

Abstract
Excitation-contraction coupling involves the faithful conversion of electrical stimuli to mechanical shortening in striated muscle cells, enabled by the ubiquitous second messenger, calcium. Crucial to this process are ryanodine receptors (RyRs), the sentinels of massive intracellular calcium stores contained within the sarcoplasmic reticulum. In response to sarcolemmal depolarization, RyRs release calcium into the cytosol, facilitating mobilization of the myofilaments and enabling cell contraction. In order for the cells to relax, calcium must be rapidly resequestered or extruded from the cytosol. The sustainability of this cycle is crucially dependent upon precise regulation of RyRs by numerous cytosolic metabolites and by proteins within the lumen of the sarcoplasmic reticulum and those directly associated with the receptors in a macromolecular complex. In addition to providing the majority of the calcium necessary for contraction of cardiac and skeletal muscle, RyRs act as molecular switchboards that integrate a multitude of cytosolic signals such as dynamic and steady calcium fluctuations, β-adrenergic stimulation (phosphorylation), nitrosylation and metabolic states, and transduce these signals to the channel pore to release appropriate amounts of calcium. Indeed, dysregulation of calcium release via RyRs is associated with life-threatening diseases in both skeletal and cardiac muscle. In this paper, we briefly review some of the most outstanding structural and functional attributes of RyRs and their mechanism of regulation. Further, we address pathogenic RyR dysfunction implicated in cardiovascular disease and skeletal myopathies.
Introduction
In striated and smooth muscle cells, fluctuations in the intracellular levels of Ca2+ ions greatly determine the magnitude and duration of contractile force. In cardiac and skeletal muscle, depolarization of the external membrane and its invaginations, the T-tubules, elicits swift and massive Ca2+ release from the sarcoplasmic reticulum (SR), which in turn causes 'flooding' of the contractile myofilaments with Ca2+ and induction of contraction. This exquisitely coordinated series of events, in which an electrical stimulus (depolarization) is converted into a mechanical contraction, is collectively termed 'excitation-contraction (EC) coupling', and it has as central players the voltage-dependent Ca2+ channels/dihydropyridine receptors (DHPRs) as the sarcolemmal voltage sensors, and the Ca2+ release channels/ryanodine receptors (RyRs) as the SR Ca2+ release conduits. The structural and functional communication between the voltage sensor and the RyR dictate the magnitude of Ca2+ release from the SR, and thus the force of contraction. In fact, genetic mutations in either of these two proteins, or alterations in the environment that promotes their functional coupling, are known to cause ventricular arrhythmias, hypercontractures and/or pathological remodeling of cellular structures. Excellent reviews on DHPRs have appeared recently [1,2]. In the current review, we focus on RyRs to discuss their most prominent structural and functional attributes, and to suggest mechanisms by which their dysfunction leads to disease.
Structural features of RyRs
RyR isoforms
RyRs are not restricted to striated muscle. This class of intracellular Ca2+ release channels is also found in the endoplasmic reticulum of neurons, exocrine cells, smooth-muscle cells, epithelial cells, lymphocytes, sea-urchin eggs, and many others [3]. In all of these cells, RyRs play a central role in the regulation of the intracellular free Ca2+ concentration ([Ca2+]i), whose elevation triggers a cascade of events that culminates in, for example, neurotransmitter release, hormone secretion, lymphocyte activation and egg fertilization. To gain the functional flexibility necessary to respond to different triggering signals, at least three isoforms of RyRs are expressed in mammals, each one encoded by a distinct gene (ryr1, ryr2 and ryr3), residing on separate chromosomes. RyR1 is expressed predominantly in fast- and slow-twitch skeletal muscle and at lower densities in cerebellar Purkinje cells, gastric smooth muscle and B lymphocytes, among others. RyR2 was originally purified from cardiac muscle (it is the major isoform expressed there), but is also robustly expressed in neurons, and in visceral and arterial smooth muscle. RyR3 is the least understood of the RyR isoforms and seems to play its most important role during development, although in mature cells RyR3 is found in diaphragm, epithelial cells, brain, and smooth muscle [3,4]. There is approximately 65% amino acid sequence identity between mammalian RyR isoforms [5]. Most prominent in sequence variation are three regions of divergence, termed D1, D2 and D3, corresponding to amino acids 4254 to 4631, 1342 to 1403, and 1872 to 1923 in RyR1, respectively to amino acids 4210 to 4562, 1353 to 1397, and 1852 to 1890 in RyR2, respectively. Because RyR2 cannot rescue EC coupling in dyspedic myoblasts (immature muscle cells lacking RyR1) [6], it is likely that these regions of divergence are responsible for the incapacity of rescuing EC coupling with either isoform. Indeed, D2 has been shown to play a crucial role in establishing mechanical coupling between RyR1 and the skeletal DHPR (Cav1.1) [7], but a complete picture of all the structural elements participating in this mode of EC coupling is still lacking. D3, by contrast, contains a low-affinity Ca2+ binding site [8], but whether this site corresponds to a Ca2+ inactivation site that operates in vivo has not been determined.
Molecular architecture of RyRs
With a molecular weight in excess of 2 MDa, RyRs are indisputably the largest known ion channels. Although structural study of RyRs has been challenging because of the channel's large size, some details of RyR structure have been obtained through cry-oelectron microscopy (cryo-EM) [9-13], comparative modeling [14] and recently, x-ray crystallography of small RyR segments [15-17]. In electron micrographs, purified RyRs are seen as quatrefoil or cloverleaf-shaped structures [18-20], or in volume-filled renderings as mushroom-shaped structures, with a large (27 × 27 × 12 nm) cytoplasmic assembly and a smaller transmembrane 'stalk' spanning approximately 6.5 nm from the base of the cytoplasmic region and extending into the SR lumen [21]. The quatrefoil structure results from the symmetrical arrangement of four identical subunits of approximately 5,000 amino acids each, thus a single tetrameric channel encompasses approximately 20,000 amino acids. As if this structural assemblage was not massive enough, an RyR in situ serves as a structural scaffold for other proteins and cofactors that add to its discrete bulkiness and distinctive shape. Indeed, RyRs and associated proteins where first observed as the 'foot' structures that bridge the gap of approximately 12 nm that separates T-tubule and SR membranes in cardiac and skeletal muscle [22].
RyR channel protein assembly
The cytoplasmic assembly of RyRs consists of 15 distinct domains per subunit, including clamp-shaped domains around the periphery of the channel, connected to 'handle' domains surrounding a central cavity [23,24]. Single-particle reconstruction studies indicate that moving between the closed and open states involves conformational changes in the cytoplasmic assembly and in the transmembrane domains, which undergo dilation and constriction around the pore, similar to the iris of a camera diaphragm [25]. Molecular sieving experiments, streaming potential experiments, and electrical distance measurements have suggested a pore approximately 3 Å wide, with a voltage drop of approximately 10.4 Å in length [26]. RyR mapping indicates that the pore of the channel consists of six to eight transmembrane segments [27]. Recent studies using single-particle three-dimensional reconstructions of cryo-EM images of RYR1 at refined resolutions (approximately 10 Å) have enabled a more detailed picture of the secondary structure of the channel pore to be pieced together. Samso et al. [13] suggested that each monomer has at least six transmembrane α-helices, with four inner helices forming a 'tepee' around the central cavity. Further, in the closed state of the channel, the conformation of the inner helices of RyR1 resembles that of the closed KcsA channel. However, Ludtke et al. [28] identified only five helix-like densities lining the pore of the RyR1 channel, which, in its closed state, more closely resembles the structure of open MthK channels rather than closed KcsA channels. The authors conceded that higher resolution was needed for further clarification.
This incongruity was addressed in a later paper [11], in which the authors compared the open and closed conformation of the channel to produce three independent 3D reconstructions, all of which upheld the previous report of six transmembrane helices. Those authors found that relocation of the ion pathway, in which the inner helices bend outward and the inner branches separate slightly, is directly related to an increase of 4 Å in diameter of the pore during channel gating. Further, by superimposing the 3D reconstructions of the open and closed channel, they were able to identify structural hinges that correspond to the previously identified binding sites of regulatory proteins, including calmodulin (CaM) and FK506 (tacrolimus)-binding protein (FKBP)12. Thus, they suggested that the hinges may be the mechanism by which the binding of a small effector is transduced to the pore region.
Two studies have recently solved high-resolution crystal structures (2.5 Å) of portions of the N-terminal region of the skeletal RyR (RyR1) associated with pathogenic mutations. The first study crystallized the N-terminal 210 residues, which were found to adopt a β-trefoil fold, similar to a suppressor domain of inositol trisphosphate receptors (IP3Rs) [15]. The disease mutations investigated in this study clustered in a region within a newly identified domain, highlighting its importance in channel regulation. The authors proposed that the β-trefoil fold, like the analogous structure in IP3Rs, may couple to the C-terminal channel domain to form a suppressor/coupling domain. The second study crystallized a larger construct, comprising N-terminal residues 1 to 559 [17]. This region folds into three distinct domains (arbitrarily termed A, B and C) per monomer (Figure 1), forming a vestibule around the pore of the channel. Fifty pathogenic RyR mutations have been mapped to this region, grouped on the basis of their proposed effect on channel function. Six of the mutations are buried deep within the domains, most probably causing protein misfolding rather than faulty interaction with channel modulators. Significantly, most mutations were mapped to inter-domain and inter-subunit interfaces, thus suggesting that RyR gating is allosterically coupled to the movement of the A, B and C domains, and agreeing well with the hypothesis proposed by Ikemoto et al. [29-31] that disruption of any of the inter-domain interfaces by disease-associated mutations may destabilize closed states, increasing the probability of the channel opening.
Figure 1.
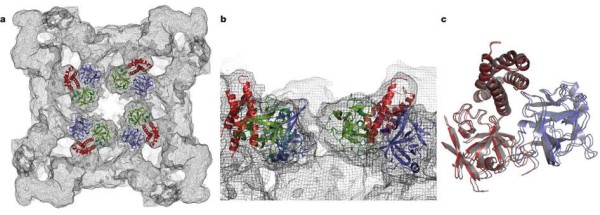
Crystallized structure of rabbit ryanodine receptor (RyR)1 for amino acid residues 1-559. This RyR1 segment folds into three distinct domains, forming a vestibule around the four-fold symmetry axis: domain A (blue; 1-205), domain B (green; 206-394) and domain C (red; 395-532). (A) Cytoplasmic view; (B) close-up lateral view from the four-fold symmetry axis, (C) Docking of domain A alone (blue) or domain BC alone (red) yields a near-perfect superposition on the docking position for ABC (grey). Adapted from Tung, et al. [17].
The RyR macromolecular complex
As mentioned above, RyRs are often complexed with several accessory proteins, forming an intricate multi-protein array [32,33]. The best known RyR-interacting proteins are CaM, which tonically inhibits RyR2 activity and produces biphasic effects on RyR1 [34,35]; FKBP12 and FKBP12.6, which stabilize RyR1 and RyR2 closures [36-38]; and the ternary complex triadin-junctin-calsequestrin, which 'senses' luminal Ca2+ content and modulates RyR activity by acting either as a Ca2+ reservoir or as a direct channel ligand [39-47]. More recently, RyR2 has been found to hold anchoring sites for protein kinase (PK)A, protein phosphatase (PP)1, the cAMP-specific phosphodiesterase (PDE)4D3 and Ca2+/calmodulin-dependent protein kinase (CaMK)II [37,48], emphasizing the importance of RyR2 regulation by phosphorylation [32]. In cardiac cells, sorcin exerts protein-protein interactions with the RyR and inhibits Ca2+ release in a Ca2+-dependent manner [49,50].
The binding sites of several regulatory proteins have been mapped near the 'clamp' region of the channels, encompassing subdomains 5 to 10, an apparent hot-spot for allosteric modulation. Phosphorylation sites for CaMKII and PKA, including Ser2808 and Ser2030, occur in different subdomains within this region [51,52]. Muscular A kinase anchoring protein (mAKAP) is believed to bind to RyRs via leucine zippers at residues 3003 to 3039, tethering PKA close to these phosphorylation sites [32]. Another scaffolding protein, Homer, purportedly interacts with RyRs via the clamp domain, where it brings Ca2+ signaling proteins in close proximity to their respective binding sites, and may facilitate crosstalk between RyRs and proteins in the surface membrane, such as DHPRs and β-adrenergic receptors [53-55].
CaM, both in its Ca2+-bound (Ca2+-CaM) and unbound forms (apoCaM), reportedly binds in a cleft between the 'clamp' and 'handle' regions [56]. This cleft occurs between domains 3 and 7 on a portion of the cytoplasmic assembly of the channel facing the SR, about 10 nm from the pore [57]. From their cryo-EM studies, Samso and Wagenknecht [57,58] proposed a model in which CaM transitions between a distinct, but overlapping apoCaM site as calcium concentration ([Ca2+]i) fluctuates. According to this model, as apoCaM binds Ca2+ ions, it loses affinity for the apoCaM site and translocates to the nearby Ca2+-CaM site. The CaM binding sites identified by these studies was corroborated by a Förster resonance energy transfer (FRET)-based approach, which identified a CaM target helix that spanned residues 3614 to 3643. However, [Ca2+]-dependent translocation of CaM between adjacent or overlapping sites could not be detected by FRET, indicating that any such movements must be subtle. The authors pointed out that such structural rearrangements could also be explained by changes in the interaction of CaM with the RyR target sequence or by rotation of CaM around its major axis [59].
Collins originally reported that a 12-kDa immunophilin (FKBP12) copurified with RyR1 [60]. Jayaraman et al. later reported that FKBP12.0 binds to RyR1 with a stoichiometry of one molecule per tetrameric channel [61]. Experimental techniques similar to those described above have shown that the binding site of FKBP12 also occurs in the clamp region of the channels; however, there is some controversy about the exact amino acid region(s) responsible for this interaction. For RyR1 and RyR3, a valyl-prolyl motif (amino acid residues 2461 and 2462 in RyR1) seems to berequired for binding of FKBP12/FKBP12.6 [62,63]. For RyR2, a variant of this motif (isoleucyl-prolyl sequence 2427 and 2428, respectively) has been implicated in binding FKBP12.6 [37]. However, Masumiya et al. reported that mutation of Ile-2427 or Pro-2428 does not alter FKBP12.6 binding [64], and further, that the RyR2 polypeptide fragment encompassing residues 1 to 1855 is sufficient for binding of FKBP12.6 [65]. Zhang et al. also found that other regions of RyR2 contribute to FKBP12.6 binding [66]. As with CaM, Wagenknecht and colleagues [57] used 3D reconstructions of cryo-EM images of purified RyRs complexed with FKBP12 to investigate the site of the interaction, which was identified on the opposite side of domain 3 from CaM, about 12 nm from the pore. A later study compared 3D reconstructions of native and FKBP12-stripped RyR1, identifying two surfaces of interaction between FKBP12 and RyR1: a portion of the β-sheet of FKBP12 interacts with domain 9 of RyR1 and the intersection of domains 3, 5 and 9, while loop 87 to 90 interacts with domain 3 of RyR1 [12].
Functional regulation of ryanodine receptors
In cardiac muscle, a small influx of external Ca2+ (ICa) through DHPRs (Cav2.1) binds to and opens RyR2, releasing a larger amount of Ca2+ from the SR. This process, known as Ca2+-induced Ca2+ release (CICR [67,68]), occurs in diads, which are specialized regions in which a segment of a T-tubule seems to be continuous with a semi-spherical extension of the SR [22]. CICR amplifies the incoming Ca2+ signal approximately 10 to 20 times, and is therefore the major component of the intracellular Ca2+ transient that induces contraction. In skeletal muscle, ICa is not required for contraction [69]. Instead, mechanical coupling between DHPRs (Cav1.1) and RyR1 (direct or facilitated by intermediary proteins) triggers Ca2+ release from the SR immediately after sarcolemmal depolarization [70,71]. At the triads, where a segment of one T-tubule seems to be continuous with two SR evaginations, not all RyRs are closely apposed to DHPRs; nearly half of RyRs remain DHPR-free and are therefore presumed to be activated by Ca2+ release from neighboring RyRs, effectively amplifying the Ca2+ signal [72]. Thus, common subcellular structures participate in EC coupling, but different processes link membrane depolarization to Ca2+ release in cardiac and skeletal muscle. In this section, we review some of the most prominent regulators of RyR1 and RyR2 and their effect in Ca2+ release in skeletal and cardiac muscle.
Ca2+ regulation
All three RyR isoforms harbor both activating and inactivating Ca2+ binding sites. Regardless of some differences (noted below), the Ca2+ affinity for those sites is roughly similar for all RyRs. Thus, Ca2+ plays a crucial role in the modulation of RyR activity, even in skeletal muscle [72].
The RyR is a Ca2+-gated channel, hence, functional assays that measure its activity ([3H]ryanodine binding, reconstitution in lipid bilayers, Ca2+ sparks) require a critical [Ca2+] in the medium to maintain the channel in its open state. However, despite the fact that RyRs in situ normally encounter fast and transient changes in [Ca2+], most of the functional characterizations of single RyR have been obtained under the continuous presence of a critical [Ca2+], that is, under steady-state conditions. How similar is this characterization to that obtained under a transient application of [Ca2+]? Fabiato, in his classic studies delineating CICR [67,68], found that the amount of Ca2+ release was strongly dependent on the velocity of the Ca2+ pulse ((d[Ca2+]/dt) applied to skinned cardiac fibers. In light of these data, it is surprising that few studies have addressed the importance of dynamic fluctuations of Ca2+ on RyR kinetics. Three such studies have revealed that a fast Ca2+ stimulus elicits only a transient increase in the Po of RyRs; however, if Ca2+ is applied rapidly but sustained for long periods, RyR Po will 'relax' to a new steady-state that follows a bell-shaped curve as a function of the magnitude of Ca2+ stimulus. Thus, Po-[Ca2+] curves of greater magnitude emerge from titration of RyR activity with fast, calibrated Ca2+ pulses than those obtained under similar but steady applications of Ca2+ (Figure 2) [73-75]. The implication of these findings is that there are components of RyR activity that remain undetected in most of the assays presently used, and these aspects of RyR regulation may explain seemingly discrepant results, as in the case of RyR phosphorylation (see below).
Figure 2.
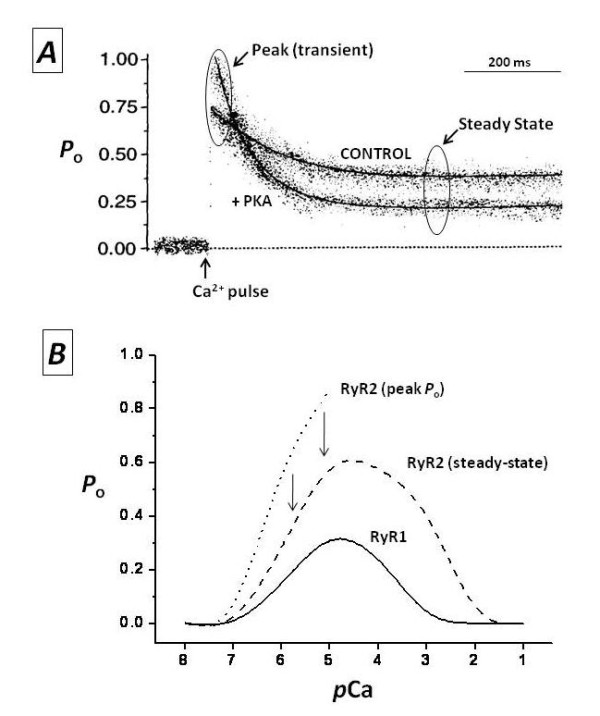
Modulation of ryanodine receptors (RyRs) by Ca2+. (A) Ensemble currents of single RyR2 show the effect of fast Ca2+ pulses on the Po of a non-phosphorylated channel ('control') or the same channel after phosphorylation with the catalytic subunit of protein kinase A ('+PKA'). Laser photolysis of 'caged Ca2+' (o-nitrophenyl EGTA, tetrapotassium salt; NP-EGTA) increased [Ca2+] from 0.1 to 10 μmol/l at the point labeled as 'Ca2+ pulse' (modified from [51]. (B) Ca2+-Po curves of RyR1 and RyR2. Channel activity was measured at the indicated stationary concentrations of Ca2+ ('RyR1' and 'RyR2 steady-state') and right after a fast Ca2+ pulse, as in (A) ('RyR2 (peak Po)') (unpublished results) (see text for details).
Under steady-state conditions, Ca2+ concentrations in the range of approximately 100 nmol/l to approximately 10 μm/l activate all three RyR isoforms [76-78]. The activation curve is sigmoidal, but the exact Hill number (cooperativity) and the plateau (maximal activation) depend greatly on the presence of other cytosolic ligands. Higher concentrations of Ca2+ in the range of 100 μmol/l to approximately 10 mmol/l also inactivate the three RyR isoforms, although there is considerable RyR heterogeneity in this process [79,80]. In general, Ca2+-dependent inactivation is most prominent in RyR1 and less important in RyR2 and RyR3 [26]. In fact, complete Ca2+-dependent inactivation in isolated RyR2 channels occurs at such high [Ca2+] (>10 mmol/l) that its physiological relevance is questionable [58,76]. The agonist effect of ATP and the inhibitory action of Mg2+ greatly influence the kinetics of calcium-dependent activation and inactivation [74], but the concentration of these two ligands is not expected to change dramatically over a short period or on a beat-to-beat basis, thus they may be considered rather as tonic modulators of RyRs whose influence becomes relevant under periods of intense muscle activity or in precarious metabolic states. Given the greater sensitivity of RyR1 to Ca2+-dependent inactivation, it is not surprising that Mg2+ also exerts a more powerful inhibition on the RyR1 isoform [74,81], with Mg2+ apparently binding to both Ca2+ activation and inactivation sites, attenuating the former and intensifying the latter.
Recently, regulation of RyRs by luminal (intra-SR) Ca2+ has been proposed as a critical control mechanism of Ca2+ release, especially in cardiac cells. The crucial observation is that, beyond a certain threshold, small changes in SR Ca2+ load result in far greater increases in Ca2+ release, with the relationship described as an inverse hyperbole with high degree of cooperativity [82,83]. Direct Ca2+ binding to sites available only from inside the SR [84], activation of cytosolic sites by the Ca2+ ions being permeated by the channel ('feed-through' mechanism [85]), and most notably, regulation by the junctin-triadin-calsequestrin ternary complex (for example, [39-47]) remain as the leading candidate mechanisms effecting luminal Ca2+ regulation of RyRs, although it has been difficult to dissect out the specific contribution of each of these phenomena.
RyR phosphorylation
RyRs are structural scaffolds for important kinases and phosphatases [32], and in the heart, RyR2 is one of the first proteins to undergo phosphorylation during β-adrenergic stimulation [86-88]. However, the functional effect of RyR phosphorylation is unclear and remains the subject of intense debate. All potential functional outcomes have been attributed to RyR phosphorylation. On one hand, Li et al. [89] found that PKA phosphorylation of RyR2 has little functional relevance for diastolic Ca2+ release if SR Ca2+ levels remain constant. On the other hand, other groups have suggested that PKA phosphorylation of RyR2 is so essential to intracellular Ca2+ homeostasis that derangement of this process may be the basis for heart failure (HF) [37,90] and catecholaminergic polymorphic ventricular tachycardia (CPVT) episodes [91]. Between these two extremes, other results, mainly from in vitro experiments, imply that PKA phosphorylation increases [86,92], decreases [74,93] or has no effect [94] on RyR2 activity.
Several factors preclude an easy interpretation of phosphorylation results. First, RyRs contain multiple phosphorylation sites that, depending on their phospho-state, may attenuate or synergize the effect of the other sites, or may require prior phosphorylation to activate the whole protein. To date, three phosphorylation sites have been recognized: Ser2809 (mouse RyR2 nomenclature) was first identified by Witcher et al. [86] as a CaMKII site, and later, Wehrens et al. suggested it as the only PKA site [95], even though RyR2 from mice with genetic ablation of this site may still be phosphorylated by PKA [88]. Ser2815 and Ser2030 seem to be exclusively phosphorylated by CaMKII [96] and PKA [97], respectively. Second, as mentioned above, RyR activity is strongly dependent on the speed of Ca2+ application, which in turn may greatly influence the overall effect of phosphorylation. For example, PKA phosphorylation of isolated RyR2 increases a transient component of activity (peak activation) but accelerates the rate of adaptation to a lower steady-state level of activity [74,88]. In cellular settings, this effect would translate into greater and faster rates of Ca2+ release in response to a given amount of Ca2+ entry. Elegant experiments in which SR Ca2+ load and ICa were kept constant showed that β-adrenergic stimulation of ventricular myocytes accelerates the rate of Ca2+ release and has little effect on the magnitude of the [Ca2+]i transient [98]. Similarly, experiments in rat and mouse skeletal muscle fibers showed little or no effect of β-adrenergic stimulation on the macroscopic Ca2+ transients [99]. A recent paper suggested that differences in the redox state of RyR could account for some the discrepancies seen [100]. Although appealing, this hypothesis still needs to be corroborated.
CaM
CaM regulates RyR activity both directly as a ligand, and indirectly as a cofactor for CaMKII. CaM binding to RyR1 increases channel activity at low [Ca2+], and decreases activity at high [Ca2+] [35]. Nevertheless, at the cellular level, this fine modulation might be negligible compared with the effect of other RyR regulators such as DHPR and calcium [101]. In skeletal muscle cells, CaM is proposed to protect RyR1 from oxidative stress and also work as an oxidative stress sensor (see below). RyR1 nitrosylation decreases CaM binding and increases channel activity [102]. The physiological importance of this mechanism is not clear, but it has been related to pathological processes [56]. In single RyR2 channels, only inhibitory effects by CaM have been shown [26]. CaM binding to both RyR1 and RyR2 can also be regulated by S100A1, a calcium-activated protein that competes with CaM for the RyR binding site [103-105]. Although the physiological beat-to-beat significance of this interaction is not yet clear, its chronic deregulation potentially plays a role in HF [106,107]. A recent study suggested that PKA phosphorylation decreases CaM binding, which would subsequently increase channel activity [108].
Oxidative stress
Oxidative modifications of thiol residues in free cysteines, such as S-nitrosylation, S-glutathionylation and disulfide oxidation, can modulate RyR1 and RyR2 [109-111]. The functional response of RyR may vary depending on the cysteine residue being modified and the type of oxidative species that targets it [110,112]. In addition, oxidative modifications can also affect the binding of accessory proteins. For instance, most data suggests that single RyR1 exposure to nitric oxide (NO) increases channel activity [102]. In skeletal muscle fibers, this effect is more evident in the presence of CaM, suggesting that s-nitrosylation of some residues produces CaM detachment and therefore reversal of the inhibitory effect of CaM over RyR [56]. Whether NO directly regulates RyR2 seems likely, but it is not completely clear. The NO donor
S-nitroso-N-acetyl penicillamine (SNAP), which targets several EC-coupling proteins, increases inotropy of cardiac myocytes at low concentrations, but decreases it at high concentrations [113,114]. It has been proposed that reduced glutathione (GSH) could quickly react with and scavenge NO in cardiac cells [111]. Under these circumstances, nitroso-GS or other small nitrosylated molecules would be responsible for RyR oxidation [111]. It is also possible that the close proximity of nitric oxide synthase (NOS)-3, xanthine oxidase and RyR2 in cardiac caveolae creates a microenvironment capable of directly nitrosylating RyR2 [114], although in physiological conditions the main targets of NOS seem to be other EC-coupling proteins [115].
RyR dysfunctions
In this section, we describe the role of RyR dysfunction in the generation of genetic and acquired diseases afflicting skeletal and cardiac muscle.
Malignant hyperthermia
Mutations in RYR1, the gene encoding the skeletal isoform of RyRs, are associated with malignant hyperthermia (MH), a pharmacogenetic disease triggered by inhalational anesthetics or depolarizing muscle relaxants [116,117]. SR Ca2+ 'leak' or excessive Ca2+ mobilization from the SR due to hyperactivity of RyR1 (especially at rest) seems to be the underlying mechanism for this syndrome [118], although increased resting [Ca2+] in human muscle has yet to be categorically demonstrated [119]. MH episodes are rare (approximately 1 in 10,000 surgeries [120]), but in their severest clinical presentation, susceptible individuals anesthetized with halothane or other volatile anesthetics suddenly develop muscle rigidity, hyperkalemia, arrhythmias, respiratory and metabolic acidosis, and an alarming increase in body temperature. Fulminant death inexorably follows unless body temperature is lowered and the excessive SR Ca2+ leak is prevented by dantrolene sodium, which presumably blocks RyR1, but not RyR2 [121]. More than 100 different point mutations in human RYR1 have been identified as potential causes of this syndrome; however, mutations in other genes, particularly in the α1 subunit of the skeletal DHPR, have also been reported to generate MH episodes [122]. This syndrome therefore emphasizes the central role that RyRs and DHPRs play in EC coupling. Interestingly, the great majority of RYR1 mutations seem to be clustered in three 'hot spots', namely, near the N-terminal (Cys35-Arg614), the central (Asp2129-Arg2458) and near the C-terminal (Ile3916-Ala4942) domains of the channel, a clustering that is repeated in an analogous RyR2-associated syndrome [123] (see below).
Porcine stress syndrome (PSS), which also results from point mutations of RYR1 (most often R615C) [124], is a similar MH syndrome, but with a wider array of triggering events (e.g. exercise, parturition, overcrowding). Susceptible pigs have been invaluable models to elucidate the molecular basis of MH in humans. Caffeine, which sensitizes RyR1 to Ca2+, elicits contractures of muscle strips of MH pigs at a lower dose than that required in control muscle. Caffeine hypersensitivity is also seen in human MH [125], and constitutes the basis for a clinical test of susceptibility. The bell-shaped Ca2+-dependence of [3H]ryanodine binding of MH-susceptible pigs is wider than that of normal pigs, suggesting greater sensitivity of RyR1 to Ca2+ activation and increased resistance to Ca2+ inactivation [77]. At the single channel level, RyR1s from MH-susceptible pigs exhibit similar conductance to 'normal' RyR1s, but they display longer mean open time and require higher [Ca2+] for inactivation. All these findings point to greater Ca2+ mobilization from RyR1-controlled pathways during resting and contracting periods, with the resultant overflowing of cytosolic compartments with Ca2+, and the ignition of multiple signaling mechanisms. Heat seems to stem from the rampant hydrolysis of ATP by the SR Ca2+ pump and other active Ca2+ transporters trying to reestablish Ca2+ homeostasis. However, the pathway to MH-associated heat production may be much more complex, as shown by the finding that MH mutations cause not only Ca2+ dysregulation, but also an increased release of cytokines (for example, interleukin 6). In sufficiently large quantities, these cytokines can be pyrogenic [126].
Central core disease
Central core disease (CCD) is another autosomal-dominant myopathy resulting from mutations in RYR1 [127,128]. Although some patients with MH may show clinical myopathies before an MH episode, histopathological lesions characterized by pale staining of the central area of muscle fibers are necessary to diagnose CCD. Non-progressive muscle weakness, hypotonia and motor deficiencies are present in most, but not all, patients with CCD [129]. Type 1 skeletal muscle fibers are preferentially affected, with central areas lacking mitochondria and their oxidative enzymes, thus staining poorly with basophilic dyes [130]. The cause for the disappearance of central mitochondria is unknown. A potential explanation is that the RYR1 mutation, as in MH, causes excessive Ca2+ leak. In the periphery of the fiber, Ca2+-extrusion mechanisms would be sufficient to remove the excess Ca2+, but the central areas would remain vulnerable to local Ca2+ gradients. Central mitochondria would, therefore, 'swallow' greater amounts of Ca2+ than they could handle, leading to swelling and death [131].
An alternative model for core formation has been proposed, based on studies using a knock-in mouse heterozygously expressing a leaky-channel mutation in the RyR1 N-terminal region [118,132-135]. According to this model, Ca2+ leak in the SR triggers the release of reactive oxygen and nitrogen species (ROS/RNS), causing RYR1 S-nitrosylation and glutathionylation, modifications that further enhance SR Ca2+ leak and RyR heat sensitivity. The authors correlated the resulting cascade of deterioration with the formation of cores that progress through a series of histopathologically distinct stages as the mice age. They proposed that oxidative damage, Ca2+-dependent proteolysis, and extreme stretching of the myofilaments due to prolonged contracture in the presence of unsequestered Ca2+ may all contribute to the progression of the disease. Importantly, these mice are prone to fulminant MH-like responses to heat challenge and halothane exposure [118,135].
An entirely different mechanism for the muscle weakness associated with CCD was revealed in recent studies using a mouse knock-in model expressing a heterozygous mutation in the RyR1 ion pore [132]. Muscle fibers from these mice exhibited reduced maximal twitch contraction, slowed rate of force development [136] and reduced global Ca2+ transients [137]. Adult mice displayed weakened upper body and grip strength [138]. These results were attributed to reduced and slowed RYR1 Ca2+ release, without change in either SR Ca2+ content or RyR1 sensitivity to activation by voltage or caffeine [138]; this pathway to CCD-associated muscle weakness is known as EC uncoupling [132]. Unlike the leaky-channel knock-ins, these mice were not prone to developing MH-like symptoms.
Surprisingly, the same mutation produced dramatically different phenotypes in different strains of mice, experimental approaches, and expression systems [136-138]. Similarly, identical RYR1 mutations cause MH in some human patients and CCD in others. The mechanism(s) that leads to the different phenotypes remains a mystery. Very interesting, however, is the observation that patients with MH who lack clinical myopathies are more likely to have mutations at the N-terminus of RYR1, whereas those with clinical myopathies are likely to have mutations at the C-terminus [128]. Although these observations seem to suggest that mutation mapping could provide an attractively simple predictor of phenotype, this notion probably oversimplifies the complexity of the problem. Transition from MH to CCD is probably a function of the severity of Ca2+ leakage, as the studies discussed above have shown that both C-terminal and N-terminal mutations are capable of producing this type of RyR1 dysfunction.
CPVT
CPVT is an autosomal-dominant inherited cardiac disease characterized by exercise- or stress-induced tachyarrhythmia episodes in the absence of apparent structural heart disease or prolonged QT interval [139,140]. The disease is rare but very malignant, often presenting for the first time in childhood and adolescence as syncopal events and/or sudden cardiac arrest. Multiple electrocardiographic irregularities (polymorphic) are simultaneously present in patients with this syndrome. More than 70 different mutations in RYR2, the gene encoding the cardiac isoform of RyRs, have been associated with CPVT [141], which is characterized by: a) more than two types of ventricular tachycardia morphologies, b) absence of underlying organic heart disease and c) absence of primary electrical disease (long QT, Brugada syndrome) [142]. CPVT usually occurs during intense exercise or acute emotional stress, and may lead to sudden cardiac death. As in MH, excessive Ca2+ release from the SR, especially during diastole, seems to be the underlying mechanism that gives rise to the VT [143,144]. Enhanced diastolic Ca2+ leak may overload the Na+/Ca2+ exchanger, which generates an inward current as it extrudes the released Ca2+. The inward current, in turn, gradually depolarizes the cell to threshold, favoring delayed after depolarizations (DADs) [145]. During depolarization, lack of Ca2+-dependent inactivation of ICa due to previous depletion of the SR leads to higher Ca2+ entry and reloading of the SR, which triggers another DAD in the next beat. Successive repetition of this altered Ca2+ cycle could result in paroxysmal tachycardia and arrhythmias even if only a few foci of ventricular cells are involved.
Although this hypothetical scheme logically relates RyR2 dysfunction with VT, it is unclear exactly what mechanism induces a group of apparently normal RyR2s to behave aberrantly and to generate sudden tachycardia. Because infusion of catecholamines also triggers CPVT, it is likely that activation of the β-adrenergic system plays an important role. In this regard, it has been suggested that phosphorylation of RyR2 by PKA, the kinase linking β1-adrenergic receptor activation to cellular effects, dissociates FKBP12.6 [37,90,91,146]), an accessory protein that presumably stabilizes RyR2 in the closed state. Phosphorylation of a mutant RyR2 would therefore remove a stabilizer from a channel on the verge of dysfunction, and would cause the pathogenic events described above. However, several studies have not found any evidence of FKBP12.6 dissociation in CPVT mutation-carrying RyR2 [147,148]. Thus, structural alterations of the RyR2 channel complex seem to be more important than dysregulation by accessory factors in the pathogenesis of CPVT. Consistent with this notion is the fact that CPVT-associated mutations of RYR2 occur in domains corresponding exactly to mutation-containing domains that give rise to MH [123,141]. Although some of these mutations are close to the apparent FKBP12.x-binding domain, the great majority are not. Recent studies point to a predominant role of Purkinje cells in the genesis of ventricular arrhythmias [149-151]. Whatever the triggering mechanism, SR Ca2+ load and release seem to be crucial, because mutations in CSQ2, the gene encoding cardiac calsequestrin, an intraluminal Ca2+ buffering protein that regulates RyR2 activity, also generate CPVT [152]. Accordingly, a prominent hypothesis, championed mainly by Chen and collaborators, proposes that cardiac myocytes have a threshold SR Ca2+ load for spontaneous Ca2+ release, and that CPVT-associated mutations decrease the sensitivity to luminal Ca2+ (that is, decrease the threshold) [143,153]. Thus, during β-adrenergic stimulation, when SR Ca2+ load increases as a result of enhanced Ca2+ entry and uptake, CPVT mutant channels reach their threshold and generate spontaneous Ca2+-release waves, creating a substrate favorable for Ca2+-dependent arrhythmias. Alternatively, CPVT-related mutations could increase the SR load, making it easier to reach the threshold upon adrenergic stimulation [154].
HF
HF is a multifactorial syndrome characterized by contractile dysfunction and pathological myocardial remodeling. There is growing consensus that Ca2+ mishandling in HF leads to altered gene transcription, resulting in maladaptive structural transformations (cardiac hypertrophy and then wall thinning) that eventually hamper basic cardiac function [155]. As with the genetic RyR dysfunctions discussed above, an enhanced diastolic Ca2+ leak seems to be central in the pathogenesis of some forms of HF [156]. According to a prominent hypothesis spearheaded by Marx et al. [37], increased catecholamine levels in patients with HF activate cAMP-dependent PKA by binding to β1-adrenergic receptors. PKA, in turn, phosphorylates RyR2-Ser2808 and dissociates FKBP12.6, increasing the probability of channel opening by inducing the appearance of long-lasting subconducting states. In this scheme, RyR2 hyperphosphorylation (up to 800% of control [37]) causes the enhanced diastolic Ca2+ leak. Unfortunately, this mechanism remains controversial. Although a few studies support some aspects of this mechanism, most groups have not been able to reproduce the central tenets of this hypothesis. Although Jiang et al. reported that PKA and the drug JTV519 both decreased FKBP12.6 binding to RyR2 [157], several other groups found that PKA phosphorylation of RyR2-S2808 neither dissociated FKBP12.6 nor substantially modified channel gating [88,89,94,97,158,159]. Ai et al. [160] reported that RyR2-S2808 phosphorylation was increased by approximately 50% in a rabbit model of HF; however, a direct test of PKA phosphorylation of RyR2 in canine and human HF yielded no difference compared with control, and a phospho-antibody against RyR2-S2808 detected equal phosphorylation levels in control and HF samples [158,161]. More recently, a mouse with genetic ablation of the RyR2-S2808 site (S2808A), supposedly stabilized in the closed state by close association with FKBP12.6, displayed normal β-adrenergic response and similar progression towards heart failure compared with wild-type mice [88,159]. Finally, FKBP12.0 dissociation of RyR1 is consistently reported to destabilize channel closure, but at least two groups failed to detect effects of FKBP12.6 dissociation of RyR2 [38,162]. Clearly, more studies are needed to resolve these conflicting results and clarify the potential role of FKBP12.6, SR Ca2+ leak, and PKA phosphorylation of RyR2-S2808 in HF.
Summary
In addition to providing the majority of the Ca2+ necessary for contraction of cardiac and skeletal muscle, RyRs act as molecular switchboards that integrate a multitude of cytosolic signals such as dynamic and steady Ca2+ fluctuations, β-adrenergic stimulation (phosphorylation), nitrosylation and metabolic states, and transduce these signals to the channel pore to release appropriate amounts of Ca2+. Furthermore, because Ca2+ release is crucially modulated by luminal factors such as SR Ca2+ content and protein-protein interactions, RyRs play an additional role as integrative switch-valves that offset cytosolic-luminal Ca2+ imbalances. Thus, there is an ample margin to interfere with the activity of RyRs. In both experimental and natural conditions, such interference results in overt contractile dysfunction and gross morphological changes. MH, CCD and CPVT are among the most studied clinical presentations of RyR dysfunction, and the elucidation of the precise molecular mechanisms affected by this dysfunction is advancing with great strides.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
EM, RL and HV contributed equally to the writing of this review. All authors read and approved the manuscript.
Contributor Information
E Michelle Capes, Email: capes@wisc.edu.
Randall Loaiza, Email: rloaiza@wisc.edu.
Héctor H Valdivia, Email: hvaldivia@wisc.edu.
References
- Benitah JP, Alvarez JL, Gomez AM. L-type Ca(2+) current in ventricular cardiomyocytes. J Mol Cell Cardiol. 2010;48:26–36. doi: 10.1016/j.yjmcc.2009.07.026. [DOI] [PubMed] [Google Scholar]
- Lorenzon NM, Beam KG. Disease causing mutations of calcium channels. Channels (Austin) 2008;2:163–179. doi: 10.4161/chan.2.3.5950. [DOI] [PubMed] [Google Scholar]
- Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010. [DOI] [PMC free article] [PubMed]
- Coronado R, Morrissette J, Sukhareva M, Vaughan DM. Structure and function of ryanodine receptors. Am J Physiol. 1994;266:C1485–1504. doi: 10.1152/ajpcell.1994.266.6.C1485. [DOI] [PubMed] [Google Scholar]
- Hakamata Y, Nakai J, Takeshima H, Imoto K. Primary structure and distribution of a novel ryanodine receptor/calcium release channel from rabbit brain. FEBS Lett. 1992;312:229–235. doi: 10.1016/0014-5793(92)80941-9. [DOI] [PubMed] [Google Scholar]
- Nakai J, Sekiguchi N, Rando TA, Allen PD, Beam KG. Two regions of the ryanodine receptor involved in coupling with L-type Ca2+ channels. J Biol Chem. 1998;273:13403–13406. doi: 10.1074/jbc.273.22.13403. [DOI] [PubMed] [Google Scholar]
- Perez CF, Mukherjee S, Allen PD. Amino acids 1-1,680 of ryanodine receptor type 1 hold critical determinants of skeletal type for excitation-contraction coupling. Journal of Biological Chemistry. 2003;278:39644–39652. doi: 10.1074/jbc.M305160200. [DOI] [PubMed] [Google Scholar]
- Hayek SM, Zhao J, Bhat M, Xu X, Nagaraj R, Pan Z, Takeshima H, Ma J. A negatively charged region of the skeletal muscle ryanodine receptor is involved in Ca(2+)-dependent regulation of the Ca(2+) release channel. FEBS Lett. 1999;461:157–164. doi: 10.1016/S0014-5793(99)01464-7. [DOI] [PubMed] [Google Scholar]
- Hamilton SL, Serysheva II. Ryanodine receptor structure: progress and challenges. J Biol Chem. 2009;284:4047–4051. doi: 10.1074/jbc.R800054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenknecht T, Samso M. Three-dimensional reconstruction of ryanodine receptors. Front Biosci. 2002;7:d1464–1474. doi: 10.2741/wagen. [DOI] [PubMed] [Google Scholar]
- Samso M, Feng W, Pessah IN, Allen PD. Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol. 2009;7:e85. doi: 10.1371/journal.pbio.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samso M, Shen X, Allen PD. Structural characterization of the RyR1-FKBP12 interaction. J Mol Biol. 2006;356:917–927. doi: 10.1016/j.jmb.2005.12.023. [DOI] [PubMed] [Google Scholar]
- Samso M, Wagenknecht T, Allen PD. Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat Struct Mol Biol. 2005;12:539–544. doi: 10.1038/nsmb938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch W, Rheault S, West DJ, Williams AJ. A model of the putative pore region of the cardiac ryanodine receptor channel. Biophys J. 2004;87:2335–2351. doi: 10.1529/biophysj.104.044180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amador FJ, Liu S, Ishiyama N, Plevin MJ, Wilson A, MacLennan DH, Ikura M. Crystal structure of type I ryanodine receptor amino-terminal β-trefoil domain reveals a disease-associated mutation "hot spot" loop. Proceedings of the National Academy of Sciences. 2009;106:11040–11044. doi: 10.1073/pnas.0905186106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo PA, Van Petegem F. Crystal structures of the N-terminal domains of cardiac and skeletal muscle ryanodine receptors: insights into disease mutations. Structure. 2009;17:1505–1514. doi: 10.1016/j.str.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Tung CC, Lobo PA, Kimlicka L, Van Petegem F. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature. 2010. [DOI] [PubMed]
- Inui M, Saito A, Fleischer S. Purification of the ryanodine receptor and identity with feet structures of junctional terminal cisternae of sarcoplasmic reticulum from fast skeletal muscle. J Biol Chem. 1987;262:1740–1747. [PubMed] [Google Scholar]
- Lai FA, Erickson HP, Rousseau E, Liu QY, Meissner G. Purification and reconstitution of the calcium release channel from skeletal muscle. Nature. 1988;331:315–319. doi: 10.1038/331315a0. [DOI] [PubMed] [Google Scholar]
- Wagenknecht T, Grassucci R, Frank J, Saito A, Inui M, Fleischer S. Three-dimensional architecture of the calcium channel/foot structure of sarcoplasmic reticulum. Nature. 1989;338:167–170. doi: 10.1038/338167a0. [DOI] [PubMed] [Google Scholar]
- Serysheva II. Structural insights into excitation-contraction coupling by electron cryomicroscopy. Biochemistry (Mosc) 2004;69:1226–1232. doi: 10.1007/s10541-005-0068-5. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Protasi F, Ramesh V. Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Ann N Y Acad Sci. 1998;853:20–30. doi: 10.1111/j.1749-6632.1998.tb08253.x. [DOI] [PubMed] [Google Scholar]
- Orlova EV, van Heel M, Hamilton SL, Chiu W. Two structural configurations of the skeletal muscle calcium release channel. Nat Struct Biol. 1996;3:547–552. doi: 10.1038/nsb0696-547. [DOI] [PubMed] [Google Scholar]
- Serysheva II, Ludtke SJ, Baker ML, Cong Y, Topf M, Eramian D, Sali A, Hamilton SL, Chiu W. Subnanometer-resolution electron cryomicroscopy-based domain models for the cytoplasmic region of skeletal muscle RyR channel. Proceedings of the National Academy of Sciences. 2008;105:9610–9615. doi: 10.1073/pnas.0803189105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz M, van Heel M, Chiu W, Hamilton SL. Structure of the skeletal muscle calcium release channel activated with Ca2+ and AMP-PCP. Biophys J. 1999;77:1936–1944. doi: 10.1016/S0006-3495(99)77035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- Du W, McMahon TJ, Zhang ZS, Stiber JA, Meissner G, Eu JP. Excitation-contraction coupling in airway smooth muscle. J Biol Chem. 2006;281:30143–30151. doi: 10.1074/jbc.M606541200. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Hamilton SL, Chiu W. The pore structure of the closed RyR1 channel. Structure. 2005;13:1203–1211. doi: 10.1016/j.str.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, El-Hayek R, Ikemoto N. Postulated role of interdomain interaction within the ryanodine receptor in Ca(2+) channel regulation. J Biol Chem. 2000;275:11618–11625. doi: 10.1074/jbc.275.16.11618. [DOI] [PubMed] [Google Scholar]
- Ikemoto N, Yamamoto T. Postulated role of inter-domain interaction within the ryanodine receptor in Ca(2+) channel regulation. Trends Cardiovasc Med. 2000;10:310–316. doi: 10.1016/S1050-1738(01)00067-6. [DOI] [PubMed] [Google Scholar]
- Ikemoto N, Yamamoto T. Regulation of calcium release by interdomain interaction within ryanodine receptors. Front Biosci. 2002;7:d671–683. doi: 10.2741/ikemoto. [DOI] [PubMed] [Google Scholar]
- Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. Journal of Molecular and Cellular Cardiology. 2004;37:417–429. doi: 10.1016/j.yjmcc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Yin C-C, D'Cruz LG, Lai FA. Ryanodine receptor arrays: not just a pretty pattern? Trends in Cell Biology. 2008;18:149–156. doi: 10.1016/j.tcb.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Bers DM, Li L, Satoh H, McCall E. Factors that control sarcoplasmic reticulum calcium release in intact ventricular myocytes. Ann N Y Acad Sci. 1998;853:157–177. doi: 10.1111/j.1749-6632.1998.tb08264.x. [DOI] [PubMed] [Google Scholar]
- Tripathy A, Xu L, Mann G, Meissner G. Calmodulin activation and inhibition of skeletal muscle Ca2+ release channel (ryanodine receptor) Biophys J. 1995;69:106–119. doi: 10.1016/S0006-3495(95)79880-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/S0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Xiao J, Tian X, Jones PP, Bolstad J, Kong H, Wang R, Zhang L, Duff HJ, Gillis AM, Fleischer S. et al. Removal of FKBP12.6 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J Biol Chem. 2007;282:34828–34838. doi: 10.1074/jbc.M707423200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonasekera SA, Beard NA, Groom L, Kimura T, Lyfenko AD, Rosenfeld A, Marty I, Dulhunty AF, Dirksen RT. Triadin binding to the C-terminal luminal loop of the ryanodine receptor is important for skeletal muscle excitation contraction coupling. J Gen Physiol. 2007;130:365–378. doi: 10.1085/jgp.200709790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Campbell KP. Association of triadin with the ryanodine receptor and calsequestrin in the lumen of the sarcoplasmic reticulum. J Biol Chem. 1995;270:9027–9030. doi: 10.1074/jbc.270.16.9027. [DOI] [PubMed] [Google Scholar]
- Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DW, Ma J, Kim DH. The asp-rich region at the carboxyl-terminus of calsequestrin binds to Ca2+ and interacts with triadin. FEBS Letters. 2000;486:178–182. doi: 10.1016/S0014-5793(00)02246-8. [DOI] [PubMed] [Google Scholar]
- Wei L, Gallant EM, Dulhunty AF, Beard NA. Junctin and triadin each activate skeletal ryanodine receptors but junctin alone mediates functional interactions with calsequestrin. The International Journal of Biochemistry & Cell Biology. 2009;41:2214–2224. doi: 10.1016/j.biocel.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, Fan GC, Dong M, Altschafl B, Diwan A, Ren X, Hahn HH, Zhao W, Waggoner JR, Jones LR. et al. Sarcoplasmic reticulum calcium overloading in junctin deficiency enhances cardiac contractility but increases ventricular automaticity. Circulation. 2007;115:300–309. doi: 10.1161/CIRCULATIONAHA.106.654699. [DOI] [PubMed] [Google Scholar]
- Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272:23389–23397. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]
- Qin J, Valle G, Nani A, Chen H, Ramos-Franco J, Nori A, Volpe P, Fill M. Ryanodine receptor luminal Ca2+ regulation: swapping calsequestrin and channel isoforms. Biophys J. 2009;97:1961–1970. doi: 10.1016/j.bpj.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer L, Rios E. Deconstructing calsequestrin. Complex buffering in the calcium store of skeletal muscle. J Physiol. 2009;587:3101–3111. doi: 10.1113/jphysiol.2009.171934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell EF, Antaramian A, Rueda A, Gomez AM, Valdivia HH. Sorcin inhibits calcium release and modulates excitation-contraction coupling in the heart. J Biol Chem. 2003;278:34660–34666. doi: 10.1074/jbc.M305931200. [DOI] [PubMed] [Google Scholar]
- Meyers MB, Pickel VM, Sheu SS, Sharma VK, Scotto KW, Fishman GI. Association of sorcin with the cardiac ryanodine receptor. J Biol Chem. 1995;270:26411–26418. doi: 10.1074/jbc.270.44.26411. [DOI] [PubMed] [Google Scholar]
- Jones PP, Meng X, Xiao B, Cai S, Bolstad J, Wagenknecht T, Liu Z, Chen SR. Localization of PKA phosphorylation site, Ser(2030), in the three-dimensional structure of cardiac ryanodine receptor. Biochem J. 2008;410:261–270. doi: 10.1042/BJ20071257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Xiao B, Cai S, Huang X, Li F, Bolstad J, Trujillo R, Airey J, Chen SRW, Wagenknecht T, Liu Z. Three-dimensional localization of serine 2808, a phosphorylation site in cardiac ryanodine receptor. Journal of Biological Chemistry. 2007;282:25929–25939. doi: 10.1074/jbc.M704474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Tu J, Pouliquin P, Cabrales E, Shen X, Dulhunty A, Worley PF, Allen PD, Pessah IN. Dynamic regulation of ryanodine receptor type 1 (RyR1) channel activity by Homer 1. Cell Calcium. 2008;43:307–314. doi: 10.1016/j.ceca.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouliquin P, Pace S, Dulhunty A. In vitro modulation of the cardiac ryanodine receptor activity by Homer1. Pflügers Archiv European Journal of Physiology. 2009;458:723–732. doi: 10.1007/s00424-009-0664-0. [DOI] [PubMed] [Google Scholar]
- Worley PF, Zeng W, Huang G, Kim JY, Shin DM, Kim MS, Yuan JP, Kiselyov K, Muallem S. Homer proteins in Ca2+ signaling by excitable and non-excitable cells. Cell Calcium. 2007;42:363–371. doi: 10.1016/j.ceca.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CP, Rodney G, Zhang JZ, Santacruz-Toloza L, Strasburg G, Hamilton SL. Apocalmodulin and Ca2+ calmodulin bind to the same region on the skeletal muscle Ca2+ release channel. Biochemistry. 1999;38:8532–8537. doi: 10.1021/bi9907431. [DOI] [PubMed] [Google Scholar]
- Wagenknecht T, Radermacher M, Grassucci R, Berkowitz J, Xin HB, Fleischer S. Locations of calmodulin and FK506-binding protein on the three-dimensional architecture of the skeletal muscle ryanodine receptor. J Biol Chem. 1997;272:32463–32471. doi: 10.1074/jbc.272.51.32463. [DOI] [PubMed] [Google Scholar]
- Samso M, Wagenknecht T. Apocalmodulin and Ca2+-calmodulin bind to neighboring locations on the ryanodine receptor. J Biol Chem. 2002;277:1349–1353. doi: 10.1074/jbc.M109196200. [DOI] [PubMed] [Google Scholar]
- Cornea RL, Nitu FR, Samso M, Thomas DD, Fruen BR. Mapping the ryanodine receptor FK506-binding protein subunit using fluorescence resonance energy transfer. J Biol Chem. 2010;285:19219–19226. doi: 10.1074/jbc.M109.066944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JH. Sequence analysis of the ryanodine receptor: possible association with a 12K, FK506-binding immunophilin/protein kinase C inhibitor. Biochem Biophys Res Commun. 1991;178:1288–1290. doi: 10.1016/0006-291X(91)91033-9. [DOI] [PubMed] [Google Scholar]
- Jayaraman T, Brillantes AM, Timerman AP, Fleischer S, Erdjument-Bromage H, Tempst P, Marks AR. FK506 binding protein associated with the calcium release channel (ryanodine receptor) Journal of Biological Chemistry. 1992;267:9474–9477. [PubMed] [Google Scholar]
- Bultynck G, Rossi D, Callewaert G, Missiaen L, Sorrentino V, Parys JB, De Smedt H. The conserved sites for the FK506-binding proteins in ryanodine receptors and inositol 1,4,5-trisphosphate receptors are structurally and functionally different. J Biol Chem. 2001;276:47715–47724. doi: 10.1074/jbc.M106573200. [DOI] [PubMed] [Google Scholar]
- Gaburjakova M, Gaburjakova J, Reiken S, Huang F, Marx SO, Rosemblit N, Marks AR. FKBP12 binding modulates ryanodine receptor channel gating. J Biol Chem. 2001;276:16931–16935. doi: 10.1074/jbc.M100856200. [DOI] [PubMed] [Google Scholar]
- Masumiya H, Wang R, Zhang J, Xiao B, Chen SR. Localization of the 12.6-kDa FK506-binding protein (FKBP12.6) binding site to the NH2-terminal domain of the cardiac Ca2+ release channel (ryanodine receptor) J Biol Chem. 2003;278:3786–3792. doi: 10.1074/jbc.M210962200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu Z, Masumiya H, Wang R, Jiang D, Li F, Wagenknecht T, Chen SR. Three-dimensional localization of divergent region 3 of the ryanodine receptor to the clamp-shaped structures adjacent to the FKBP binding sites. J Biol Chem. 2003;278:14211–14218. doi: 10.1074/jbc.M213164200. [DOI] [PubMed] [Google Scholar]
- Zissimopoulos S, Lai FA. Interaction of FKBP12.6 with the cardiac ryanodine receptor C-terminal domain. J Biol Chem. 2005;280:5475–5485. doi: 10.1074/jbc.M412954200. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1–14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla FM, Horowicz P. Twitches in the presence of ethylene glycol bis(-aminoethyl ether)-N,N'-tetracetic acid. Biochim Biophys Acta. 1972;267:605–608. doi: 10.1016/0005-2728(72)90194-6. [DOI] [PubMed] [Google Scholar]
- Rios E, Ma JJ, Gonzalez A. The mechanical hypothesis of excitation-contraction (EC) coupling in skeletal muscle. J Muscle Res Cell Motil. 1991;12:127–135. doi: 10.1007/BF01774031. [DOI] [PubMed] [Google Scholar]
- Knudson CM, Chaudhari N, Sharp AH, Powell JA, Beam KG, Campbell KP. Specific absence of the alpha 1 subunit of the dihydropyridine receptor in mice with muscular dysgenesis. J Biol Chem. 1989;264:1345–1348. [PubMed] [Google Scholar]
- Stern MD, Pizarro G, Rios E. Local control model of excitation-contraction coupling in skeletal muscle. J Gen Physiol. 1997;110:415–440. doi: 10.1085/jgp.110.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorke S, Fill M. Ryanodine receptor adaptation: control mechanism of Ca(2+)-induced Ca2+ release in heart. Science. 1993;260:807–809. doi: 10.1126/science.8387229. [DOI] [PubMed] [Google Scholar]
- Valdivia HH, Kaplan JH, Ellis-Davies GC, Lederer WJ. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez P, Gyorke S, Escobar AL, Vergara J, Fill M. Adaptation of single cardiac ryanodine receptor channels. Biophys J. 1997;72:691–697. doi: 10.1016/S0006-3495(97)78705-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu A, Fill M, Stefani E, Entman ML. Cytoplasmic Ca2+ does not inhibit the cardiac muscle sarcoplasmic reticulum ryanodine receptor Ca2+ channel, although Ca(2+)-induced Ca2+ inactivation of Ca2+ release is observed in native vesicles. J Membr Biol. 1993;135:49–59. doi: 10.1007/BF00234651. [DOI] [PubMed] [Google Scholar]
- Fill M, Coronado R, Mickelson JR, Vilven J, Ma JJ, Jacobson BA, Louis CF. Abnormal ryanodine receptor channels in malignant hyperthermia. Biophys J. 1990;57:471–475. doi: 10.1016/S0006-3495(90)82563-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner A, Conti A, Bertocchini F, Schindler H, Sorrentino V. Functional properties of the ryanodine receptor type 3 (RyR3) Ca2+ release channel. EMBO J. 1998;17:2790–2798. doi: 10.1093/emboj/17.10.2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull R, Marengo JJ. Sarcoplasmic reticulum release channels from frog skeletal muscle display two types of calcium dependence. FEBS Lett. 1993;331:223–227. doi: 10.1016/0014-5793(93)80341-Q. [DOI] [PubMed] [Google Scholar]
- Copello JA, Barg S, Onoue H, Fleischer S. Heterogeneity of Ca2+ gating of skeletal muscle and cardiac ryanodine receptors. Biophys J. 1997;73:141–156. doi: 10.1016/S0006-3495(97)78055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner G, Darling E, Eveleth J. Kinetics of rapid Ca2+ release by sarcoplasmic reticulum. Effects of Ca2+, Mg2+, and adenine nucleotides. Biochemistry. 1986;25:236–244. doi: 10.1021/bi00349a033. [DOI] [PubMed] [Google Scholar]
- Gyorke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res. 2008;77:245–255. doi: 10.1093/cvr/cvm038. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Bers DM. Assessment of intra-SR free [Ca] and buffering in rat heart. Biophys J. 1997;73:1524–1531. doi: 10.1016/S0006-3495(97)78184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoso P, Prieto H, Hidalgo C. Luminal calcium regulates calcium release in triads isolated from frog and rabbit skeletal muscle. Biophys J. 1995;68:507–515. doi: 10.1016/S0006-3495(95)80212-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver DR, O'Neill ER, Lamb GD. Luminal Ca2+-regulated Mg2+ inhibition of skeletal RyRs reconstituted as isolated channels or coupled clusters. J Gen Physiol. 2004;124:741–758. doi: 10.1085/jgp.200409092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher DR, Kovacs RJ, Schulman H, Cefali DC, Jones LR. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991;266:11144–11152. [PubMed] [Google Scholar]
- Yoshida A, Takahashi M, Imagawa T, Shigekawa M, Takisawa H, Nakamura T. Phosphorylation of ryanodine receptors in rat myocytes during beta-adrenergic stimulation. J Biochem. 1992;111:186–190. doi: 10.1093/oxfordjournals.jbchem.a123735. [DOI] [PubMed] [Google Scholar]
- Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, Powers PA, Valdivia HH. Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–829. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–316. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C, Huang F, Gaburjakova M, Gaburjakova J, Rosemblit N. et al. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol. 2003;160:919–928. doi: 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N. et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/S0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Hain J, Onoue H, Mayrleitner M, Fleischer S, Schindler H. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J Biol Chem. 1995;270:2074–2081. doi: 10.1074/jbc.270.5.2074. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Gyorke S. Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes. J Physiol. 2003;552:109–118. doi: 10.1113/jphysiol.2003.046367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stange M, Xu L, Balshaw D, Yamaguchi N, Meissner G. Characterization of recombinant skeletal muscle (Ser-2843) and cardiac muscle (Ser-2809) ryanodine receptor phosphorylation mutants. J Biol Chem. 2003;278:51693–51702. doi: 10.1074/jbc.M310406200. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci USA. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304:292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- Xiao B, Zhong G, Obayashi M, Yang D, Chen K, Walsh MP, Shimoni Y, Cheng H, Ter Keurs H, Chen SR. Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem J. 2006;396:7–16. doi: 10.1042/BJ20060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg KS, Bers DM. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol. 2004;556:463–480. doi: 10.1113/jphysiol.2003.055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazev R, Hussain M, Bakker AJ, Head SI, Lamb GD. Effects of the PKA inhibitor H-89 on excitation-contraction coupling in skinned and intact skeletal muscle fibres. J Muscle Res Cell Motil. 2001;22:277–286. doi: 10.1023/A:1012289526618. [DOI] [PubMed] [Google Scholar]
- Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375–4387. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell KM, Yamaguchi N, Meissner G, Dirksen RT. Calmodulin binding to the 3614-3643 region of RyR1 is not essential for excitation-contraction coupling in skeletal myotubes. J Gen Physiol. 2002;120:337–347. doi: 10.1085/jgp.20028617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/S0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- Wright NT, Prosser BL, Varney KM, Zimmer DB, Schneider MF, Weber DJ. S100A1 and calmodulin compete for the same binding site on ryanodine receptor. J Biol Chem. 2008;283:26676–26683. doi: 10.1074/jbc.M804432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser BL, Wright NT, Hernandez-Ochoa EO, Varney KM, Liu Y, Olojo RO, Zimmer DB, Weber DJ, Schneider MF. S100A1 binds to the calmodulin-binding site of ryanodine receptor and modulates skeletal muscle excitation-contraction coupling. J Biol Chem. 2008;283:5046–5057. doi: 10.1074/jbc.M709231200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev K, Ackermann GE, Heizmann CW, Niggli E. Ca2+ signaling in mouse cardiomyocytes with ablated S100A1 protein. Gen Physiol Biophys. 2009;28:371–383. doi: 10.4149/gpb_2009_04_371. [DOI] [PubMed] [Google Scholar]
- Remppis A, Most P, Loffler E, Ehlermann P, Bernotat J, Pleger S, Borries M, Reppel M, Fischer J, Koch WJ. et al. The small EF-hand Ca2+ binding protein S100A1 increases contractility and Ca2+ cycling in rat cardiac myocytes. Basic Res Cardiol. 2002;97(Suppl 1):I56–62. doi: 10.1007/s003950200031. [DOI] [PubMed] [Google Scholar]
- Most P, Seifert H, Gao E, Funakoshi H, Volkers M, Heierhorst J, Remppis A, Pleger ST, DeGeorge BR Jr, Eckhart AD. et al. Cardiac S100A1 protein levels determine contractile performance and propensity toward heart failure after myocardial infarction. Circulation. 2006;114:1258–1268. doi: 10.1161/CIRCULATIONAHA.106.622415. [DOI] [PubMed] [Google Scholar]
- Ono M, Yano M, Hino A, Suetomi T, Xu X, Susa T, Uchinoumi H, Tateishi H, Oda T, Okuda S. et al. Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca(2+) release in heart failure. Cardiovasc Res. 2010;87:609–617. doi: 10.1093/cvr/cvq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- Aracena P, Sanchez G, Donoso P, Hamilton SL, Hidalgo C. S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. J Biol Chem. 2003;278:42927–42935. doi: 10.1074/jbc.M306969200. [DOI] [PubMed] [Google Scholar]
- Sun J, Yamaguchi N, Xu L, Eu JP, Stamler JS, Meissner G. Regulation of the cardiac muscle ryanodine receptor by O(2) tension and S-nitrosoglutathione. Biochemistry. 2008;47:13985–13990. doi: 10.1021/bi8012627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aracena-Parks P, Goonasekera SA, Gilman CP, Dirksen RT, Hidalgo C, Hamilton SL. Identification of cysteines involved in s-nitrosylation, s-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. Journal of Biological Chemistry. 2006;281:40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- Gonzalez DR, Fernandez IC, Ordenes PP, Treuer AV, Eller G, Boric MP. Differential role of S-nitrosylation and the NO-cGMP-PKG pathway in cardiac contractility. Nitric Oxide. 2008;18:157–167. doi: 10.1016/j.niox.2007.09.086. [DOI] [PubMed] [Google Scholar]
- Ziolo MT. The fork in the nitric oxide road: cyclic GMP or nitrosylation? Nitric Oxide. 2008;18:153–156. doi: 10.1016/j.niox.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Viatchenko-Karpinski S, Sun J, Gyorke I, Benkusky NA, Kohr MJ, Valdivia HH, Murphy E, Gyorke S, Ziolo MT. Regulation of myocyte contraction via neuronal nitric oxide synthase: role of ryanodine receptor S-nitrosylation. J Physiol. 2010;588:2905–2917. doi: 10.1113/jphysiol.2010.192617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickelson JR, Louis CF. Malignant hyperthermia: excitation-contraction coupling, Ca2+ release channel, and cell Ca2+ regulation defects. Physiol Rev. 1996;76:537–592. doi: 10.1152/physrev.1996.76.2.537. [DOI] [PubMed] [Google Scholar]
- Maclennan DH, Zvaritch E. Mechanistic models for muscle diseases and disorders originating in the sarcoplasmic reticulum. Biochim Biophys Acta. 2010. [DOI] [PubMed]
- Chelu MG, Goonasekera SA, Durham WJ, Tang W, Lueck JD, Riehl J, Pessah IN, Zhang P, Bhattacharjee MB, Dirksen RT, Hamilton SL. Heat- and anesthesia-induced malignant hyperthermia in an RyR1 knock-in mouse. FASEB J. 2006;20:329–330. doi: 10.1096/fj.05-4497fje. [DOI] [PubMed] [Google Scholar]
- Benca J, Hogan K. Malignant hyperthermia, coexisting disorders, and enzymopathies: risks and management options. Anesth Analg. 2009;109:1049–1053. doi: 10.1213/ane.0b013e3181adca28. [DOI] [PubMed] [Google Scholar]
- Gurnaney H, Brown A, Litman RS. Malignant hyperthermia and muscular dystrophies. Anesth Analg. 2009;109:1043–1048. doi: 10.1213/ane.0b013e3181aa5cf6. [DOI] [PubMed] [Google Scholar]
- Zhao F, Li P, Chen SR, Louis CF, Fruen BR. Dantrolene inhibition of ryanodine receptor Ca2+ release channels. Molecular mechanism and isoform selectivity. J Biol Chem. 2001;276:13810–13816. doi: 10.1074/jbc.M006104200. [DOI] [PubMed] [Google Scholar]
- Ali SZ, Taguchi A, Rosenberg H. Malignant hyperthermia. Best Pract Res Clin Anaesthesiol. 2003;17:519–533. doi: 10.1016/j.bpa.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Yano M, Yamamoto T, Ikeda Y, Matsuzaki M. Mechanisms of Disease: ryanodine receptor defects in heart failure and fatal arrhythmia. Nat Clin Pract Cardiovasc Med. 2006;3:43–52. doi: 10.1038/ncpcardio0419. [DOI] [PubMed] [Google Scholar]
- Gillard EF, Otsu K, Fujii J, Khanna VK, de Leon S, Derdemezi J, Britt BA, Duff CL, Worton RG, MacLennan DH. A substitution of cysteine for arginine 614 in the ryanodine receptor is potentially causative of human malignant hyperthermia. Genomics. 1991;11:751–755. doi: 10.1016/0888-7543(91)90084-R. [DOI] [PubMed] [Google Scholar]
- Valdivia HH, Hogan K, Coronado R. Altered binding site for Ca2+ in the ryanodine receptor of human malignant hyperthermia. Am J Physiol. 1991;261:C237–245. doi: 10.1152/ajpcell.1991.261.2.C237. [DOI] [PubMed] [Google Scholar]
- Ducreux S, Zorzato F, Muller C, Sewry C, Muntoni F, Quinlivan R, Restagno G, Girard T, Treves S. Effect of ryanodine receptor mutations on interleukin-6 release and intracellular calcium homeostasis in human myotubes from malignant hyperthermia-susceptible individuals and patients affected by central core disease. J Biol Chem. 2004;279:43838–43846. doi: 10.1074/jbc.M403612200. [DOI] [PubMed] [Google Scholar]
- Klingler W, Rueffert H, Lehmann-Horn F, Girard T, Hopkins PM. Core myopathies and risk of malignant hyperthermia. Anesth Analg. 2009;109:1167–1173. doi: 10.1213/ANE.0b013e3181b5ae2d. [DOI] [PubMed] [Google Scholar]
- Mathews KD, Moore SA. Multiminicore myopathy, central core disease, malignant hyperthermia susceptibility, and RYR1 mutations: one disease with many faces? Arch Neurol. 2004;61:27–29. doi: 10.1001/archneur.61.1.27. [DOI] [PubMed] [Google Scholar]
- Jungbluth H. Central core disease. Orphanet J Rare Dis. 2007;2:25. doi: 10.1186/1750-1172-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno C, Minetti C. Congenital myopathies. Curr Neurol Neurosci Rep. 2004;4:68–73. doi: 10.1007/s11910-004-0015-7. [DOI] [PubMed] [Google Scholar]
- Loke J, MacLennan DH. Malignant hyperthermia and central core disease: disorders of Ca2+ release channels. Am J Med. 1998;104:470–486. doi: 10.1016/S0002-9343(98)00108-9. [DOI] [PubMed] [Google Scholar]
- Avila G, Dirksen RT. Functional effects of central core disease mutations in the cytoplasmic region of the skeletal muscle ryanodine receptor. The Journal of General Physiology. 2001;118:277–290. doi: 10.1085/jgp.118.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompagni S, Rossi AE, Micaroni M, Hamilton SL, Dirksen RT, Franzini-Armstrong C, Protasi F. Characterization and temporal development of cores in a mouse model of malignant hyperthermia. Proc Natl Acad Sci USA. 2009;106:21996–22001. doi: 10.1073/pnas.0911496106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen RT, Avila G. Altered ryanodine receptor function in central core disease: leaky or uncoupled Ca2+ release channels? Trends in Cardiovascular Medicine. 2002;12:189–197. doi: 10.1016/S1050-1738(02)00163-9. [DOI] [PubMed] [Google Scholar]
- Durham WJ, Aracena-Parks P, Long C, Rossi AE, Goonasekera SA, Boncompagni S, Galvan DL, Gilman CP, Baker MR, Shirokova N. et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53–65. doi: 10.1016/j.cell.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvaritch E, Kraeva N, Bombardier E, McCloy RA, Depreux F, Holmyard D, Kraev A, Seidman CE, Seidman JG, Tupling AR, MacLennan DH. Ca2+ dysregulation in Ryr1(I4895T/wt) mice causes congenital myopathy with progressive formation of minicores, cores, and nemaline rods. Proc Natl Acad Sci USA. 2009;106:21813–21818. doi: 10.1073/pnas.0912126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompagni S, Loy RE, Dirksen RT, Franzini-Armstrong C. The I4895T mutation in the type 1 ryanodine receptor induces fiber-type specific alterations in skeletal muscle that mimic premature aging. Aging Cell. 2010;9:958–970. doi: 10.1111/j.1474-9726.2010.00623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loy RE, Orynbayev M, Xu L, Andronache Z, Apostol S, Zvaritch E, MacLennan DH, Meissner G, Melzer W, Dirksen RT. Muscle weakness in Ryr1I4895T/WT knock-in mice as a result of reduced ryanodine receptor Ca2+ ion permeation and release from the sarcoplasmic reticulum. The Journal of General Physiology. 2011;137:43–57. doi: 10.1085/jgp.201010523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano C, Priori SG, Bloise R, (Eds) Catecholaminergic polymorphic ventricular tachycardia. Seattle, WA: University of Washington, Seattle; 2004. [PubMed] [Google Scholar]
- Ylanen K, Poutanen T, Hiippala A, Swan H, Korppi M. Catecholaminergic polymorphic ventricular tachycardia. Eur J Pediatr. 2010;169:535–542. doi: 10.1007/s00431-010-1154-2. [DOI] [PubMed] [Google Scholar]
- Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP, Mannens MM, Wilde AA, Ackerman MJ. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J Am Coll Cardiol. 2009;54:2065–2074. doi: 10.1016/j.jacc.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen PJ, Swan H, Piippo K, Viitasalo M, Toivonen L, Kontula K. Genes, exercise and sudden death: molecular basis of familial catecholaminergic polymorphic ventricular tachycardia. Ann Med. 2004;36(Suppl 1):81–86. doi: 10.1080/17431380410032599. [DOI] [PubMed] [Google Scholar]
- Jiang D, Xiao B, Zhang L, Chen SR. Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res. 2002;91:218–225. doi: 10.1161/01.RES.0000028455.36940.5E. [DOI] [PubMed] [Google Scholar]
- MacLennan DH, Chen SR. Store overload-induced Ca2+ release as a triggering mechanism for CPVT and MH episodes caused by mutations in RYR and CASQ genes. J Physiol. 2009;587:3113–3115. doi: 10.1113/jphysiol.2009.172155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannankeril PJ, Mitchell BM, Goonasekera SA, Chelu MG, Zhang W, Sood S, Kearney DL, Danila CI, De Biasi M, Wehrens XH. et al. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci USA. 2006;103:12179–12184. doi: 10.1073/pnas.0600268103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Cornea RL, Huke S, Camors E, Yang Y, Picht E, Fruen BR, Bers DM. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ Res. 2010;106:1743–1752. doi: 10.1161/CIRCRESAHA.110.219816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003;93:531–540. doi: 10.1161/01.RES.0000091335.07574.86. [DOI] [PubMed] [Google Scholar]
- Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O'Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C. et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101:1039–1048. doi: 10.1161/CIRCRESAHA.107.148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron TJ, Milstein ML, Anumonwo J, Priori SG, Jalife J. Purkinje cell calcium dysregulation is the cellular mechanism that underlies catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2010;7:1122–1128. doi: 10.1016/j.hrthm.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Giovannone SF, Liu N, Liu FY, Zhang J, Priori SG, Fishman GI. Purkinje cells from RyR2 mutant mice are highly arrhythmogenic but responsive to targeted therapy. Circ Res. 2010;107:512–519. doi: 10.1161/CIRCRESAHA.110.221481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen PJ, Swan H, Kontula K. Molecular genetics of exercise-induced polymorphic ventricular tachycardia: identification of three novel cardiac ryanodine receptor mutations and two common calsequestrin 2 amino-acid polymorphisms. Eur J Hum Genet. 2003;11:888–891. doi: 10.1038/sj.ejhg.5201061. [DOI] [PubMed] [Google Scholar]
- Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR) Proc Natl Acad Sci USA. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashimura T, Briston SJ, Trafford AW, Napolitano C, Priori SG, Eisner DA, Venetucci LA. In the RyR2(R4496C) mouse model of CPVT, beta-adrenergic stimulation induces Ca waves by increasing SR Ca content and not by decreasing the threshold for Ca waves. Circ Res. 2010;107:1483–1489. doi: 10.1161/CIRCRESAHA.110.227744. [DOI] [PubMed] [Google Scholar]
- Dorn GW, Molkentin JD. Manipulating cardiac contractility in heart failure: data from mice and men. Circulation. 2004;109:150–158. doi: 10.1161/01.CIR.0000111581.15521.F5. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- Blayney LM, Jones JL, Griffiths J, Lai FA. A mechanism of ryanodine receptor modulation by FKBP12/12.6, protein kinase A, and K201. Cardiovasc Res. 2010;85:68–78. doi: 10.1093/cvr/cvp273. [DOI] [PubMed] [Google Scholar]
- Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res. 2002;91:1015–1022. doi: 10.1161/01.RES.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- MacDonnell SM, Garcia-Rivas G, Scherman JA, Kubo H, Chen X, Valdivia H, Houser SR. Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ Res. 2008;102:e65–72. doi: 10.1161/CIRCRESAHA.108.174722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- Xiao B, Sutherland C, Walsh MP, Chen SR. Protein kinase A phosphorylation at serine-2808 of the cardiac Ca2+-release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP12.6) Circ Res. 2004;94:487–495. doi: 10.1161/01.RES.0000115945.89741.22. [DOI] [PubMed] [Google Scholar]
- Barg S, Copello JA, Fleischer S. Different interactions of cardiac and skeletal muscle ryanodine receptors with FK-506 binding protein isoforms. Am J Physiol. 1997;272:C1726–1733. doi: 10.1152/ajpcell.1997.272.5.C1726. [DOI] [PubMed] [Google Scholar]