

Abstract
The increasing use of the anti-microbial triclocarban (TCC) in personal care products (PCPs) has resulted in concern regarding environmental pollution. TCC is a potent inhibitor of soluble epoxide hydrolase (sEH). Inhibitors of sEH (sEHIs) are anti-inflammatory, anti-hypertensive and cardio-protective in multiple animal models. However, the in vivo effects anticipated from a sEHI have not been reported for TCC. Here we demonstrated the anti-inflammatory effects in vivo of TCC in a murine model. TCC was employed in a lipopolysaccharide (LPS)-challenged murine model. Systolic blood pressure, plasma levels of several inflammatory cytokines and chemokine, and metabolomic profile of plasma oxylipins were determined. TCC significantly reversed LPS-induced morbid hypotension in a time-dependent manner. TCC significantly repressed the increased release of inflammatory cytokines and chemokine caused by LPS. Furthermore, TCC significantly shifted the oxylipin profile in vivo in a time-dependent manner towards resolution of inflammation as expected from a sEHI. These results demonstrated that at the doses used TCC is anti-inflammatory in the murine model. This study suggests that TCC may provide some benefits in humans in addition to its antimicrobial activities due to its potent inhibition of sEH. It may be a promising starting point for developing new low volume high value applications of TCC. However these biological effects also caution against the general over use of TCC in PCPs.
Keywords: hypotension, lipopolysaccharide, metabolomics, metabolic profiling, mice
Introduction
Triclocarban (3,4,4′-trichlorocarbanilide, TCC) is an antimicrobial agent that has been used over 40 years in personal care products (PCPs) such as soaps and disinfectants (Roman et al., 1957; Black et al., 1975). TCC and other PCPs are currently used in very large amounts in the United States and Europe. Pharmacokinetics of TCC has been investigated in multiple species including mouse (Demetrulias et al., 1984), rat (Howes and Black, 1976; Jeffcoat et al., 1977; Warren et al., 1977; Birch et al., 1978), monkey (Birch et al., 1978; Hiles et al., 1978) and human (Scharpf et al., 1975; Howes and Black, 1976; Birch et al., 1978; Hiles and Birch, 1978; Northroot et al., 1984; Schebb et al., 2011). Specifically, 1.1% of TCC was deposited on hairless mice skin by dermal exposure to TCC-containing soap bar for 10 min (Demetrulias et al., 1984), which is compared to the amount deposited onto the skin of human (Northroot et al., 1984). Parenteral injection or dermal use of TCC resulted in low levels of TCC in rat blood, indicating rapid metabolism of TCC in rodent models (Howes and Black, 1976). The principal metabolites of TCC common to rat, monkey and human were the sulphate and glucuronide conjugates of 2′-, 3′- and 6-hydroxy-TCC while the major urinary metabolites of TCC found in human and monkey were N- and N’-TCC glucuronides (Jeffcoat et al., 1977; Warren et al., 1977; Birch et al., 1978; Schebb et al., 2011). Oral administration of TCC to human at a dose of 2.2 umol/kg body weight resulted in the maximum plasma level of 3.7 uM occurred 2.8 h after dosing. And the same study demonstrated that TCC was mainly excreted via fecal elimination (70% of dose) and urinary excretion (27% of dose) completed in 120 hr and 80 hr after dosing respectively (Hiles and Birch, 1978). In short, TCC is rapidly metabolized in both human and animal models. However, TCC biodegrades much more slowly in the environment than it does in humans and other animals (Gledhill, 1975; Ying et al., 2007). As an antimicrobial agent in PCPs, TCC enters the environment mainly by discharge of effluent from wastewater treatment plants and disposal of biosolids on land. In addition, TCC is a hydrophobic chemical, capable of forming multiple hydrogen bonds. These physical properties lead TCC to partition from a waste water stream where it binds to sludge and becomes recalcitrant to microbial degradation (Heidler et al., 2006; Ying et al., 2007). This results in an accumulation of TCC in not only aqueous effluent but also the resulting biosolids. Thus TCC has been found to be one of the serious contaminants of water resources nationwide in the United States (Halden and Paull, 2005). Furthermore, TCC was recently found at higher concentrations to enhance the bioactivity of several hormones in vitro and in vivo, but it alone has little agonist activity (Ahn et al., 2008; Chen et al., 2008). Although the bioactivity and toxicity of TCC are still being explored, a general concern has been raised regarding the extensive use of TCC.
TCC, an N,N′-disubstituted urea, has been reported to be a potent inhibitor of recombinant murine and human soluble epoxide hydrolases (sEH) in vitro (Table 1) (Morisseau et al., 1999; McElroy et al., 2003; Morisseau et al., 2009). The sEH is the main enzyme that catalyzes the metabolism of epoxyeicosatrienoic acids (EETs) and other fatty acid epoxides into the more polar and usually less potent metabolites, dihydroxyeicosatrienoic acids (DHETs) and other corresponding diols (Morisseau and Hammock, 2005; Newman et al., 2005). EETs and other epoxy fatty acids have been demonstrated to be potent anti-inflammatory and cardio-protective mediators (Node et al., 1999; Spector et al., 2004; Dhanasekaran et al., 2006; Larsen et al., 2006a; Nithipatikom et al., 2006; Seubert et al., 2007; Li et al., 2009). The inhibitors of sEH (sEHIs) stabilize the endogenous EET levels by inhibiting sEH and thus reduce inflammation, cardiovascular diseases, nephrotoxicity and other diseases in multiple animal models (Jung et al., 2005; Schmelzer et al., 2005; Larsen et al., 2006b; Morisseau et al., 2006; Xu et al., 2006; Parrish et al., 2008). In addition, sEHIs synergize the anti-inflammatory effect of non-steroidal anti-inflammatory drugs (indomethacin, celecoxib, and rofecoxib) (Schmelzer et al., 2006), aspirin and the 5-lipooxygenase activation protein inhibitor MK886 (Liu et al., 2010). Thus, we hypothesized that TCC may be beneficial in several models of inflammatory diseases in human and/or animals due to its sEH inhibition activity. Of course the unintentional or inappropriate inhibition of the sEH may lead to undesirable effects.
Table 1.
Structures and inhibition potency against the murine and human sEHs
| Inhibitors structure | Name | Inhibition against the Murine sEH | Inhibition against the Human sEH | |
|---|---|---|---|---|
| IC50 (nM) | IC50 (nM) | KI (nM) | ||
|
|
TCC | 370 ± 40 | 13 ± 1 | 96 ± 5 |
|
|
t-AUCB | 4 ± 1 | 2 ± 1 | 1.5 ± 0.2 |
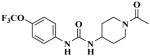
|
TPAU | 44 ± 3 | 12 ± 3 | 57 ± 6 |
Data are presented as mean ± SD (N = 3). IC50 values were determined with the fluorescent substrate CMNPC. The data of t-AUCB and TPAU are from (Liu et al., 2009a). The Ki was determined using t-DPPO assay. TCC, triclocarban; t-AUCB, trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid; TPAU, 1-trifluoromethoxyphenyl-3-(1-acetylpiperidin-4-yl) urea.
In this study, we address a possible health outcome via exposure either through the environment or personal use of products containing TCC by directly investigating its in vivo effects in a murine model of inflammation by lipopolysaccharide (LPS) challenge. The sEH inhibition demonstrated by TCC shows the expected alterations in oxylipin profile in vivo. Moreover, TCC resulted in the reversal in LPS-challenged hypotension as well as the amelioration of the increased release of the inflammatory cytokines similar to other sEHIs. Thus, TCC exposure may result in beneficial effects on human health such as anti-inflammation, reduced hypertension, and cardiovascular protection based on its sEH inhibitory activity. These effects should be particularly dramatic near the site of application of PCPs.
Materials and methods
Animals and Chemicals
All procedures and animal care were performed in accordance with the protocols approved by Institutional Animal Care and Use Committee of the University of California, Davis. Male Swiss Webster mice were purchased from Charles River Laboratories. 13C-labeled TCC (4′-chlorophenyl-13C6, 99%) used as an internal standard in drug level determination was purchased from Cambridge Isotope Laboratories Inc. (Andover, MA); Acetonitrile and methanol used for liquid chromatography tandem mass spectrometry (LC/MS/MS) were purchased from Fisher Scientific (Pittsburgh, PA). Oleic oil (a mixture of oleic ester rich triglycerides) was purchased from Adams Vegetable Oils, Inc. (Arbuckle, CA). TCC, LPS (Escherichia coli serotype, L4130, 0111:B4), and polyethylene glycol (PEG400) were purchased from Sigma–Aldrich (St. Louis, MO). EDTA (K3) was purchased from Tyco Health Group LP (Mansfield, MA). Water (>18.0 MΩ) was purified by a NANO pure system (Barnstead, Newton, MA). Cytometric Bead Array (CBA) mouse inflammation kit was purchased from BD Biosciences (San Jose, CA). Oxylipin standards were purchased from Cayman Chemical (Ann Arbor, MI). The sEHIs, t-AUCB (trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid) and TPAU (1-trifluoromethoxyphenyl-3-(1-acetylpiperidin-4-yl) urea) were synthesized previously in house (Jones et al., 2006; Hwang et al., 2007).
Measurement of sEH Inhibition
The inhibitory activities of TCC against recombinant human and murine sEHs shown in Table 1 were measured by using the fluorescent cyano(6-methoxy-naphthalen-2-yl)methyl trans-[(3-phenyloxyran-2-yl)methyl] carbonate (CMNPC) (Jones et al., 2005) or racemic [3H]-trans-1,3-diphenylpropene oxide (t-DPPO) (Borhan et al., 1995) as substrate. Results are presented as the average ± standard deviation (SD) of three separate measurements.
Kinetic studies
Dissociation constants (Ki) were determined following a method (Dixon, 1972) for competitive tight binding inhibitors, using [3H]-1,3-diphenyl-trans-propene oxide (t-DPPO) as substrate (Borhan et al., 1995). Inhibitor concentrations between 0 and 500 nM for TCC were incubated in triplicate for 5 min in pH 7.4 sodium phosphate buffer at 30 °C with 100 μL of the enzyme (1 nM of human sEH). Substrate (7.5 ≤ [S]final ≤ 30 μM) was then added. Velocity was measured as described (Borhan et al., 1995). For each substrate concentration, the plots of the velocity as a function of the inhibitor concentration allowed the determination of an apparent inhibition constant (KIapp) (Dixon, 1972). The KIapp was calculated by non-linear fitting of the Dixon’s equation to the obtained results using Sigma Plot version 11.0 (Systat Software Inc.; Chicago, IL). The linear plot of these KIapp as a function of the substrate concentration allows the determination of KI when [S] = 0. Results are presented as average ± standard error of KI calculation.
Molecular modelling
Molecular modelling was performed using “Scigress Explorer Standard ver. 7.7.0.49” software (Fujitsu Computer Systems Corporation). The atomic coordinates of the crystal structure of human sEH complex with CIU (N-cyclohexyl-N′-(4-iodophenyl)urea) were retrieved from Protein Data Bank (entry 1VJ5) (Gomez et al., 2004). TCC was docked into the ligand-binding pocket manually by superposition with the parent molecule (CIU)) and minimized on MM geometry (MM2). The image was produced using freewares VMD 1.8.6 (www.ks.uiuc.edu/Research/vmd) and POV-Ray 3.6 (www.povray.org).
Anti-hypotensive studies
LPS (4 mg) was dissolved in saline (2 mL) to give an opaque solution after strong mixing for 1 min. TCC (2 mg) was dissolved in oleic acid rich triglycerides containing 10% PEG400 (v/v) and 10% ethanol (v/v) (2 mL) to give a clear solution after strong mixing for 3-h. Specifically, 16 male Swiss Webster mice (8 weeks) were randomly assigned to 4 equal groups (N = 4). Mice in group 1 were injected intraperitoneally (ip) with LPS saline solution at a dose of 10 mg/kg and immediately treated with the vehicle solution per oral (po) as a positive control. Mice in group 2 were ip injected with LPS saline solution at the same dose as group 1 and immediately administered with TCC (po) at a dose of 5 mg/kg. Mice in group 3 were ip injected with saline and treated with vehicle solution (po) as a negative control. The mice in group 4 were received saline (ip) and TCC (20 mg/kg, po) to investigate the possible toxicity of TCC at high dose. The systolic blood pressure was measured with the method described previously (Liu et al., 2009a) to characterize the anti-hypotensive effect of TCC. Systolic blood pressure was determined before, 8- and 24- h after treatment with non-invasive tail cuff methods using a CODA™-6 system (Kent Scientific Co., CT). The instrument CODA™-6 was set up in the animal room where the mice were housed to measure blood pressure. For each measurement set, 3 acclimation cycles and 25 data cycles were used. The reported systolic blood is presented as a mean of at least 5 cycles of the measurement set. If the systolic blood pressure was under the detection limit, we recorded it as the detection limit (60 mmHg) for further analysis. The heart rate and tail volume were also recorded.
Anti-inflammatory studies
Swiss Webster mice (8 weeks old, 36 males) were assigned randomly to 6 equal groups (N=6) for the treatments described in Table 2. LPS and TCC were formulated and treated as same as those in the anti-hypotensive studies. Animals were sacrificed at 8- or 24-h after the treatments indicated in the Table 2. Blood was collected and the plasma was separated as in the previously described method (Liu et al., 2009b). Plasma was stored in 3 separate eppendorf tubes for analysis of cytokine, drug level, and oxylipin. Tissue was removed and immediately frozen with liquid nitrogen. All the samples were stored at -80°C until analysis.
Table 2.
Murine plasma level of key oxylipin mediators, cytokines and chemokine (MCP-1) in current study
| Group | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| LPS (mg/kg) | - | 10 | 10 | 10 | 10 | - |
| TCC (mg/kg) | - | - | - | 5 | 5 | 20 |
| Treatment time (h) | 24 | 8 | 24 | 8 | 24 | 24 |
| 6-keto-PGF1a (nM) | 2.2 ± 0.9 | 12.6 ± 2.8* a | 6.6 ± 2.6* a | 11.9 ± 2.9* a | 4.4 ± 1.2 a | 3.6 ± 1.1 a |
| TXB2 (nM) | 0.8 ± 0.9 | 2.7 ± 1.0 b | 3.9 ± 2.0* a | 5.1 ± 2.9* b | 3.0 ± 2.6 a | 2.0 ± 1.7 a |
| PGE2 (nM) | 0.05 ± 0.02 | 0.20 ± 0.08* a | 0.16 ± 0.02* a | 0.20 ± 0.08* a | 0.11 ± 0.04‡ a | 0.08 ± 0.06 a |
| PGD2 (nM) | 0.25 ± 0.07 | 1.0 ± 0.2* a | 0.79 ± 0.24* b | 0.77 ± 0.18* a | 0.52 ± 0.13* b | 0.29 ± 0.07 a |
| 14,15-DHET (nM) | 2.8 ± 0.5 | 4.3 ± 2.4 b | 1.0 ± 0.1* a | 3.0 ± 0.6 b | 1.0 ± 0.1* a | 1.9 ± 0.6 a |
| 11,12-DHET (nM) | 1.8 ± 0.4 | 2.0 ± 1.4 a | 0.5 ± 0.1* a | 1.9 ± 0.3 a | 0.5 ± 0.1* | 1.1 ± 0.4 a |
| 8,9-DHET (nM) | 1.4 ± 0.3 | 1.5 ± 0.8 a | 0.5 ± 0.1* a | 1.4 ± 0.2 a | 0.6 ± 0.1* | 1.1 ± 0.3 a |
| 5,6-DHET (nM) | 1.4 ±0.2 | 1.0 ± 0.2 a | 0.5 ± 0.1* b | 1.3 ± 0.2 a | 0.7 ± 0.1* b | 1.2 ± 0.2 a |
| 15-HETE (nM) | 5.6 ± 2.4 | 4.7 ± 1.6 a | 3.1 ± 0.9 a | 5.6 ± 1.0 a | 2.9 ± 0.8 a | 5.0 ± 2.0 a |
| 11-HETE (nM) | 2.5 ± 0.8 | 2.7 ± 1.1 a | 1.8 ± 0.6 a | 3.0 ± 0.6 a | 1.7 ± 0.4 a | 2.0 ± 0.5 a |
| 12-HETE (nM) | 300 ±110 | 410 ± 175 b | 435 ± 165 a | 615 ± 280 b | 205 ± 145 a | 250 ± 190 a |
| 8-HETE (nM) | 6.0 ± 4.1 | 4.5 ± 1.4 b | 3.6 ± 0.5 a | 6.4 ± 1.4 b | 3.4 ± 1.2 a | 5.0 ± 3.2 a |
| 9-HETE (nM) | 6.0 ± 4.5 | 6.8 ± 2.3 a | 3.4 ± 0.6 a | 5.6 ± 2.3 a | 5.4 ± 2.2 a | 5.5 ± 1.9 a |
| 5-HETE (nM) | 5.3 ± 1.0 | 3.8 ± 1.4 a | 2.4 ± 0.3* a | 4.6 ± 1.0 a | 3.1 ± 1.1* a | 1.1 ± 1.9* a |
| 14,15-EET (nM) | 3.2 ± 1.5 | 1.3 ± 0.3* a | 0.7 ± 0.1* b | 1.8 ± 0.4 a | 1.0 ± 0.3* b | 1.9 ± 0.5 a |
| 11,12-EET (nM) | 2.1 ± 1.0 | 1.1 ± 0.5 b | 0.9 ± 0.1* a | 1.7 ± 0.2 b | 1.1 ± 0.2 a | 1.7 ± 0.5 a |
| 8,9-EET (nM) | 1.6 ± 1.0 | 0.4 ± 0.2* b | 0.1± 0.0* a | 0.9 ± 0.2† b | 0.5 ± 0.1‡ a | 1.1 ± 0.4 a |
| 5,6-EET (nM) | 8.3 ± 3.9 | 3.7 ± 2.6 a | 2.9 ± 0.6* a | 6.6 ± 1.1 a | 4.0 ± 1.2 a | 7.1 ± 2.9 a |
| TNFα (pg/mL) | 16 ± 2 | 200 ± 40* b | 60 ± 10*, † a | 175 ± 75* b | 30 ± 10*, ‡, § a | 17 ± 5 a |
| IL-6 (pg/mL) | 11 ± 3 | 4210 ± 775* b | 190 ± 45*, † a | 4090 ± 2460* b | 65 ± 30*, ‡, § a | 9 ± 3 a |
| MCP-1(pg/mL) | 80 ± 15 | 11900 ± 1330* b | 3240 ± 715*, † a | 12400 ±1285* b | 2080 ± 425*,§ a | 85 ± 35 a |
| IFN-γ (pg/mL) | 6 ± 2 | 2805 ± 755* a | 9 ± 2† a | 1820 ± 540* a | 10 ±5§ a | 6 ± 2 a |
Data are presented as mean ± SD (N=6).
Significantly different (P < 0.05) from group 10,
significantly different (P < 0.05) from group 2,
significantly different from Group 3, and
significantly different (P < 0.05) from Group 4 were determined by ANOVA followed with Tukey’s a or Games-Howell’s b test.
Plasma cytokine levels were analyzed using a CBA mouse inflammation kit. Briefly, thawed plasma samples (30 μL each) were mixed for 2 hours at room temperature with florescence-labelled capture beads with the PE detection reagents to measure the concentrations of interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor-α (TNF-α) and Interferon-gamma (IFN-γ). Samples were then washed with washing buffer and analyzed on a FACScan flow cytometer (BD Immunocytometry Systems); Data were analyzed using BD CBA Analysis software (BD Immunocytometry Systems).
Measurement of oxylipins in plasma
Thawed plasma (200 μL) was extracted for oxylipin analysis as previously described (Liu et al., 2009b) and was analyzed by LC/MS/MS as described (Yang et al., 2009). Epoxides such as epoxyeicosatrienoic acids (EETs) and their corresponding sEH-mediated metabolites diols such as dihydroxyeicosatrienoic acids (DHETs) were quantified to evaluate the in vivo action of sEH.
Measurement of plasma level of TCC
TCC was extracted from the plasma samples by a small modification of the previous method (Liu et al., 2009b). Specifically, surrogate solution (10 uL of 500 nM TCC-13C6 in MeOH) was added to each thawed plasma sample. After mixing on a Vortex mixer for 30 s, ethyl acetate (200 μL) was added to each sample. Samples were mixed on a Vortex mixer for 1 min followed by centrifugation at 20,817 × g. for 5 min. The supernatant was transferred to a new tube and the residue was extracted with a second 200 μL of ethyl acetate. The supernatants were then combined and the mixture was evaporated to dryness using a centrifugal vacuum concentrator. Finally, 50 μL of internal standard solution [200 nM of 12-(3-cyclohexan-1-yl-ureido)-dodecanoic acid (CUDA) in methanol] was used to reconstitute the residue for high-performance liquid chromatography-tandem mass analysis.
Analysis of data
Data are presented as mean ± SD unless noted. Statistic analyses were performed by ANOVA followed with Tukey’s (variance homogeneity) or Games-Howell’s (variance heterogeneity) test using the software SPSS 10.0 (SPSS Inc., Chicago, IL) with P < 0.05 as the significant level.
Results
In vitro inhibitory activity of TCC against sEH
As shown in Table 1 for the murine sEH, TCC is roughly 100-fold less potent than trans-4-(4-(3-adamantan-1-yl-ureido)-cyclohexyloxy)-benzoic acid (t-AUCB) and 10-fold less potent than 1-trifluoromethoxyphenyl-3-(1-acetylpiperidin-4-yl) urea (TPAU). However, for the human sEH the difference in potency is greatly reduced; TCC has the same potency as TPAU and is only about 6-fold less potent than t-AUCB. In addition, to better define the potency of TCC against human sEH, we determined its Ki (Figure 1) using a radioactivity-based assay. TCC has a Ki of 96 nM, which is 60-fold less potent than that of t-AUCB, but close to that of TPAU (Liu et al., 2009a).
Figure 1.
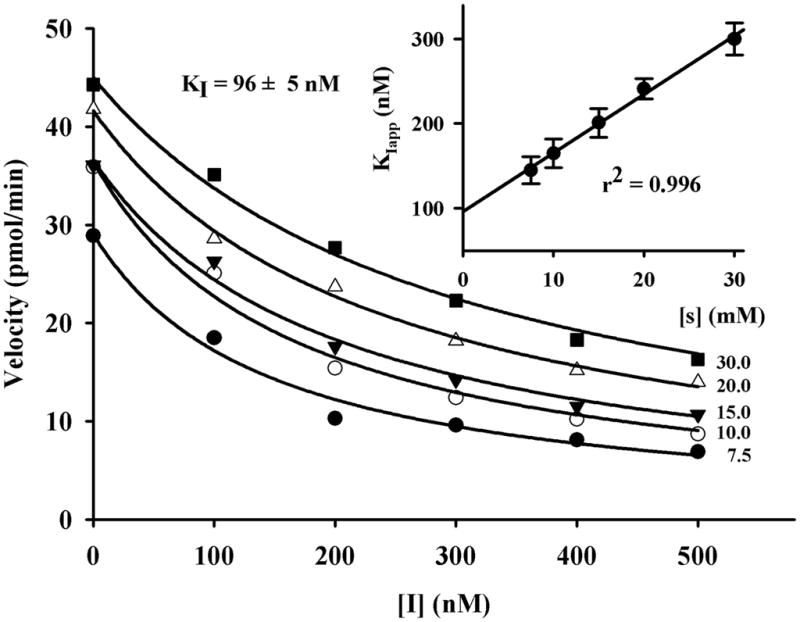
Determination of the KI of TCC with the human sEH (1 nM) using [3H]-trans-1,3-diphenyl-trans-propene oxide ([3H]-t-DPPO) as substrate (7.5 ≤ [S]final ≤ 30 μM). For [S] = 0, a Ki value of 96 nM was found.
In order to understand how TCC binds to this enzyme, the structure of TCC was manually docked at the active site of the human sEH using an X-ray crystal structure of human sEH complex with the urea-based ligand (CIU) (Gomez et al., 2004; Gomez et al., 2006). All plausible binding modes for TCC bound well at the active site of the enzyme. Among the plausible binding modes for TCC, the orientation given in Figure 2 was the most stable (ΔE ranging from 0.04 to 6.74 kcal mol−1), and thus it is the most probable. As seen in Figure 2, TCC was modelled to bind to sEH primarily through the interactions of its urea pharmacophore with Tyr381, Tyr465 and Asp333 as expected from a potent sEHI.
Figure 2.
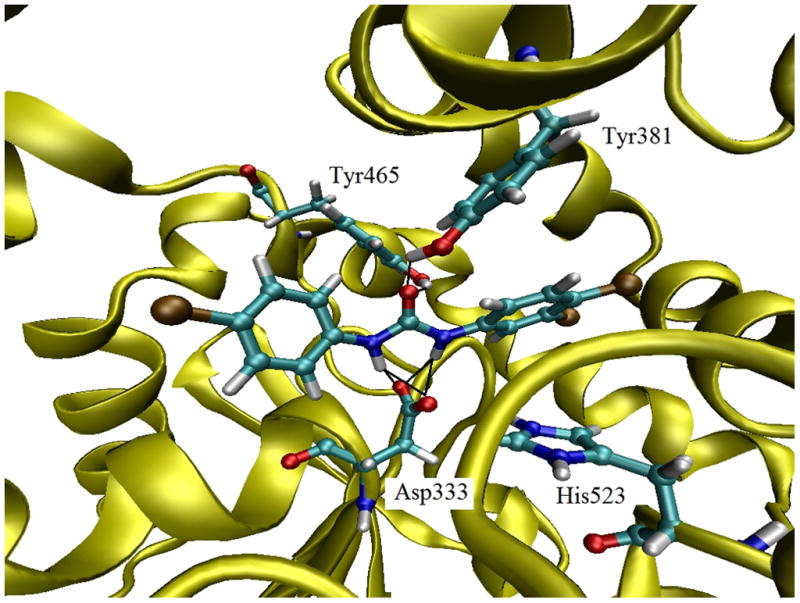
Docking TCC with the human sEH enzyme. TCC was manually docked into the active site of human sEH. The amino acids are shown as ball and stick structures on the backbone ribbon diagram.
TCC reverses LPS-challenged hypotension
Hypotension resulting from LPS-challenge is a hallmark of septic shock (Weinberg et al., 1992). The anti-hypotensive effect of TCC is illustrated in Figure 3. Specifically, the systolic blood pressure dramatically fell below the detection limit of the equipment (60 mmHg) 8-h and 24-h after LPS dosing. Oral administration of TCC at a dose of 5 mg/kg significantly reversed LPS-induced hypotension at 24-h after treatment while it did not show a detectable effect on blood pressure 8-h after treatment. On the other hand, TCC (20 mg/kg, po) did not change the systolic blood pressure of control mice either 8- or 24-h after treatment (Liu et al., 2009b). In addition to the significant alteration in systolic blood pressure, LPS administration slightly increased the heartbeat of mice (790 ± 80 vs. 880 ± 120 beat·min−1, p = 0.22) and significantly decreased the tail volume (65 ± 20 μL vs. 30 ±12 μL, p = 0.01), and administration of TCC reversed the decreased tail volume from LPS-challenged towards normal level at 8- and 24-h after treatment. Tail volume is the total volume going in and out of the tail, essentially based on the amount of blood moving through the tail at each measurement cycle.
Figure 3.
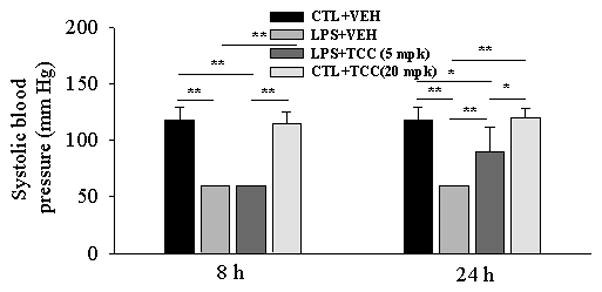
The anti-hypotensive effect of TCC in a LPS-challenged murine model. LPS (10 mg/kg) and TCC (5 mg/kg) were administered to male Swiss-Webster mice (8-week old) with i.p. injection and oral gavage, respectively. Systolic blood pressure was recorded 8-h and 24-h after treatment with a non-invasive tail cuff method. Data represent mean ± SD of 4 mice. If the systolic blood pressure was under the detection limit of instrument (60 mmHg), the value was recorded as 60 mmHg. Data represent mean ± SD of 6 mice. Animals were sacrificed 8 and 24-h after treatment, respectively. Control (CTL) was saline, and vehicle (VEH) was oleic acid rich triglycerides containing 10% PEG400 (v/v) and 10% ethanol (v/v); mpk: mg/kg. Statistically significant differences (*P<0.05, **P<0.01)) were determined by ANOVA followed with Tukey’s.
TCC suppresses the production of cytokines and a chemokine
Table 2 presents the plasma levels of the cytokines (TNF-α, IL-6, IFN-γ) and a chemokine (MCP-1). Table 2 shows the effects of TCC on the release of TNF-α and IL-6, respectively. After LPS challenge, the plasma levels of TNF-α and IL-6 dramatically increased 8- and 24-h after treatment. TCC (5 mg/kg) significantly suppressed the plasma levels of TNF-α and IL-6 at 24-h after treatment while it had poor effects at 8-h after treatment, which correlates with the anti-inflammatory effect of TCC. On the other hand, TCC (20 mg/kg, po) did not alter the release of detected cytokines and a chemokine in control mice 24-h after treatment (Liu et al., 2009b).
Plasma level of TCC
The plasma concentration of TCC at 8- and 24-h after treatment was 400 ± 100 nM and 200 ± 15 nM, respectively, in the LPS-challenged mice orally treated with 5 mg/kg of TCC. The plasma TCC level at 8 h after treatment is significantly higher than those at 24-h after treatment. The plasma levels of TCC at 24-h after treatment were 540 ± 180 nM in the mice orally treated with 20 mg/kg of TCC.
TCC shifts oxylipin profile like other sEHIs in vivo
The efficacy of TCC in vivo as a sEHI or target engagement was evaluated by the plasma levels of total EETs and DHETs, as well as the plasma ratio of EETs to DHETs (Figure 4). Specifically, upon LPS challenge, plasma levels of EETs dramatically decreased after 8- and 24-h compared to the normal control. The plasma level of DHETs dramatically decreased 24-h after LPS treatment only in comparison with the normal control. These changes resulted in a significant decrease in the plasma ratio of EETs to DHETs at 8- and 24-h after treatment in comparison with the vehicle control. When compared with the LPS group, treatment with TCC (5 mg/kg) significantly increased the plasma levels of EETs at 8- and 24-h after treatment, while it had little effect on plasma level of DHETs 8- and 24-h after treatment, resulting in significant increase in the ratio of EETs to DHETs at 8-h and 24-h after treatment. This reversal of the expected increase in the ratio of EETs to DHETs has been observed in the same animal model for several sEHIs (Liu et al., 2009b). However, without LPS challenge TCC (20 mg/kg) only slightly changes the plasma levels of EETs and DHETs, as well as the plasma ratio of EETs to DHETs (Table 2).
Figure 4.

Administration of TCC alters the plasma oxylipins levels of EETs (A) and DHETs (B) in vivo, resulting in an alteration in the ratio of EETs to DHETs (C). LPS and TCC were administered to male Swiss-Webster mice (8-week old) with ip injection and oral gavage, respectively. Data represent mean ± SD of 6 mice. The total EETs column sums 14, 15-, 11, 12-, 8, 9- and 5, 6-EETs and their corresponding diols compose the total DHETs column. Control (CTL) and vehicle (VEH) are the same as those in Figure 3. The full oxylipin data are presented in Table 2. Statistically significant differences (*P < 0.05, **P < 0.01) were determined by ANOVA followed with Tukey’s test.
Discussion
TCC can inhibit human and murine recombinant sEH in vitro (Morisseau et al., 1999; McElroy et al., 2003; Morisseau et al., 2009). Such sEHIs have therapeutic utility for the treatment of inflammation, cardiovascular diseases, and nephrotoxicity in multiple animal models (Jung et al., 2005; Larsen et al., 2006b; Morisseau et al., 2006; Schmelzer et al., 2006; Xu et al., 2006; Parrish et al., 2008; Liu et al., 2010). In addition, sEHIs have been moved into clinical trials for treatment of diabetes and hypertension (Whitcomb et al., 2009). As shown in Table 1, in vitro potency of TCC approaches the one of some of the more potent sEHIs known for the latter enzyme. In addition, the KIs obtained support the IC50s results that TCC has a similar potency to TPAU for the human sEH but it is less potent than t-AUCB. Thus it seems pivotal to investigate the potential in vivo effects of TCC as a sEHI. Because TCC is 10-fold less potent than TPAU for the mouse sEH, this antimicrobial could be less effective in perturbing sEH activity in a murine model than in human. Thus, the effects of TCC on human health through sEH inhibition could be expected to be more severe than ones observed in mice.
Here, we reported the in vivo effect of TCC in a LPS-challenged murine model of inflammation. TCC at the dose of 5 mg/kg showed a significant anti-hypotensive effect 24-h after treatment. This suggests TCC reverses the LPS-induced hypotension in a time-dependent manner. In addition, the more potent sEHI t-AUCB (1 mg/kg, po) showed similarly significant anti-hypotensive effects in the same murine model at 24-h post treatment. These observations suggest that anti-hypotensive effect of sEHIs is complex and time dependent. In addition to the significant alteration in systolic blood pressure, while LPS administration did not significantly alter the heartbeat of mice, it significantly deteriorated the vascular function (tail volume dropped significantly). The administration of TCC reversed the decreased blood output from LPS-challenged towards normal level at 8- and 24-h after treatment. These data indicated that LPS-challenge is involved in the physiological process of vascular function, and inhibition of sEH plays a beneficial role in modulation of vascular function. However, in absence of LPS challenge treatement with either TCC (20 mg/kg) or t-AUCB (1 mg/kg) result in no significant change in the systolic blood pressure.
The plasma levels of pro-inflammatory TNF-α (Dinarello, 2000), MCP-1 (Melgarejo et al., 2009), anti-inflammatory IFN-γ (Schroder et al., 2004) and pleiotropic IL-6 (Ludat et al., 1994; Xing et al., 1998) at 8-h after LPS treatment are dramatically higher than those of the control mice and those at 24-h after LPS treatment. The plasma cytokine levels at 24-h after LPS treatment are significantly higher than control mice levels. This suggests that the LPS-challenge elicited inflammation in a time-dependent process that is more intense at 8-h than 24-h post treatment. In this LPS-challenged murine model, TCC significantly decreased the release of cytokines and a chemokine 24-h after treatment while it had no significant effect at 8-h after treatment, also showing a time-dependence for the anti-inflammatory effect of TCC. In addition, in a separate study, t-AUCB (1 mg/kg) also significantly reversed the LPS-induced increased plasma levels of TNF-α and IL-6 to 20 ± 6 and 90 ± 40 pg/mL, respectively at 24-h after treatment. Thus a 1 mg/kg dose of t-AUCB altered cytokines to a similar degree to the 5 mg/kg dose of TCC. This indicates that t-AUCB is more potent than TCC in this anti-inflammatory animal model, reflecting their respective in vitro potency (Table 1). On the other hand, in absence of LPS challenge, neither TCC (20 mg/kg) nor t-AUCB (1 mg/kg) dramatically altered the plasma cytokines and chemokines tested (Table 2). This is consistent with the observations from the anti-hypotensive studies (Figure 3).
LPS-challenge significantly decreased the production of EETs in plasma, resulting in a significant decrease in the plasma ratio of EETs to DHETs, a biomarker of sEH action. TCC reversed the LPS-induced decreases in both the level of EETs and the ratio of EETs to DHETs (Figure 4). These effects on blood pressure, release of cytokines and chemokines, EETs level and plasma ratio of EETs to DHETs from TCC administration are similar to those from the treatment of previous sEHI t-AUCB (1 mg/kg) in the same model (Liu et al., 2009a). These data also suggest that inhibition of sEH by TCC does contribute to the anti-hypotensive and anti-inflammatory effect observed. Furthermore, TCC at 8-h after treatment with the oral dose of 5 mg/kg reversed the significant decrease in EET levels and the plasma ratio of EETs to DHETs caused by LPS administration while no significant beneficial effect was observed in hypotension and cytokine release. However, all the beneficial effects expected from a sEH inhibitor could be seen at 24-h after treatment. These data also suggest that the TCC acts on sEH enzyme resulting in the alteration in EETs earlier than its regulation of blood pressure and the release of cytokines and chemokine. An alternate hypothesis which might be expected given the physical properties of TCC is that blood levels do not reflect the tissue levels at the sites of action. In addition, when administered to normal mice, TCC (20 mg/kg) did not significantly affect the plasma levels of EETs, DHETs and the ratio of EETs to DHETs, while t-AUCB (1 mg/kg) significantly decreased the plasma level of DHETs, did not change the plasma levels of EETs, resulting in a significantly increases the ratio of EET/DHET. This is consistent with the greater potency of t-AUCB on sEH. However, in addition to sEH inhibition, other characteristics of TCC may also contribute to the in vivo anti-inflammatory effect of TCC. This possibility needs further study.
The plasma levels of TCC are above its murine IC50 value at 8-h but below its murine IC50 value at 24-h after treatment, however the dramatic reverse of LPS induced symptoms was far greater at 24-h after treatment. These observations indicate that the TCC action in the current model is a time-dependent process. This suggests that the drug action is not simply correlated with the drug level in circulation however adequate blood levels of TCC are needed to trigger the cascade of events leading to the reduction of sepsis-induced hypotension.
A previous percutaneous study in human showed that blood levels of TCC were below the detection limit of 80 nM (25 ppb) in the test samples (Howes and Black, 1976), however, this detection limit is much higher than its in vitro IC50 against human sEH. In another human study, oral administration of TCC at a low dose of 2.2 μmol/kg (~0.7 mg/kg) resulted in a plasma level of 3.7 nmol/g, almost 280 fold higher than its in vitro IC50 at 2.8-h after treatment (Hiles and Birch, 1978). A recent study showed that percutaneous use of TCC-containing soap bar resulted in a blood level of TCC higher than its in vitro IC50 for over 30 hours (Figure S1). This underlines the urgency of investigation of TCC exposure. In this study, a significant beneficial effects of TCC expected from its sEHI property was observed at 24-h after treatment, which provides a direct linkage between use of TCC-containing PCPs and its effects expected as a sEHI. These data indicate the possibility that endogenous EET concentrations can be elevated in humans through use of TCC in PCPs. Generally, increased blood levels of EETs seem beneficial to reduce inflammation, and other deleterious symptoms due to the beneficial function of the endogenous EETs. These levels are more likely to have a local dermal effect than a general systemic effect. However, caution should be paid to the unanticipated side effects due to the increase in EETs and other epoxy fatty acids. For example, EETs has been found to promote endothelial cell proliferation, migration, and angiogenesis (Michaelis et al., 2005; Pozzi et al., 2005; Wang et al., 2005; Michaelis and Fleming, 2006; Webler et al., 2008; Yan et al., 2008), which have been associated with various cardiovascular diseases and cancers.
In summary, both in vitro and in vivo studies demonstrated the properties expected from a sEH inhibitor for the antimicrobial TCC, and this inhibition contributes to the anti-inflammatory effect of TCC. This study not only confirms the pharmacological effect of a sEH inhibitor in vivo, but also discloses a novel pharmacological action of the common antimicrobial agent TCC that is present at high concentration in many PCPs. Based on our study and literature data on TCC exposure from environmental contamination, we conclude that systemic inhibition of the sEH by TCC at levels which could lead to significant alterations in the immune system in a normal human is unlikely. However, the use of PCPs with TCC added as an antimicrobial could lead to significant sEH inhibition and the resulting downstream biological effects. These effects are anticipated to be greatest near the site of TCC application. Some individuals (e. g. dental and dermal inflammation, as well as other inflammatory diseases) may benefit from use of TCC-containing PCPs due to its sEH inhibitory potency. Although local reduction of inflammation is often beneficial to human health, use of TCC in PCPs could result in unwanted local effects. Given the environmental persistence of TCC, possibly the benefits and risks of the use of this common anti-microbial agent should be re-evaluated with its use moving to more high value and limited specific applications where it is beneficial to reduce both microbial density and inflammation.
Supplementary Material
Acknowledgments
This work was supported in part by NIEHS grant R01 ES002710, NIEHS Superfund P42 ES004699, and NIH R01 HL085727. BDH is a George and Judy Marcus Senior Fellow of the American Asthma Foundation.
Abbreviations
- APAU
1-(1-acetypiperidin-4-yl)-3-adamantanylurea
- t-AUCB
trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid
- CBA
Cytometric Bead Array
- CIU
N-cyclohexyl-N′-(4-iodophenyl)urea
- COX
cyclooxygenase
- CUDA
12-(3-cyclohexan-1-yl-ureido)-dodecanoic acid
- DHET
dihydroxyeicosatrienoic acid
- t-DPPO
[3H]-trans-1,3-diphenyl-trans-propene oxide
- EET
epoxyeicosatrienoic acid
- IFN-γ
Interferon-gamma
- IL-6
interleukin-6
- ip
intraperitoneally
- KI
dissociation constant
- KIapp
apparent inhibition constant
- LC/MS/MS
liquid chromatography/tandem mass spectrometry
- LOX
lipoxygenase
- LPS
lipopolysaccharide
- MCP-1
monocyte chemoattractant protien-1
- PCP
personal care products
- po
per oral
- sEH
soluble epoxide hydrolase
- sEHI
inhibitor of soluble epoxide hydrolase
- TCC
triclocarban
- TNF-α
tumor necrosis factor-alpha
- TPAU
1-trifluoromethoxyphenyl-3-(1-acetylpiperidin-4-yl) urea
Footnotes
Conflict of Interest
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the National Institute of Health, National Institute of Environmental Health Sciences, or American Asthma Foundation. CM, SHH and BDH are inventors of composition of matter and use patents in the area of soluble epoxide hydrolase inhibitors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn KC, Zhao B, Chen J, Cherednichenko G, Sanmarti E, Denison MS, Lasley B, Pessah IN, Kultz D, Chang DPY, Gee SJ, Hammock BD. In vitro biologic activities of the antimicrobials triclocarban, its analogs, and triclosan in bioassay screens: Receptor-based bioassay screens. Environ Health Persp. 2008;116:1203–1210. doi: 10.1289/ehp.11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch CG, Hiles RA, Eichhold TH, Jeffcoat AR, Handy RW, Hill JM, Willis SL, Hess TR, Wall ME. Biotransformation Products of 3,4,4′-Trichlorocarbanilide in Rat, Monkey, and Man. Drug Metab Dispos. 1978;6:169–176. [PubMed] [Google Scholar]
- Black JG, Howes D, Rutherford T. Skin Deposition and Penetration of Trichlorocarbanilide. Toxicology. 1975;3:253–264. doi: 10.1016/0300-483x(75)90089-x. [DOI] [PubMed] [Google Scholar]
- Borhan B, Mebrahtu T, Nazarian S, Kurth MJ, Hammock BD. Improved radiolabeled substrates for soluble epoxide hydrolase. Analytical biochemistry. 1995;231:188–200. doi: 10.1006/abio.1995.1520. [DOI] [PubMed] [Google Scholar]
- Chen JG, Ahn KC, Gee NA, Ahmed MI, Duleba AJ, Zhao L, Gee SJ, Hammock BD, Lasley BL. Triclocarban enhances testosterone action: A new type of endocrine disruptor? Endocrinology. 2008;149:1173–1179. doi: 10.1210/en.2007-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetrulias J, Corbin N, Northroot H. The Hairless Mouse as a Model for Quantitating Skin Deposition of 3,4,4′-Trichlorocarbanilide in Bar Soap. Toxicol Lett. 1984;22:241–248. doi: 10.1016/0378-4274(84)90073-0. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran A, Al-Saghir R, Lopez B, Zhu DL, Gutterman DD, Jacobs ER, Medhora M. Protective effects of epoxyeicosatrienoic acids on human endothelial cells from the pulmonary and coronary vasculature. Am J Physiol-Heart C. 2006;291:H517–H531. doi: 10.1152/ajpheart.00953.2005. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–508. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- Dixon M. Graphical Determination of Km and Ki. Biochem J. 1972;129:197. doi: 10.1042/bj1290197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gledhill WE. Biodegradation of 3,4,4′-Trichlorocarbanilide, Tcc, in Sewage and Activated-Sludge. Water Res. 1975;9:649–654. [Google Scholar]
- Gomez GA, Morisseau C, Hammock BD, Christianson DW. Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry-Us. 2004;43:4716–4723. doi: 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- Gomez GA, Morisseau C, Hammock BD, Christianson DW. Human soluble epoxide hydrolase: Structural basis of inhibition by 4-(3-cyclohexylureido)-carboxylic acids. Protein Sci. 2006;15:58–64. doi: 10.1110/ps.051720206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halden RU, Paull DH. Co-occurrence of triclocarban and triclosan in US water resources. Environ Sci Technol. 2005;39:1420–1426. doi: 10.1021/es049071e. [DOI] [PubMed] [Google Scholar]
- Heidler J, Sapkota A, Halden RU. Partitioning, persistence, and accumulation in digested sludge of the topical antiseptic triclocarban during wastewater treatment. Environ Sci Technol. 2006;40:3634–3639. doi: 10.1021/es052245n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiles RA, Birch CG. Absorption, Excretion, and Biotransformation of 3,4,4′-Trichlorocarbanilide in Humans. Drug Metab Dispos. 1978;6:177–183. [PubMed] [Google Scholar]
- Hiles RA, Caudill D, Birch CG, Eichhold T. Metabolism and Disposition of 3,4,4′-Trichlorocarbanilide in the Intact and Bile Duct-Cannulated Adult and in the Newborn Rhesus-Monkey (M Mulatta) Toxicol Appl Pharm. 1978;46:593–608. doi: 10.1016/0041-008x(78)90306-x. [DOI] [PubMed] [Google Scholar]
- Howes D, Black JG. Percutaneous Absorption of Triclocarban in Rat and Man. Toxicology. 1976;6:67–76. doi: 10.1016/0300-483x(76)90008-1. [DOI] [PubMed] [Google Scholar]
- Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffcoat AR, Handy RW, Francis MT, Willis S, Wall ME, Birch CG, Hiles RA. Metabolism and Toxicity of Halogenated Carbanilides - Biliary Metabolites of 3,4,4′-Trichlorocarbanilide and 3-Trifluoromethyl-4,4′-Dichlorocarbanilide in Rat. Drug Metab Dispos. 1977;5:157–166. [PubMed] [Google Scholar]
- Jones PD, Tsai HJ, Do ZN, Morisseau C, Hammock BD. Synthesis and SAR of conformationally restricted inhibitors of soluble epoxide hydrolase. Bioorg Med Chem Lett. 2006;16:5212–5216. doi: 10.1016/j.bmcl.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Analytical biochemistry. 2005;343:66–75. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45:759–765. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- Larsen BT, Gutterman DD, Hatoum OA. Emerging role of epoxyeicosatrienoic acids in coronary vascular function. Eur J Clin Invest. 2006a;36:293–300. doi: 10.1111/j.1365-2362.2006.01634.x. [DOI] [PubMed] [Google Scholar]
- Larsen BT, Miura H, Hatoum OA, Campbell WB, Hammock BD, Zeldin DC, Falck JR, Gutterman DD. Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BKCa channels: implications for soluble epoxide hydrolase inhibition. Am J Physiol-Heart C. 2006b;290:H491–H499. doi: 10.1152/ajpheart.00927.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Liu JY, Timofeyev V, Qiu H, Hwang SH, Tuteja D, Lu L, Yang J, Mochida H, Low R, Hammock BD, Chiamvimonvat N. Beneficial effects of soluble epoxide hydrolase inhibitors in myocardial infarction model: Insight gained using metabolomic approaches. J Mol Cell Cardiol. 2009;47:835–845. doi: 10.1016/j.yjmcc.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JY, Park SH, Morisseau C, Hwang SH, Hammock BD, Weiss RH. Sorafenib has soluble epoxide hydrolase inhibitory activity, which contributes to its effect profile in vivo. Molecular cancer therapeutics. 2009a;8:2193–2203. doi: 10.1158/1535-7163.MCT-09-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JY, Tsai HJ, Hwang SH, Jones PD, Morisseau C, Hammock BD. Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation. Br J Pharmacol. 2009b;156:284–296. doi: 10.1111/j.1476-5381.2008.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JY, Yang J, Inceoglu B, Qiu H, Ulu A, Hwang SH, Chiamvimonvat N, Hammock BD. Inhibition of soluble epoxide hydrolase enhances the anti-inflammatory effects of aspirin and 5-lipoxygenase activation protein inhibitor in a murine model. Biochemical Pharmacology. 2010;79:880–887. doi: 10.1016/j.bcp.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludat K, Lang J, Gahl GM, Frei U, Jorres A. The Production of Pro-Inflammatory Cytokines (Il-6, Il-8) in Human Peritoneal Fibroblasts Is up-Regulated Following Stimulation with Il-1-Beta and or Tnf-Alpha. J Am Soc Nephrol. 1994;5:463–463. [Google Scholar]
- McElroy NR, Jurs PC, Morisseau C, Hammock BD. QSAR and classification of murine and human soluble epoxide hydrolase inhibition by urea-like compounds. J Med Chem. 2003;46:1066–1080. doi: 10.1021/jm020269o. [DOI] [PubMed] [Google Scholar]
- Melgarejo E, Medina MA, Sanchez-Jimenez F, Urdiales JL. Monocyte chemoattractant protein-1: A key mediator in inflammatory processes. Int J Biochem Cell B. 2009;41:998–1001. doi: 10.1016/j.biocel.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Michaelis UR, Fisslthaler B, Barbosa-Sicard E, Falck JR, Fleming I, Busse R. Cytochrome P450 epoxygenases 2C8 and 2C9 are implicated in hypoxia-induced endothelial cell migration and angiogenesis. J Cell Sci. 2005;118:5489–5498. doi: 10.1242/jcs.02674. [DOI] [PubMed] [Google Scholar]
- Michaelis UR, Fleming I. From endothelium-derived hyperpolarizing factor (EDHF) to angiogenesis: Epoxyeicosatrienoic acids (EETs) and cell signaling. Pharmacol Therapeut. 2006;111:584–595. doi: 10.1016/j.pharmthera.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. P Natl Acad Sci USA. 1999;96:8849–8854. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annual review of pharmacology and toxicology. 2005;45:311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- Morisseau C, Merzlikin O, Lin A, He G, Feng W, Padilla I, Denison MS, Pessah IN, Hammock BD. Toxicology in the Fast Lane: Application of High-Throughput Bioassays to Detect Modulation of Key Enzymes and Receptors. Environ Health Persp. 2009;117:1867–1872. doi: 10.1289/ehp.0900834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Schmelzer K, Pedersen T, Eiserich J, Pinkerton K, Imig J, Hammock B. Physiological roles of soluble epoxide hydrolase and therapeutic prospects of its inhibition. Faseb J. 2006;20:A669–A669. [Google Scholar]
- Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Progress in lipid research. 2005;44:1–51. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Nithipatikom K, Moore JM, Isbell MA, Falck JR, Gross GJ. Epoxyeicosatrienoic acids in cardioprotection: ischemic versus reperfusion injury. Am J Physiol-Heart C. 2006;291:H537–H542. doi: 10.1152/ajpheart.00071.2006. [DOI] [PubMed] [Google Scholar]
- Node K, Huo YQ, Ruan XL, Yang BC, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northroot H, Demetrulias J, Wester R, Maibach H, Corbin N. Deposition of 3,4,4′-Trichlorocarbanilide on Human-Skin. Toxicol Lett. 1984;22:235–239. doi: 10.1016/0378-4274(84)90072-9. [DOI] [PubMed] [Google Scholar]
- Parrish AR, Chen G, Burghardt RC, Watanabe T, Morisseau C, Hammock BD. Attenuation of cisplatin nephrotoxicity by inhibition of soluble epoxide hydrolase. Cell biology and toxicology. 2008 doi: 10.1007/s10565-008-9071-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi A, Macias-Perez I, Abair T, Wei SZ, Su Y, Zent R, Falck JR, Capdevila JH. Characterization of 5,6-and 8,9-epoxyeicosatrienoic acids (5,6-and 8,9-EET) as potent in vivo angiogenic lipids. J Biol Chem. 2005;280:27138–27146. doi: 10.1074/jbc.M501730200. [DOI] [PubMed] [Google Scholar]
- Roman DP, Barnett EH, Balske RJ. Cutaneous antiseptic activity of 3,4,4′-trichlorocarbanilide. Proc Scientific Section of Toilet Goods Association. 1957;28:1213–1214. [Google Scholar]
- Scharpf LG, Hill ID, Maibach HI. Percutaneous Penetration and Disposition of Triclocarban in Man - Body Showering. Arch Environ Health. 1975;30:7–14. doi: 10.1080/00039896.1975.10666624. [DOI] [PubMed] [Google Scholar]
- Schebb NH, Inceoglu B, Ahn KC, Morisseau C, Gee SJ, Hammock BD. Investigation of Human Exposure to Triclocarban after Showering and Preliminary Evaluation of Its Biological Effects. Environ Sci Technol. 2011 doi: 10.1021/es103650m. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, Hammock BD. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. P Natl Acad Sci USA. 2006;103:13646–13651. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. P Natl Acad Sci USA. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukocyte Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Seubert JM, Zeldin DC, Nithipatikom K, Gross GJ. Role of epoxyeicosatrienoic acids in protecting the myocardium following ischemia/reperfusion injury. Prostag Oth Lipid M. 2007;82:50–59. doi: 10.1016/j.prostaglandins.2006.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wei X, Xiao X, Hui RT, Card JW, Carey MA, Wang DW, Zeldin DC. Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. J Pharmacol Exp Ther. 2005;314:522–532. doi: 10.1124/jpet.105.083477. [DOI] [PubMed] [Google Scholar]
- Warren JT, Allen R, Carter DE. Identification of Metabolites of Trichlorocarbanilide in Rat. Drug Metab Dispos. 1977;6:38–44. [PubMed] [Google Scholar]
- Webler AC, Michaelis UR, Popp R, Barbosa-Sicard E, Murugan A, Falck JR, Fisslthaler B, Fleming I. Epoxyeicosatrienoic acids are part of the VEGF-activated signaling cascade leading to angiogenesis. Am J Physiol-Cell Ph. 2008;295:C1292–C1301. doi: 10.1152/ajpcell.00230.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg JR, Boyle P, Meager A, Guz A. Lipopolysaccharide, Tumor-Necrosis-Factor, and Interleukin-1 Interact to Cause Hypotension. J Lab Clin Med. 1992;120:205–211. [PubMed] [Google Scholar]
- Whitcomb R, Chen D, Wang YX, Anandan SK, Gless R, Webb H. AR9281, a Soluble Epoxide Hydrolase Inhibitor-Efficacy in a DIO Mouse Model Plus Pharmacokinetics and Pharmacodynamics in Mice and Men. Diabetes. 2009;58:A165–A165. [Google Scholar]
- Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, Achong MK. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–320. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RKP, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. P Natl Acad Sci USA. 2006;103:18733–18738. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan GJ, Chen SP, You B, Sun JX. Activation of sphingosine kinase-1 mediates induction of endothelial cell proliferation and angiogenesis by epoxyeicosatrienoic acids. Cardiovasc Res. 2008;78:308–314. doi: 10.1093/cvr/cvn006. [DOI] [PubMed] [Google Scholar]
- Yang J, Schmelzer K, Georgi K, Hammock BD. Quantitative Profiling Method for Oxylipin Metabolome by Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry. Anal Chem. 2009;81:8085–8093. doi: 10.1021/ac901282n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying GG, Yu XY, Kookana RS. Biological degradation of triclocarban and triclosan in a soil under aerobic and anaerobic conditions and comparison with environmental fate modelling. Environ Pollut. 2007;150:300–305. doi: 10.1016/j.envpol.2007.02.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.