in the problem. For these cases a hierarchical version of the sketch-map approach, which makes use of multiple sigmoid function with different σ parameters could be very useful.
The most successful approaches for performing dimensionality reduction on trajectory data do not assume that the low-dimensionality manifold, which contains the low-energy configurations, is isometric with a low-dimensional linear space. Instead these methods distort the distances between the high-dimensionality data points so that the essential features in the data can be represented in a low-dimensionality space. Sketch-map works in a similar manner and has this observation at its core. In addition, sketch-map produces an embedding from a very small number of landmark frames and is able to embed further configurations after the projection of this initial training set. This means that one can feasibly imagine combining sketch-map projections with enhanced sampling techniques to calculate the free-energy landscapes for systems in which the interesting events are not observable on the simulation time scale. Consequentially, we are currently working on ensuring this mapping is continuous so that the embedding can be used as CVs for biased MD.
In all of this work we focus on the data output by simulation trajectories, which presents a particular set of problems to manifold learning algorithms. However, the ubiquity of high-dimensionality data in disciplines of science, from chemistry and physics to social sciences and psychology, suggests that there is an abundance of potential applications of sketch-map.
Materials and Methods
Reconnaissance Metadynamics Simulations.
All simulations of polyalanine were run using gromacs-4.5.1 (38), the amber96 forcefield (39) and a distance dependent dielectric. A time step of 2 fs was used, all bonds were kept rigid using the LINCS algorithm, and the van der Waals and electrostatic interactions were calculated without any cutoff. The global thermostat of Bussi et al. (40) was used to maintain the system at a temperature of 300 K. Recently we introduced an accelerated sampling method, reconnaissance metadynamics (27), which can be used with large numbers of collective variables. This method uses a self-learning algorithm to examine the trajectory and to construct an adaptive simulation bias that accelerates the exploration of phase space. We chose to use this method to perform the enhanced sampling calculations in this work and in particular the implementation of it in PLUMED (41). In previous work (27) we have shown that a 50-ns reconnaissance metadynamics simulation started from a random configuration can be used to find the alpha-helical, folded state of polyalanine-12. However, in this work, so as to have an extensive exploration of the region of phase space about the folded state, we took our trajectory data from four reconnaissance metadynamics simulations started from the folded state. In these simulations CVs were stored every 250 steps, whereas cluster analysis was done every 5 × 105 steps. The expansion parameter was set equal to 0.05, only basins with a weight greater than 0.2 were considered, and attempts were made every 1,000 steps to add to these basins hills of height 1.0 kJ mol-1 and width 1.5. During all calculations we stored frames every 8 ps for later analysis but discarded the first nanosecond of all simulations so as to ensure that our trajectories were independent. Hence, in this work all analysis is based on a set of 46,182 trajectory frames.
Optimization strategy.
Eq. 1 is a nonconvex function and is thus very difficult to optimize. Moreover, the problem becomes stiffer as the sigmoid function becomes steeper at the inflection point. Hence, we have found that a combination of strategies is required to minimize χ2 effectively. During the early stages of the minimization we introduce the better-behaved, although still nonconvex, merit function for least-squares distance matching: 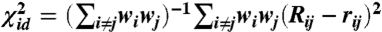 . Iterative minimization of this function can be initialized using the result from classical MDS, which will minimize a
. Iterative minimization of this function can be initialized using the result from classical MDS, which will minimize a 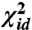 -like stress in which all weights are equal to one. Once the minimum for the weighted
-like stress in which all weights are equal to one. Once the minimum for the weighted 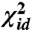 is found, we introduce the sigmoid function by performing a series of minimizations of a stress function given by
is found, we introduce the sigmoid function by performing a series of minimizations of a stress function given by 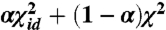 in which we progressively reduce α from 1 to 0. During our experiments we found that the most effective strategy for iteratively minimizing stress functions like
in which we progressively reduce α from 1 to 0. During our experiments we found that the most effective strategy for iteratively minimizing stress functions like 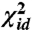 and χ2 is to perform 20–50 steps of conjugate gradient optimization followed by a “pointwise-global” scheme in which the minimum stress position for each landmark point is found by minimizing Eq. 3, while keeping the positions of the other landmarks fixed. Sweeping through all the landmarks a few times performing this second procedure allows one to quickly find an optimal configuration for all the projected points. This global optimization step becomes costly when the target dimensionality d is increased. However, χ2 is a relatively smooth function, so an adaptive grid search or a more sophisticated global minimization algorithm could be used to reduce the overhead.
and χ2 is to perform 20–50 steps of conjugate gradient optimization followed by a “pointwise-global” scheme in which the minimum stress position for each landmark point is found by minimizing Eq. 3, while keeping the positions of the other landmarks fixed. Sweeping through all the landmarks a few times performing this second procedure allows one to quickly find an optimal configuration for all the projected points. This global optimization step becomes costly when the target dimensionality d is increased. However, χ2 is a relatively smooth function, so an adaptive grid search or a more sophisticated global minimization algorithm could be used to reduce the overhead.
Supplementary Material
Acknowledgments.
The authors thank Michael Bronstein and Davide Branduardi for useful discussions and also acknowledge funding from the European Union (Grant ERC-2009-AdG-247075), the Royal Society, and the Swiss National Science Foundation.
Footnotes
The authors declare no conflict of interest.
See Commentary on page 12969.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1108486108/-/DCSupplemental.
*The distribution of distances between uniformly distributed points in a periodic space is related to the surface area of a diced hypersphere (30). In contrast to the distribution in a nonperiodic space there is a maximum in this function after which the function decays to zero at 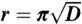 .
.
†|·| does not have to be a Euclidean distance—here, for instance, we apply the minimum image convention to take account of the periodicity of the space.
‡Distance matching was performed by linear multidimensional scaling followed by iterative minimization of χ2 with both of the sigmoid functions set to be the identity.
References
- 1.Wales DJ. Energy Landscapes. Cambridge, UK: Cambridge Univ Press; 2003. [Google Scholar]
- 2.Garcia AE. Large-amplitude nonlinear motions in proteins. Phys Rev Lett. 1992;68:2696–2699. doi: 10.1103/PhysRevLett.68.2696. [DOI] [PubMed] [Google Scholar]
- 3.Amadei A, Linssen ABM, Berendsen HJC. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 4.Hegger R, Altis A, Nguyen PH, Stock G. How complex is the dynamics of peptide folding? Phys Rev Lett. 2007;98:028102. doi: 10.1103/PhysRevLett.98.028102. [DOI] [PubMed] [Google Scholar]
- 5.Zhuravlev PI, Materese CK, Papoian GA. Deconstructing the native state: Energy landscapes, function and dynamics of globular proteins. J Phys Chem B. 2009;113:8800–8812. doi: 10.1021/jp810659u. [DOI] [PubMed] [Google Scholar]
- 6.Das P, Moll M, Stamati H, Kavraki LE, Clementi C. Low-dimensional, free-energy landscapes of protein-folding reactions by nonlinear dimensionality reduction. Proc Natl Acad Sci USA. 2006;103:9885–9890. doi: 10.1073/pnas.0603553103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferguson AL, Panagiotopoulos AZ, Debenedetti PG, Kevrekidis IG. Systematic determination of order parameters for chain dynamics using diffusion maps. Proc Natl Acad Sci USA. 2010;107:13597–13602. doi: 10.1073/pnas.1003293107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rohrdanz MA, Zheng W, Maggioni M, Clementi C. Determination of reaction coordinates via locally scaled diffusion map. J Chem Phys. 2011;134:124116. doi: 10.1063/1.3569857. [DOI] [PubMed] [Google Scholar]
- 9.Shaw DE, et al. Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 10.Zheng W, Rohrdanz MA, Maggioni M, Clementi C. Polymer reversal rate calculated via locally scaled diffusion map. J Chem Phys. 2011;134:144109. doi: 10.1063/1.3575245. [DOI] [PubMed] [Google Scholar]
- 11.Piana S, Laio A. Advillin folding takes place on a hypersurface of small dimensionality. Phys Rev Lett. 2008;101:208101. doi: 10.1103/PhysRevLett.101.208101. [DOI] [PubMed] [Google Scholar]
- 12.Frenkel D, Smit B. Understanding Molecular Simulation. London: Academic; 2002. [Google Scholar]
- 13.Roweis ST, Saul LK. Nonlinear dimensionality reduction by locally linear embedding. Science. 2000;290:2323–2326. doi: 10.1126/science.290.5500.2323. [DOI] [PubMed] [Google Scholar]
- 14.Plaku E, Stamati H, Clementi C, Kavraki LE. Fast and reliable analysis of molecular motion using proximity relations and dimensionality reduction. Proteins. 2007;67:897–907. doi: 10.1002/prot.21337. [DOI] [PubMed] [Google Scholar]
- 15.Stamati H, Clementi C, Kavraki LE. Application of nonlinear dimensionality reduction to characterize the conformational landscape of small peptides. Proteins. 2010;78:223–235. doi: 10.1002/prot.22526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singer A, Erban R, Kevrekidis IG, Coifman RR. Detecting intrinsic slow variables in stochastic dynamical systems by anisotropic diffusion maps. Proc Natl Acad Sci USA. 2009;106:16090–16095. doi: 10.1073/pnas.0905547106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou C, Wang J, Wu Y, Yi D. Local linear transformation embedding. Neurocomputing. 2009;72:2368–2378. [Google Scholar]
- 18.Cox TF, Cox MAA. Multidimensional Scaling. London: Chapman and Hall; 1994. [Google Scholar]
- 19.Tenenbaum JB, Silva Vd, Langford JC. A global geometric framework for nonlinear dimensionality reduction. Science. 2000;290:2319–2323. doi: 10.1126/science.290.5500.2319. [DOI] [PubMed] [Google Scholar]
- 20.Bronstein AM, Bronstein MM, Kimmel R, Mahmoudi M, Sapiro G. A Gromov–Hausdorff framework with diffusion geometry for topologically-robust non-rigid shape matching. Int J Comput Vis. 2010;89:266–286. [Google Scholar]
- 21.Donoho DL, Grimes C. When Does Isomap Recover the Natural Parameterization of Familes of Articulated Images? Stanford University; 2002. pp. 2002–27. [Google Scholar]
- 22.Donoho DL, Grimes C. Hessian eigenmaps: Locally linear embedding techniques for high-dimensional data. Proc Natl Acad Sci USA. 2003;100:5591–5596. doi: 10.1073/pnas.1031596100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coifman RR, et al. Geometric diffusions as a tool for harmonic analysis and structure definition of data: Multiscale methods. Proc Natl Acad Sci USA. 2005;102:7432–7437. doi: 10.1073/pnas.0500896102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coifman RR, Lafon S. Diffusion maps. Appl Comput Harmon Anal. 2006;21:5–30. [Google Scholar]
- 25.Belkin M, Niyogi P. Laplacian eigenmaps for dimensionality reduction and data representation. Neural Comput. 2003;15:1373–1396. [Google Scholar]
- 26.Mortenson PN, Evans DA, Wales DJ. Energy landscapes of model polyalanines. J Chem Phys. 2002;117:1363–1376. [Google Scholar]
- 27.Tribello GA, Ceriotti M, Parrinello M. A self-learning algorithm for biased molecular dynamics. Proc Natl Acad Sci USA. 2010;107:17509–17514. doi: 10.1073/pnas.1011511107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sims GE, Choi IG, Kim SH. Protein conformational space in higher order ϕ - ψ maps. Proc Natl Acad Sci USA. 2005;102:618–621. doi: 10.1073/pnas.0408746102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bellman R. Adaptive Control Processes: A Guided Tour. Princeton, NJ: Princeton Univ Press; 1961. [Google Scholar]
- 30.Langerholc J. Volumes of diced hyperspheres: resuming the tam-zardecki formula. Appl Math Comput. 1989;30:1–18. [Google Scholar]
- 31.Schölkopf B, Smola A, Muller KR. Advances in Kernel Methods: Support Vector Learning. Cambridge, MA: MIT Press; 1999. pp. 327–352. [Google Scholar]
- 32.Rosman G, Bronstein MM, Bronstein AM, Kimmel R. Nonlinear dimensionality reduction by topologically constrained isometric embedding. Int J Comput Vis. 2010;89:56–58. [Google Scholar]
- 33.Sammon JW. A nonlinear mapping for data structure analysis. IEEE Trans Comput. 1969;18:401–409. [Google Scholar]
- 34.Bronstein MM, Bronstein AM, Kimmel R, Yavneh I. A multigrid approach for multi-dimensional scaling; Proceedings of the Copper Mountain Conference on Multigrid Methods; Philadelphia: Society for Industrial and Applied Mathematics; 2005. [Google Scholar]
- 35.Hochbaum DS, Shmoys DB. A best possible heuristic for the k-center problem. Math Oper Res. 1985;10:180–184. [Google Scholar]
- 36.de Silva V, Tenenbaum B. Sparse multidimensional scaling using landmark points. Technical Report, Stanford University. 2004. [Accessed September 10, 2010]. Available at http://graphics.stanford.edu/courses/cs468-05-winter/Papers/Landmarks/Silva_landmarks5.pdf.
- 37.Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 38.Hess B, Kutzner C, van der Spoel D, Lindahl E. Gromacs 4: Algorithms for highly efficient, load-balanced and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 39.Kollman PA. Advances and continuing challeges in achieving realistic and predictive simulations of the properties of organic and biological molecules. Acc Chem Res. 1996;29:461–469. [Google Scholar]
- 40.Bussi G, Donadio D, Parrinello MJ. Cannonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 41.Bonomi M, et al. Plumed: A portable plugin for free-energy calculations with molecular dynamics. Comput Phys Commun. 2009;180:1961–1972. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.