

Abstract
The transcription factors Pea3, Erm, and Er81 can promote cancer initiation and progression in various types of solid tumors. However, their role in esophageal squamous cell carcinoma (ESCC) has not been elucidated. In this study, we found that the expression levels of Pea3 and Erm, but not that of Er81, were significantly higher in ESCC compared with nontumor esophageal epithelium. A high level of Pea3 expression was significantly correlated with a shorter overall survival in a cohort of 81 patients with ESCC and the subgroup with N1 stage tumor (Wilcoxon-Gehan test, P = 0.016 and P = 0.001, respectively). Pea3 was overexpressed in seven ESCC cell lines compared with two immortalized esophageal cell lines. Pea3 knockdown reduced cell proliferation and suppressed nonadherent growth, migration, and invasion in ESCC cells in vitro. In addition, Pea3 knockdown in ESCC cells resulted in a down-regulation of phospho-Akt and matrix metalloproteinase 13, whereas a significant positive correlation in the expression levels was observed between Pea3 and phospho-Akt (r = 0.281, P < 0.013) and between Pea3 and matrix metalloproteinase 13 in the human specimens (r = 0.462, P < 0.001). Moreover, Pea3 modulated the sensitivity of EC109 cells to doxorubicin, probably via reduced activity of the phosphatidylinositol 3-kinase–Akt–mammalian target of Rapamycin complex 1 pathway on Pea3 knockdown. In conclusion, our results suggest that Pea3 plays an important role in the progression of ESCC.
Esophageal squamous cell carcinoma (ESCC) is common among Asian populations.1 Despite recent advances in the detection of the premalignant lesions and the development of combination therapies, its incidence is increasing, and its outcome remains poor.2–4 Given the poor prognosis of ESCC and its high incidence rate, it is increasingly important to understand the initiation and progression of this type of cancer and to identify the associated prognostic factors.
Pea3, Erm, and Er81 belong to the Pea3 subgroup of the Ets transcription factor family. This group of proteins contains several functional domains, and the individual members demonstrate extensive amino acid sequence similarities.5 The roles of these proteins in mammary gland development and tumorigenesis have also been extensively studied and reviewed.6–8 Pea3 group transcription factors promote metastatic development and cancer progression through transcriptional activation of metastasis-related genes, such as matrix metalloproteinases (MMPs)9–13 and cyclooxygenase (COX)-2.14,15 Overexpression of Pea3 also increases the motility and invasiveness of lung cancer cells via activation of the ρ pathway and an increase in COX-2 expression.16–18
The prognostic significance of Pea3 has also been demonstrated in various solid tumors. Pea3 is overexpressed in mouse metastatic mammary adenocarcinoma19 and in human breast cancer, in which its overexpression is also correlated with HER-2 expression and poor prognosis.20–23 A high level of Pea3 expression correlates with poor survival in patients with ovarian,24,25 colorectal,26 oral,27 lung,28 and gastric cancers.29 The other two members of the Pea3 subgroup, Erm and Er81, are also overexpressed in mammary tumors.21 Erm knockdown reduces the tumorigenicity of mouse mammary cancer cells, and a high Erm expression level also acts as an independent adverse prognostic factor in patients with breast cancer.30,31 Moreover, Erm overexpression enhances the aggressiveness of cancer cells in vitro and correlates with disease progression in endometrial carcinoma.32–34
To the best of our knowledge, the roles of Pea3 group transcription factors in ESCC have not been studied. In the present study, we investigated the expression of the three transcription factors in an ESCC patient cohort and found that Pea3 overexpression was associated with poor prognosis. Our findings for the role of Pea3 in ESCC suggest that Pea3 is required for ESCC progression by enhancing proliferation, increasing tumor cell invasiveness, promoting drug resistance, and activating phosphatidylinositol 3-kinase (PI3K)—Akt signaling.
Materials and Methods
Patients and Specimens
The ESCC patient cohort has been previously described.35 Formalin-fixed, paraffin-embedded (FFPE) esophagectomy specimens from 81 Chinese patients with ESCC (mean follow-up, 14.5 months; range, 0.7 to 65.2 months) were collected from Queen Mary Hospital, Hong Kong, China, from January 1998 to December 2005. The specimens were collected consecutively, exclusive of patients who had prior treatment directed against ESCC. The tumor specimens were then incorporated into six different TMAs, as previously described.35 Specimens for which there was not sufficient tumor tissue available for incorporation into the TMA block were excluded. Thirty-three paired nonneoplastic esophageal epithelia were selected from the upper resection margin of the respective esophagectomy specimens. The clinicopathological data are summarized in Table 1.
Table 1.
Patient Clinical and Pathological Features
| Features | No. (%) of patients |
|---|---|
| Sex | |
| Male | 62 (77) |
| Female | 19 (23) |
| T stage | |
| 1 | 2 (3) |
| 2 | 13 (16) |
| 3 | 49 (60) |
| 4 | 17 (21) |
| N stage | |
| 0 | 30 (37) |
| 1 | 51 (63) |
| M stage | |
| 0 | 68 (84) |
| 1 | 13 (16) |
| pTNM stage | |
| II | 32 (40) |
| III | 36 (44) |
| IV | 13 (16) |
| Histological grade | |
| Well differentiated | 13 (16) |
| Moderately differentiated | 48 (59) |
| Poorly differentiated | 20 (25) |
The median (range) age of the 81 patients was 67 (41 to 87) years.
IHC Staining and Evaluation
Immunohistochemical (IHC) staining was performed as previously described using the Dako EnVision+ system-HRP (Dako, Glostrup, Denmark).35 Pea3 monoclonal, Er81 polyclonal, and Erm polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) and MMP13 antibodies (Abcam, Cambridge, MA) were applied to the TMA sections at 1:50, 1:100, or 1:150 dilutions, respectively, overnight at 4°C. The primary antibody was omitted in the negative control. The stained sections were reviewed by two independent observers (Y.-P.C. and K.-W.C.) with no prior knowledge of the clinicopathological data for the patients. All three cores of each specimen were individually scored from 0 to 3, which indicated negative, weak, moderate, and strong staining, respectively. The highest score was selected for later statistical analysis. Negative and weak staining was further classified as low-level expression, whereas moderate and strong staining was classified as high-level expression for Kaplan-Meier analysis. The number of samples varied slightly between the detected markers because of the variability in the number of interpretable tissue cores in the TMA sections. The results for phospho-Akt (pAkt) staining in the same cohort have been previously described.35
Statistical Analysis of IHC Staining
The statistical analysis was performed using SPSS 16.0 (IBM Corporation, Armonk, NY). Differences in expression levels among the different pathological stages were analyzed using the χ2 or Fisher's exact tests, where applicable. Correlations between the expression levels of different proteins and the tumor staging were estimated by the Spearman's rank correlation test. Kaplan-Meier analysis was performed, and the results were compared using the Wilcoxon-Gehan test to correlate the protein expression levels with patient survival.
Cell Lines
The NE1 and NE3 immortalized esophageal cell lines were maintained in keratinocyte serum-free medium (Invitrogen, Paisley, UK). The ESCC cancer cell lines HKESC-1, HKESC-2, and HKESC-3 were maintained in α-modified Eagle's medium (Invitrogen), supplemented with 10% fetal bovine serum; whereas SLMT, EC1, EC18, and EC109 were maintained in RPMI 1640 medium (Invitrogen), supplemented with 10% fetal bovine serum. The Phoenix Ampho retroviral packaging cell line and the 293T Lentiviral packaging cell line were maintained in Dulbecco's modified Eagle's medium (Invitrogen), supplemented with 10% fetal bovine serum. Stable pools of NE1 or NE3 vector control and Flag-Pea3 cells were generated using the retroviral infection of pBabe empty vector and pBabe-Flag-Pea3, respectively. The scramble short hairpin (shScr)/shPea3-1/shPea3-2 cancer cells were generated by lentiviral infection using the pLKO.1 plasmid expressing a shScr RNA and two Pea3 shRNAs, respectively. We were unable to stably maintain the shPea3-1 and shPea3-2 cells; therefore, Western blot analyses and different biological assays were performed using transiently infected cells at 72 hours after infection.
Plasmids
The pLKO.1 shScr plasmid (ID:17920) was obtained from Addgene (Cambridge, MA; donated by Professor Sheila A. Stewart), and the pLKO.1-shPea3-1 and shPea3-2 plasmids (ID: NM_001986.1-736s1c1 and NM_001986.1-1264s1c1) were obtained from Sigma (St Louis, MO). The pBabe-vector was obtained from Addgene (ID: 1764), and the pBabe-FlagPea3 plasmid was generated by cloning of the Flag-Pea3 from pCMV7.1-Pea3 (a gift from Prof. John A. Hassell) into the pBabe vector.
RT-PCR
RNA was extracted by TRIzol (Invitrogen), and reverse transcription was performed using the SuperScript III first strand synthesis system (Invitrogen), according to the manufacturer's instructions. PCR was performed using Phusion High Fidelity Taq polymerase (New England Biolabs, Ipswich, UK), according to the manufacturer's instructions. The primers for different PCR products are listed in Table 2. Quantitative RT-PCR was performed according to the manufacturer's instructions (Applied Biosystems, Foster City, CA) using Pea3 (Hs00385910_m1) and MMP13 (Hs00233992_m1) Taqman assays.
Table 2.
Details of Primers for PCRs
| RT-PCR products | Primers |
|---|---|
| MMP1 | |
| Forward | 5′-TTACATCGTGTTGCGGCTCATGAA-3′ |
| Reverse | 5′-CGGACTTCATCTCTGTCGGCAAAT-3′ |
| MMP2 | |
| Forward | 5′-ACCGCGACAAGAAGTATGGCTTCT-3′ |
| Reverse | 5′-CAAGCCTCGTATACCGCATCAATCTTT-3′ |
| MMP7 | |
| Forward | 5′-CAGGCTCAGGACTATCTCAAGAGA-3′ |
| Reverse | 5′-GTTCATCCTCATCGAAGTGAGCAT-3′ |
| MMP9 | |
| Forward | 5′-CCAACTCGGTTTGGAAACGCAGAT-3′ |
| Reverse | 5′-TCCGGCACTGAGGAATGATCTAAG-3′ |
| MMP13 | |
| Forward | 5′-GGAGCACTCATGTTTCCTATCTAC-3′ |
| Reverse | 5′-GCGAACAATACGGTTACTCCAGAT-3′ |
| MMP14 | |
| Forward | 5′-CATCCAGGGTCTCAAATGGCAACA-3′ |
| Reverse | 5′-GACAAACATCTCCCCTCGGAGCAT-3′ |
| Actin⁎ | |
| Forward | 5′-GTGGGGCGCCCCAGGCACCA-3′ |
| Reverse | 5′-CTCCTTAATGTCACGCACGATTTC-3′ |
Data are from Yuen et al.36
Western Blot Analysis
Western blot analysis was performed as previously described.37 Pea3 (Santa Cruz Biotechnology) and actin (Sigma) monoclonal antibodies were used at a dilution of 1:150 and 1:10,000, respectively. pAkt, total Akt, phospho—extracellular signal-regulated kinase (ERK) 1/2, total ERK1/2, poly (ADP-ribose) polymerase, caspase 3, cleaved caspase 3, caspase 9, and cleaved caspase 9 antibodies (Cell Signaling, Danvers, MA) were used at a dilution of 1:1000.
Colony Formation
Five hundred cells were seeded in a 6-well plate and allowed to grow for 10 days. The cells were then fixed with 70% ethanol, and the colonies were stained with 0.1% crystal violet. The plate was photographed, and the number of colonies was counted.
Crystal Violet Proliferation Assay
One thousand cells were seeded in a 24-well plate and allowed to grow for 24 hours. The cells were fixed with 70% ethanol and stained with 0.1% crystal violet. After washing, the retained crystal violet was extracted using 70% ethanol and measured by spectrophotometric analysis at 590 nm.
BrdU Cell Proliferation Analysis
5-Bromo-2′-deoxyuridine (BrdU) incorporation was performed using the BrdU cell proliferation assay (Calbiochem, Darmstadt, Germany), according to the manufacturer's instructions.
Soft Agar Nonadherent Growth Assay
Five thousand EC109 cells resuspended in medium containing 0.35% (w/v) low melting point agarose were laid on top of a solidified bottom layer of 0.7% (w/v) agarose in normal medium. The top layer was allowed to solidify, and a few drops of medium were added to the solidified agarose. The cells were incubated at 37°C, and a few drops of medium were added to the cells every 2 days until formed colonies could be visualized. The plate was photographed, and the formed colonies were counted.
Migration and Invasion Assays
EC109 cells were washed twice with 1× PBS and resuspended at densities of 5 × 104 and 2.5 × 105 cells per 200 μL in serum- and phenol red—free medium for the migration and invasion assays, respectively. The cells were added to the upper chamber of a Boyden chamber with or without Matrigel (1 mg/mL) for the invasion and migration assays, respectively. Phenol red—free medium, supplemented with 10% fetal bovine serum, was used as the chemoattractant. Migration and invasion were allowed to proceed for 16 and 24 hours, respectively, at 37°C.
Flow Cytometry Analysis of Propidium Iodide Staining
Trypsinized and floating cells were collected and fixed with 70% ethanol overnight at 4°C. Fixed cells were incubated with propidium iodide (PI) (10 μg/mL) and RNase A (10 μg/mL) for 30 minutes at 37°C, and the stained cells were analyzed by flow cytometry using an LSRII flow cytometer (BD Biosciences, Oxford, UK).
MTT Assay
MTT reagent (Melford, Ipswich, UK) was added to the cells at a concentration of 0.2 mg/mL. The cells were incubated at 37°C for 3 hours. The crystalized MTT was then allowed to dissolve in dimethyl sulfoxide. Absorbance at 570 nm was measured. Background reading was subtracted, and the data were presented as percentage of control wells.
Statistical Analysis of the in Vitro Assays
The Student's t-test was used to determine the statistical significance between different groups of samples in our in vitro studies.
Results
Pea3 and Erm, but Not Er81, Are Overexpressed in ESCC
Pea3 was either moderately or strongly expressed in 54.5% (42 of 77) of the ESCC specimens in both nuclear and cytoplasmic regions, whereas all of the nontumor esophageal epithelium specimens (32 of 32) demonstrated either negative or weak staining for Pea3 (Figure 1, A and B; χ2 test, P < 0.001). High expression levels of Erm were detected in 56.6% (43 of 76) of the ESCC specimens, predominantly in the nuclear region, but only 30% (9 of 30) of the nontumor esophageal epithelia expressed similar levels of Erm (Figure 1, A and B; χ2 test, P = 0.013). In contrast, the expression levels of Er81 were not significantly different between nontumor esophageal epithelium and ESCC specimens; the predominantly stained region is the cytoplasm in both types of specimens (Figure 1, A and B; χ2 test, P = 0.183). These results show that Pea3 and Erm, but not Er81, were overexpressed in our ESCC cohort. To avoid the significant differences in Pea3 staining because of interpatient variations of Pea3 expression, the Pea3 staining was compared between the paired nontumor and ESCC specimens. Of the 32 nontumor esophageal epitheliums, 21 of the paired ESCC specimens had interpretable spots of Pea3 staining in the TMA section. The mean scores of Pea3 staining were 0.57 and 1.67 in the 21 nontumor and ESCC paired specimens, respectively (P < 0.001, paired t-test for paired specimens; Figure 1C).
Figure 1.
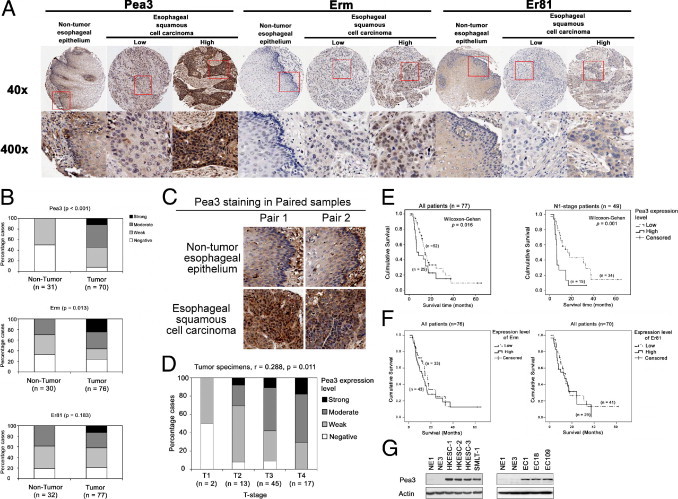
IHC staining of Pea3, Erm, and Er81 and their correlation with clinicopathological parameters. A: Representative images of nontumor esophageal epithelium and ESCC specimens stained for Pea3, Erm, and Er81. Original magnification: ×40 (top); ×400 (bottom). B: Bar charts showing the percentage of cases that demonstrated staining with different levels of Pea3, Erm, and Er81 in nontumor and tumor specimens. C: Representative images of paired nontumor and tumor specimens stained for Pea3. D: Bar chart showing the percentage of cases that demonstrated staining with different levels of Pea3 in specimens with different T stages; the result of Spearman's rank correlation test for all tumor specimens is shown. E: Kaplan-Meier analyses of Pea3 in the whole ESCC patient cohort (left) and in patients with N1 tumors (right). F: Kaplan-Meier analyses of Erm (left) and Er81 (right) in the whole ESCC patient cohort. All Kaplan-Meier analyses were compared using the Wilcoxon-Gehan test. G: Pea3 expression was undetectable in the NE1 and NE3 immortalized esophageal epithelial cell lines and was highly expressed in the esophageal cancer cell lines HKESC-1, HKESC-2, HKESC-3, SMLT-1, EC1, EC18, and EC109. A representative image from three independent experiments is shown.
Pea3 Expression Level Correlates with Tumor Progression and the Overall Survival of Patients with ESCC
The expression levels of Pea3, Erm, and Er81 were correlated with clinicopathological parameters, including age, TNM stage, and histological grade of the tumors. The expression level of Pea3 in the primary tumors showed a significant positive correlation with the T stage (Figure 1D; Spearman's rank test, r = 0.288, P = 0.011) but not with the other clinicopathological parameters. On the other hand, the expression levels of Erm and Er81 did not demonstrate a significant correlation with any of the clinicopathological parameters tested. A high level of Pea3 expression was associated with a shorter survival in the entire cohort (Figure 1E, left; Wilcoxon-Gehan test, P = 0.016) and in patients with N1 stage disease (Figure 1E, right; Wilcoxon-Gehan test, P = 0.001). No association with survival was observed for Erm and Er81 (Figure 1F). Our results suggest that Pea3 overexpression may promote ESCC progression and, therefore, may be a useful prognostic marker in ESCC.
Pea3 Is Expressed in ESCC Cells but Not in Immortalized Esophageal Epithelial Cells
Pea3 expression in two immortalized esophageal epithelial cell lines (ie, NE1 and NE3) and in seven ESCC cell lines (ie, HKESC-1, HKESC-2, HKESC-3, SMLT-1, EC1, EC18, and EC109) was assessed by Western blot analysis. We found that Pea3 was highly expressed in the seven ESCC cell lines but was not detectable in the NE1 and NE3 cell lines (Figure 1G). This result is consistent with our findings in human ESCC specimens, which demonstrated that Pea3 was overexpressed in tumors compared with nontumor esophageal epithelia.
Pea3 Knockdown Reduces Proliferation and Colony Formation in Noninvasive HKESC-1 and HKESC-2 Cells
Pea3 expression in HKESC-1 and HKESC-2 cells was knocked down by lentiviral infection using two Pea3 shRNA sequences, which were designated as shPea3-1 and shPea3-2. As demonstrated by Western blot analysis, Pea3 expression was reduced in cells expressing shPea3-1 or shPea3-2 compared with those expressing shScr RNA in both HKESC-1 and HKESC-2 cells (Figure 2, A and E). HKESC-1 shPea3-1 or shPea3-2 cells formed significantly fewer colonies (mean ± SD, 24 ± 4 and 23 ± 6, respectively; P < 0.01 for both sequences) than HKESC-1 shScr cells (mean ± SD, 58 ± 8) in the colony formation assay (Figure 2B). Similar results were obtained using HKESC-2 cells; HKESC-2 shScr cells formed significantly more colonies (mean ± SD, 75 ± 15) than HKESC-2 shPea3-1 and shPea3-2 cells (mean ± SD, 32 ± 4 and 28 ± 4, respectively; P < 0.01 for both sequences; Figure 2F). HKESC-1 shPea3-1 and shPea3-2 cells also exhibited a significantly lower rate of proliferation compared with HKESC shScr cells, as demonstrated using the crystal violet (Figure 2C) and the BrdU proliferation (Figure 2D) assays. Similar results were obtained using HKESC-2 cells (Figure 2, G and H). These findings suggest that Pea3 knockdown reduced the proliferation rate and two-dimensional clonogenicity ability in noninvasive ESCC cells.
Figure 2.
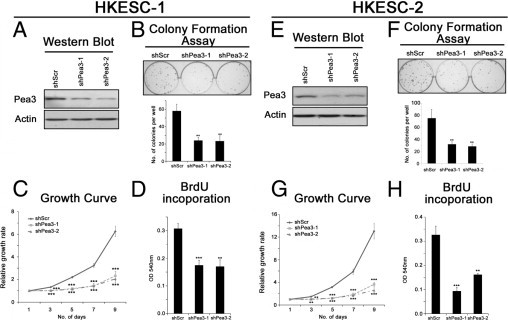
The effects of Pea3 knockdown in HKESC-1 (A–D) and HKESC-2 (E–H) cells. A and E: A representative image from three independent Western blot analyses of Pea3 expression; actin was used as a control to normalize the data. B and F: Colony formation assay to test the clonogenic potential of the cells in two-dimensional culture plates. C and G: The relative growth rates of the cells were determined by crystal violet staining during a 9-day period. D and H: BrdU incorporation was performed during an 8-hour period to determine the rate of DNA synthesis. Mean ± SD values were obtained from three independent experiments. **P < 0.01 and ***P < 0.001 compared with the Scr control.
Pea3 Knockdown Reduces the Proliferation, Nonadherent Growth, Migration, and Invasion of Invasive EC109 Cells
Similar to HKESC-1 and HKESC-2 cells, Pea3 knockdown in EC109 invasive ESCC cells reduced the amount of colony formation and cell proliferation (Figure 3, A–D). To test whether Pea3 knockdown correlates with reduced metastatic potential, the nonadherent growth and migratory and invasive abilities of EC109 cells expressing various levels of Pea3 were tested. As shown in Figure 3E, Pea3 knockdown resulted in a significant reduction of the nonadherent growth of EC109 cells, as reflected by a decrease in the number of colonies formed in the soft agar assay (mean ± SD; shScr, 345 ± 40; shPea3-1, 47 ± 20; shPea3-2, 27 ± 18). Pea3 knockdown also resulted in a significant reduction of both migration (mean ± SD, 66% ± 7% and 72% ± 9% reduction in shPea3-1 and shPea-2, respectively; Figure 3F) and invasion (mean ± SD, 55% ± 4% and 71% ± 4% reduction in shPea3-1 and shPea3-2, respectively; Figure 3G). The migration and invasion assays were performed for 16 and 24 hours, respectively; as shown in Figure 4C, the proliferation of EC109 cells was not significantly affected by Pea3 knockdown at the 24-hour point, indicating that the effects seen in the migration and invasion assays were not an artifact of the inhibition of proliferation cased by Pea3 knockdown. Therefore, our results suggest that Pea3 knockdown suppresses the migration and invasion of invasive ESCC cells.
Figure 3.
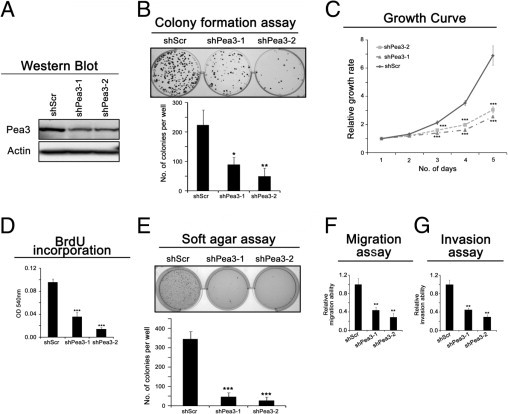
The effect of Pea3 on the invasive esophageal cancer cell line EC109. A: A representative image of Pea3 expression from three independent Western blot analyses; actin was used as a control to normalize the data. B: Colony formation assay to test the clonogenic potential of the cells in two-dimensional culture plates. C: The relative growth rate of the cells was determined by crystal violet staining during a 5-day period. D: BrdU incorporation was performed during a 1-hour period to determine the rate of DNA synthesis. E: Soft agar assay to test the nonadherent growth ability of the cells. F: The migration assay was performed in a Boyden chamber during a 16-hour period. G: The invasion assay was performed in a Matrigel-coated Boyden chamber during a 24-hour period. Mean ± SD values were obtained from three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the Scr control.
Figure 4.
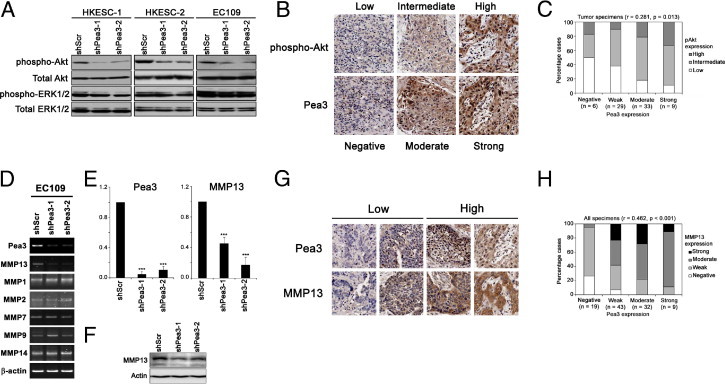
Correlation of Pea3 and pAkt expression. A: A representative Western blot analysis image from three independent experiments showing pAkt, total Akt, phospho-ERK, and total ERK expression. The expression levels of total Akt and total ERK were used as a control. B: Representative IHC staining images showing pAkt and Pea3 staining of three tissue cores. C: A bar chart showing the percentage of specimens that demonstrated different expression levels of pAkt and Pea3 by IHC staining. The result of the Spearman's rank correlation test for all of the tumor specimens is presented. D: RT-PCR of different MMPs that were regulated by Pea3 in other cellular contexts. E: Quantitative real-time PCR showing that EC109 shScr cells express significantly higher levels of mRNA of Pea3 (left) and MMP13 (right) compared with EC109 shPea3-1 and shPea3-2 cells. F: The protein expression of MMP13 was reduced on Pea3 knockdown in EC109 cells. A representative image from three independent experiments is shown. G: Representative IHC staining images for specimens stained with low or high levels of Pea3 and MMP13. The same core is shown in the same column. H: A bar chart showing the percentage of specimens that demonstrated different expression levels of MMP13 and Pea3 by IHC staining. The result of Spearman's rank correlation test for all of the specimens is presented. ***P < 0.001 compared with the shScr control.
Pea3 Expression Level Is Associated with pAkt Expression Level in ESCC Cell Lines and Human Specimens
The activity of Akt and ERK plays an important role in cell proliferation, migration, and invasion. We investigated the expression levels of the active phosphorylated forms of Akt and ERK in Pea3 knockdown cells and found that Pea3 knockdown reduced the phosphorylation of Akt, but not that of ERK, in the three ESCC cell lines tested (Figure 4A). These results demonstrate that Pea3 might interrupt signaling upstream of Akt but not the global phosphorylation of other proteins and suggest that Pea3 is required for the full activation of Akt in ESCC cells. We confirmed this in vitro finding in our ESCC specimens, in which the expression level of Pea3 demonstrated a significant positive correlation (Spearman's rank correlation, r = 0.281, P = 0.013; Figure 4, B and C). Taken together, our results suggest that Pea3 might promote ESCC progression via activation of the PI3K-Akt pathway.
MMP13 May Be Involved in Pea3 Knockdown-Mediated Decreases in Invasiveness of EC109 Cells
Pea3 transcriptionally activates different MMP expression in different cellular contexts. In this study, we found that Pea3 knockdown resulted in down-regulation of the MMP13 mRNA level but not of the other MMPs tested (Figure 4D). The decrease in Pea3 and MMP13 mRNA levels in shPea3-infected EC109 cells was further confirmed by quantitative RT-PCR (Figure 4E). The protein level of MMP13 was also down-regulated in EC109 infection with either shPea3-1 or shPea3-2. To confirm the clinical significance of our in vitro findings, we performed IHC staining in our patient specimens. The MMP13 expression pattern was similar to that of Pea3. It was significantly overexpressed in ESCC compared with nontumor esophageal epithelium specimens (P < 0.001). Pea3 and MMP13 expression levels were significantly positively correlated in all patient specimens (Spearman's rank test, r = 0.462, P < 0.001; Figure 4, G and H). These results suggest that Pea3 may regulate MMP13 to promote the invasiveness of the invasive EC109 cells.
Pea3 Knockdown Sensitizes EC109 Cells to Doxorubicin Treatment
In addition to Pea3-enhanced aggressiveness in ESCC cell lines, we also found that Pea3 knockdown sensitized EC109 cells to doxorubicin treatment. EC109 cells were relatively resistant to doxorubicin treatment compared with the NE1 immortalized esophageal cell line. The treatment of EC109 cells with up to 2 μg/mL doxorubicin for 48 hours resulted in only 5% sub-G1 apoptotic cells, whereas the treatment of NE1 cells with 200 ng/mL doxorubicin resulted in 64% sub-G1 apoptotic cells (Figure 5A). EC109 cells lacking Pea3 were significantly more susceptible to treatment with 1 μg/mL doxorubicin (mean ± SD, 12% ± 2% and 15% ± 4%, respectively) compared with EC109 shScr control cells (mean ± SD, 4% ± 2%; Figure 5, B and C). Similarly, EC109 shPea3-1 and shPea3-2 cells demonstrated higher levels of cleaved caspase 9, caspase 3, and poly (ADP-ribose) polymerase than EC109 shScr cells when treated with the same concentration of doxorubicin (Figure 5D). As shown in Figure 5E, inhibition of the PI3K-Akt-mammalian target of Rapamycin complex 1 (mTORC1) pathway by PI103 resulted in sensitization of EC109 shScr cells to doxorubicin treatment for 48 hours, as measured by the MTT assay (Figure 5E). Increased susceptibility of EC109 shPea3-1 and shPea3-2 cells to doxorubicin compared with EC109 shScr control cells was also observed using the MTT assay at 48 hours (Figure 5E). In addition, we found that EC109 shPea3-1 and shPea3-2 cells, treated by a combination of PI103 and doxorubicin, had a significant reduction in cell survival compared with EC109 shScr control cells (Figure 5E). Our results were further confirmed by Western blot analysis of apoptotic markers, including cleavages of poly (ADP-ribose) polymerase, caspase 3, and caspase 9 (Figure 5F). These results suggest that the PI3K-Akt-mTORC1 pathway may be an important survival determinant in EC109 cells in the presence of doxorubicin and that the down-regulation of pAkt levels in Pea3 knocked down EC109 cells may explain the increased sensitivity of the cells to doxorubicin.
Figure 5.
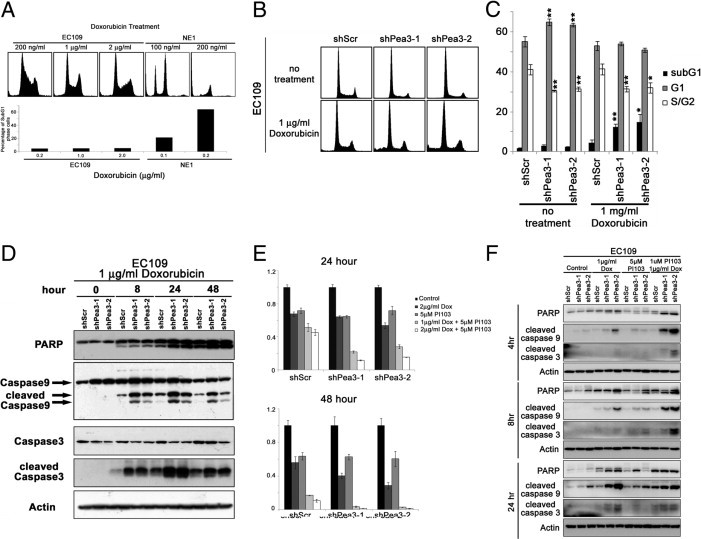
Pea3 modulates the sensitivity of EC109 cells to doxorubicin treatment. A: PI-stained cells showing that the esophageal cancer cell line EC109 is highly resistant to doxorubicin treatment compared with immortalized NE1 cells. The results are representative of two independent experiments. B: Representative histograms of PI-stained cells under different conditions. C: A bar chart showing the percentage of cells in different phases of the cell cycle. D: Western blot analysis was used to detect apoptotic protein markers in cells that were treated with doxorubicin at different points during a 48-hour period. An MTT assay (E) and Western blot analysis (F) were used to detect apoptotic protein markers of EC109 cells expressing various levels of Pea3 that were treated with PI103 and doxorubicin, alone or in combination. *P < 0.05, **P < 0.01 compared with the shScr control.
Knockdown of Pea3 Enhances Paclitaxel Resistance in EC109 Cells
EC109 shPea3-1 and shPea3-2 cells were more resistant to paclitaxel compared with EC109 shScr control cells. Because of the higher proliferation rate of EC109 shScr cells compared with EC109 shPea3-1 and shPea3-2 cells, the higher sensitivity of EC109 shScr cells to paclitaxel treatment was not unexpected. As shown in Figure 6, A and B, actively cycling EC109 shScr cells demonstrated a higher percentage of S-phase cells compared with actively cycling EC109 shPea3-1 and shPea3-2 cells, which is in accordance with the higher proliferation rate of EC109 shScr cells. The EC109 shScr cells were more efficiently arrested in the G2/M phase at 24 hours after treatment with paclitaxel than the EC109 shPea3-1 and shPea3-2 cells (Figure 6B). Moreover, EC109 shScr cells showed a higher percentage of G1-phase cells at 2 hours after release from nocodazole-mediated G2/M arrest compared with EC109 shPea3-1 and shPea3-2 cells (Figure 6C). These results imply that the increased proliferation of EC109 shScr cells might be because of the increased rate of cell cycling, and this increase may be an explanation for the higher sensitivity of EC109 shScr cells to paclitaxel compared with EC109 shPea3 cells. Our results also suggest that the increased sensitivity of EC109 cells to doxorubicin on Pea3 knockdown might not be the result of a general modulation of the apoptotic machinery.
Figure 6.
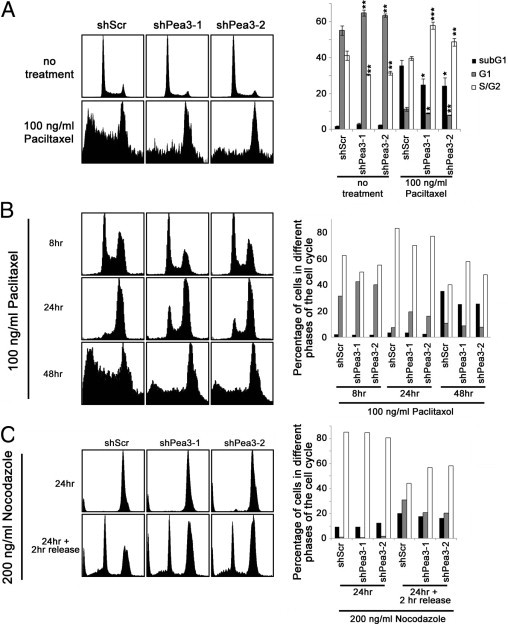
Pea3 modulates the sensitivity of EC109 cells to paclitaxel treatment. A: Representative histograms of PI-stained cells with or without treatment of paclitaxel (right). Representative images are from three independent experiments. A bar chart showing the percentage of cells in different phases of the cell cycle (left). Mean ± SD values from three independent experiments were obtained. Pea3 knockdown results in a decreased rate of cell cycle progression. B: More cells were arrested in the G2/M phase after 24 hours of paclitaxel treatment, and an elevated apoptotic response was detected in EC109 shScr cells compared with EC109 shPea3-1 and shPea3-2 cells. The results are representative of two independent experiments. C: More cells passed through the G2/M phase 2 hours after release from nocodazole-induced G2/M arrest in EC109 shScr cells compared with EC109 shPea3-1 and shPea3-2 cells. The results are representative of two independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the shScr control.
Overexpression of Pea3 in NE1 and NE3 Immortalized Esophageal Epithelial Cells
By using retroviral infection, Flag-Pea3 was overexpressed in NE1 and NE3 immortalized esophageal epithelial cells (Figure 7A). The expression of Pea3 was not persistent in these two cell lines and became low or undetectable by Western blot analysis after three passages (data not shown). By using the cells within the three passages, we found that NE1 and NE3 cells overexpressing Flag-Pea3 were unable to grow in soft agar or invade through Matrigel. This may be because of the transience of Pea3 overexpression. On the other hand, Pea3 may also be essential, but not sufficient, to drive ESCC progression. Nevertheless, overexpression of Flag-Pea3 significantly reduced doxorubicin-induced cell death in both NE1 and NE3 cells compared with their vector controls (Figure 7, B and C), suggesting that Pea3 may be an important factor in governing the sensitivity of esophageal epithelial cells to doxorubicin treatment.
Figure 7.
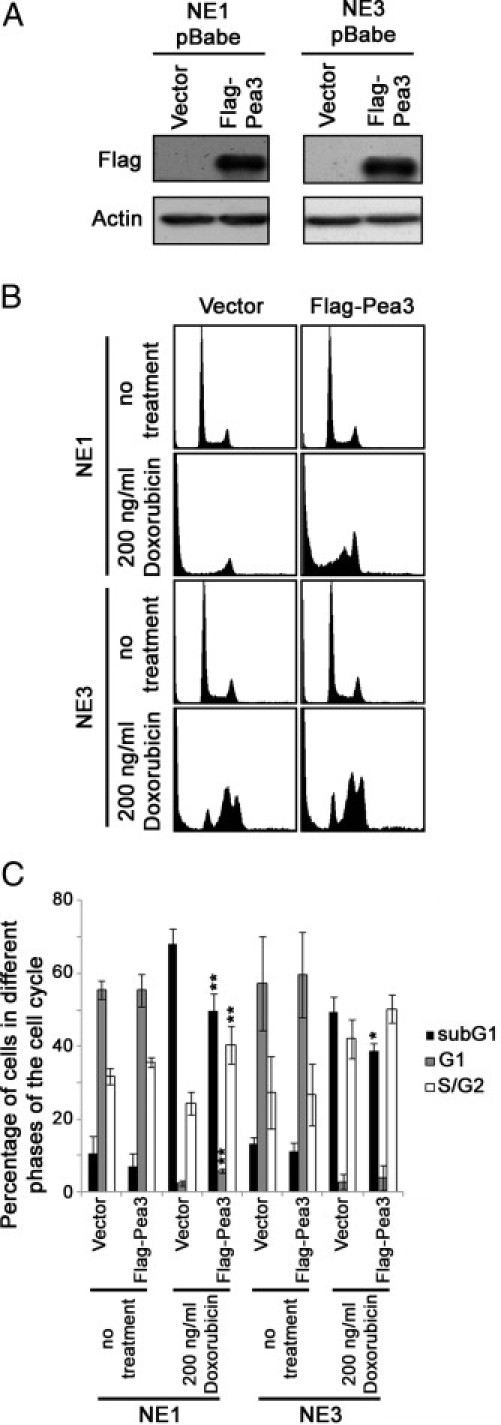
NE1 and NE3 cells expressing various levels of Pea3 and their sensitivity to doxorubicin treatment. A: Western blot analyses were used to detect Flag-Pea3 overexpression. B: Representative histograms of PI-stained cells under different conditions. C: A bar chart showing the percentage of cells in various phases of the cell cycle. Mean ± SD values were obtained from three independent experiments. *P < 0.05 and **P < 0.01 compared with the Scr/empty vector control.
Discussion
In the present study, IHC was performed to investigate the expression levels of the transcription factor members of the Pea3 subgroup of the Ets transcription factor family. Pea3 and Erm were highly expressed in human ESCC specimens. A high expression level of Pea3 correlated with a higher T stage and a shorter patient survival. In addition, Pea3 was highly expressed in ESCC cell lines but was not detectable in immortalized esophageal epithelial cells. Pea3 knockdown in ESCC cells resulted in a decrease in aggressiveness and an increase in doxorubicin sensitivity. Our results suggest that Pea3 is a potential prognostic factor and therapeutic target in ESCC.
In a recent study38 on abnormal changes in chromosomal regions in ESCC development, chromosomal region 17q21 was amplified in 86.3%, 44 of 51, patients with ESCC. Although Pea3 is located in 17q21, it is tempting to speculate that 17q21 amplification might explain for the Pea3 overexpression in ESCC. In the same study,38 patients with 17q21 amplification had a worse prognosis in terms of survival, which is in line with our present study showing a significant correlation between a high level of Pea3 expression and shorter patient survival. Further investigation on whether Pea3 accounts for the aggressive phenotype observed in ESCC with 17q21 amplification is highly warranted.
Pea3 transcriptionally up-regulates various metastasis-promoting proteins, such as MMPs and COX-2. MMPs are enzymes that assist cancer cells in metastasis via digestion of the extracellular matrix.39 Indeed, EC109 shPea3-1 and shPea3-2 cells that expressed a lower level of Pea3 demonstrated a lower expression level of MMP13 compared with EC109 shScr cells (Figure 4, D–F), which has been regulated by Pea3 in other cellular contexts.12 These observations suggest that MMP13 might be an MMP that functions downstream of Pea3, which modulates Pea3-mediated metastatic progression in EC109 cells. COX-2 plays important roles in the progression of ESCC by enhancing cellular proliferation and invasion and inhibiting apoptosis.40–44 In the present study, we found that Pea3 knockdown reduced the Akt phosphorylation in cancer cells, whereas Pea3 expression correlated significantly with pAkt expression in human ESCC specimens. These findings suggest that the PI3K-Akt pathway may be a downstream target of Pea3. PI3K-Akt pathway activity has been an important prognostic determinant of ESCC.45–47 COX-2 inhibition reduces Akt phosphorylation in ovarian, prostate, and esophageal cancers,48–50 implying that COX-2 might be a key intermediate between Pea3 and Akt. However, COX-2 mRNA expression levels were not significantly different between EC109 shScr, shPea3-1, and shPea3-2 cells. This result suggests that COX-2 may not be involved in the Pea3-mediated down-regulation of pAkt in EC109 cells and that further experiments are required to delineate the mechanisms.
Pea3 expression has modulated the sensitivity to different chemotherapeutic drugs; Pea3 expression is increased in cisplatin-resistant ovarian cancer cells.51 cGMP-dependent protein kinase protects cells against oxidative stress via up-regulation of thioredoxin in a Pea3 binding site—dependent manner.52 On the other hand, Pea3 is required for apoptosis induced by γ-linolenic acid53 and etoposide.54 The differential effects of Pea3 knockdown on doxorubicin and paclitaxel treatments suggest that Pea3-mediated resistance to doxorubicin in EC109 may not represent a general modulation of the apoptosis or survival machinery of the cells. The mechanisms that lead to enhanced resistance to doxorubicin require further investigation. The use of doxorubicin55,56 and taxane-based drugs57 for the treatment of esophageal cancer has been described; the potential use of Pea3 as a biomarker to predict drug efficacy may warrant further investigation. Inhibition of PI3K-Akt-mTORC1–sensitized EC109 cells to doxorubicin treatment suggests that this pathway may be important in governing doxorubicin resistance. Indeed, EC109 shPea3-1 and shPea3-2 cells had lower levels of pAkt; they were also more sensitive to treatment of doxorubicin alone or in combination with PI103 compared with EC109 shScr control cells. These results suggest that the down-regulation of the activity of the PI3K-Akt-mTORC1 pathway in EC109 cells on Pea3 knockdown may lead to the increase in the sensitivity of the cells to doxorubicin. Further investigation is required for a better understanding. Because of the heterogeneity of postesophagectomy chemotherapy in our patient cohort, no useful information could be obtained on reviewing the clinical data. Further investigations using randomized controlled clinical trials would be required to confirm these in vitro findings.
The progression-elevated gene 3 promoter is active in cancer cells but not in healthy cells.58 The cancer-specific activity of this promoter is governed by Pea3 binding sites on the promoter, and this Pea3–progression-elevated gene 3 link promotes the transformation of rat embryonic cells.59,60 The importance of Pea3 group transcription factors in tumorigenesis is further strengthened by a recent study demonstrating that Pea3 and Erm are required for the tumorigenesis of mammary cancer cells. In that study,30 the regulatomes of the two transcription factors were different, suggesting that Pea3 and Erm regulate distinct, but complementary, molecular networks. In the present study, we found that both Erm and Pea3 were overexpressed in human ESCC compared with their nontumor epithelia. Although Erm expression did not correlate with any clinicopathological data, Erm might play a distinct role from Pea3 in ESCC initiation, which warrants further investigation. Whether Pea3 and Erm regulate different gene networks during ESCC onset and whether their regulatomes are complementary to each other for tumor onset would be a focus of great interest.
In this study, we also attempted to stably overexpress Pea3 in both NE1 and NE3 cells by using pBabe-Pea3 and pBabe-FlagPea3. Although Pea3 overexpression could be achieved, it was lost after two to three passages (data not shown). NE1 and NE3 cells overexpressing Pea3 were more resistant to doxorubicin, but they were unable to form colonies in soft agar or invade through Matrigel (data not shown). This result suggests that a transient increase in Pea3 may not be sufficient to transform immortalized esophageal epithelial cells.
In conclusion, we found that a high level of Pea3 is associated with a shorter survival of patients with ESCC and that Pea3 promotes cancer cell aggressiveness in vitro. Our results indicate that Pea3 may be a useful prognostic marker and therapeutic target for this common cancer that demonstrates a generally poor clinical outcome.
Acknowledgment
We thank Professor George S.W. Tsao for providing the NE1 and NE3 immortalized esophageal cell lines and the EC1, EC18, and EC109 ESCC cell lines.
Footnotes
Supported by a postdoctoral fellowship from Cancer Research UK (H.-F.Y.).
CME Disclosure: The authors did not disclose any relevant financial relationships.
Contributor Information
Kwok-Wah Chan, Email: kwchan@pathology.hku.hk.
Mohamed El-Tanani, Email: m.el-tanani@qub.ac.uk.
References
- 1.Hongo M., Nagasaki Y., Shoji T. Epidemiology of esophageal cancer: Orient to Occident: effects of chronology, geography and ethnicity. J Gastroenterol Hepatol. 2009;24:729–735. doi: 10.1111/j.1440-1746.2009.05824.x. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M.J. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 3.Klein C.A., Stoecklein N.H. Lessons from an aggressive cancer: evolutionary dynamics in esophageal carcinoma. Cancer Res. 2009;69:5285–5288. doi: 10.1158/0008-5472.CAN-08-4586. [DOI] [PubMed] [Google Scholar]
- 4.Bird-Lieberman E.L., Fitzgerald R.C. Early diagnosis of oesophageal cancer. Br J Cancer. 2009;101:1–6. doi: 10.1038/sj.bjc.6605126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Launoit Y., Baert J.L., Chotteau A., Monte D., Defossez P.A., Coutte L., Pelczar H., Leenders F. Structure-function relationships of the PEA3 group of Ets-related transcription factors. Biochem Mol Med. 1997;61:127–135. doi: 10.1006/bmme.1997.2605. [DOI] [PubMed] [Google Scholar]
- 6.de Launoit Y., Baert J.L., Chotteau-Lelievre A., Monte D., Coutte L., Mauen S., Firlej V., Degerny C., Verreman K. The Ets transcription factors of the PEA3 group: transcriptional regulators in metastasis. Biochim Biophys Acta. 2006;1766:79–87. doi: 10.1016/j.bbcan.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Kurpios N.A., Sabolic N.A., Shepherd T.G., Fidalgo G.M., Hassell J.A. Function of PEA3 Ets transcription factors in mammary gland development and oncogenesis. J Mammary Gland Biol Neoplasia. 2003;8:177–190. doi: 10.1023/a:1025948823955. [DOI] [PubMed] [Google Scholar]
- 8.de Launoit Y., Chotteau-Lelievre A., Beaudoin C., Coutte L., Netzer S., Brenner C., Huvent I., Baert J.L. The PEA3 group of ETS-related transcription factors: role in breast cancer metastasis. Adv Exp Med Biol. 2000;480:107–116. doi: 10.1007/0-306-46832-8_13. [DOI] [PubMed] [Google Scholar]
- 9.Crawford H.C., Fingleton B., Gustavson M.D., Kurpios N., Wagenaar R.A., Hassell J.A., Matrisian L.M. The PEA3 subfamily of Ets transcription factors synergizes with beta-catenin-LEF-1 to activate matrilysin transcription in intestinal tumors. Mol Cell Biol. 2001;21:1370–1383. doi: 10.1128/MCB.21.4.1370-1383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bièche I., Tozlu S., Girault I., Onody P., Driouch K., Vidaud M., Lidereau R. Expression of PEA3/E1AF/ETV4, an Ets-related transcription factor, in breast tumors: positive links to MMP2, NRG1 and CGB expression. Carcinogenesis. 2004;25:405–411. doi: 10.1093/carcin/bgh024. [DOI] [PubMed] [Google Scholar]
- 11.Cowden Dahl K.D., Zeineldin R., Hudson L.G. PEA3 is necessary for optimal epidermal growth factor receptor-stimulated matrix metalloproteinase expression and invasion of ovarian tumor cells. Mol Cancer Res. 2007;5:413–421. doi: 10.1158/1541-7786.MCR-07-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan J., Erdem H., Li R., Cai Y., Ayala G., Ittmann M., Yu-Lee L.Y., Tsai S.Y., Tsai M.J. Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res. 2008;68:5460–5468. doi: 10.1158/0008-5472.CAN-08-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin L., Liao L., Redmond A., Young L., Yuan Y., Chen H., O'Malley B.W., Xu J. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol Cell Biol. 2008;28:5937–5950. doi: 10.1128/MCB.00579-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howe L.R., Crawford H.C., Subbaramaiah K., Hassell J.A., Dannenberg A.J., Brown A.M. PEA3 is up-regulated in response to Wnt1 and activates the expression of cyclooxygenase-2. J Biol Chem. 2001;276:20108–20115. doi: 10.1074/jbc.M010692200. [DOI] [PubMed] [Google Scholar]
- 15.Subbaramaiah K., Norton L., Gerald W., Dannenberg A.J. Cyclooxygenase-2 is overexpressed in HER-2/neu-positive breast cancer: evidence for involvement of AP-1 and PEA3. J Biol Chem. 2002;277:18649–18657. doi: 10.1074/jbc.M111415200. [DOI] [PubMed] [Google Scholar]
- 16.Upadhyay S., Liu C., Chatterjee A., Hoque M.O., Kim M.S., Engles J., Westra W., Trink B., Ratovitski E., Sidransky D. LKB1/STK11 suppresses cyclooxygenase-2 induction and cellular invasion through PEA3 in lung cancer. Cancer Res. 2006;66:7870–7879. doi: 10.1158/0008-5472.CAN-05-2902. [DOI] [PubMed] [Google Scholar]
- 17.Hiroumi H., Dosaka-Akita H., Yoshida K., Shindoh M., Ohbuchi T., Fujinaga K., Nishimura M. Expression of E1AF/PEA3, an Ets-related transcription factor in human non-small-cell lung cancers: its relevance in cell motility and invasion. Int J Cancer. 2001;93:786–791. doi: 10.1002/ijc.1410. [DOI] [PubMed] [Google Scholar]
- 18.Hakuma N., Kinoshita I., Shimizu Y., Yamazaki K., Yoshida K., Nishimura M., Dosaka-Akita H. E1AF/PEA3 activates the Rho/Rho-associated kinase pathway to increase the malignancy potential of non-small-cell lung cancer cells. Cancer Res. 2005;65:10776–10782. doi: 10.1158/0008-5472.CAN-05-0060. [DOI] [PubMed] [Google Scholar]
- 19.Trimble M.S., Xin J.H., Guy C.T., Muller W.J., Hassell J.A. PEA3 is overexpressed in mouse metastatic mammary adenocarcinomas. Oncogene. 1993;8:3037–3042. [PubMed] [Google Scholar]
- 20.Myers E., Hill A.D., Kelly G., McDermott E.W., O'Higgins N.J., Young L.S. A positive role for PEA3 in HER2-mediated breast tumour progression. Br J Cancer. 2006;95:1404–1409. doi: 10.1038/sj.bjc.6603427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galang C.K., Muller W.J., Foos G., Oshima R.G., Hauser C.A. Changes in the expression of many Ets family transcription factors and of potential target genes in normal mammary tissue and tumors. J Biol Chem. 2004;279:11281–11292. doi: 10.1074/jbc.M311887200. [DOI] [PubMed] [Google Scholar]
- 22.Benz C.C., O'Hagan R.C., Richter B., Scott G.K., Chang C.H., Xiong X., Chew K., Ljung B.M., Edgerton S., Thor A., Hassell J.A. HER2/Neu and the Ets transcription activator PEA3 are coordinately upregulated in human breast cancer. Oncogene. 1997;15:1513–1525. doi: 10.1038/sj.onc.1201331. [DOI] [PubMed] [Google Scholar]
- 23.Kinoshita J., Kitamura K., Tanaka S., Sugimachi K., Ishida M., Saeki H. Clinical significance of PEA3 in human breast cancer. Surgery. 2002;131:S222–S225. doi: 10.1067/msy.2002.119792. [DOI] [PubMed] [Google Scholar]
- 24.Davidson B., Goldberg I., Gotlieb W.H., Kopolovic J., Ben-Baruch G., Reich R. PEA3 is the second Ets family transcription factor involved in tumor progression in ovarian carcinoma. Clin Cancer Res. 2003;9:1412–1419. [PubMed] [Google Scholar]
- 25.Davidson B., Goldberg I., Tell L., Vigdorchik S., Baekelandt M., Berner A., Kristensen G.B., Reich R., Kopolovic J. The clinical role of the PEA3 transcription factor in ovarian and breast carcinoma in effusions. Clin Exp Metastasis. 2004;21:191–199. doi: 10.1023/b:clin.0000037703.37275.35. [DOI] [PubMed] [Google Scholar]
- 26.Horiuchi S., Yamamoto H., Min Y., Adachi Y., Itoh F., Imai K. Association of ets-related transcriptional factor E1AF expression with tumour progression and overexpression of MMP-1 and matrilysin in human colorectal cancer. J Pathol. 2003;200:568–576. doi: 10.1002/path.1387. [DOI] [PubMed] [Google Scholar]
- 27.Hida K., Shindoh M., Yoshida K., Kudoh A., Furaoka K., Kohgo T., Fujinaga K., Totsuka Y. Expression of E1AF, an ets-family transcription factor, is correlated with the invasive phenotype of oral squamous cell carcinoma. Oral Oncol. 1997;33:426–430. doi: 10.1016/s0964-1955(97)00047-x. [DOI] [PubMed] [Google Scholar]
- 28.Sloan K.A., Marquez H.A., Li J., Cao Y., Hinds A., O'Hara C.J., Kathuria S., Ramirez M.I., Williams M.C., Kathuria H. Increased PEA3/E1AF and decreased Net/Elk-3, both ETS proteins, characterize human NSCLC progression and regulate caveolin-1 transcription in Calu-1 and NCI-H23 NSCLC cell lines. Carcinogenesis. 2009;30:1433–1442. doi: 10.1093/carcin/bgp129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto H., Horiuchi S., Adachi Y., Taniguchi H., Nosho K., Min Y., Imai K. Expression of ets-related transcriptional factor E1AF is associated with tumor progression and over-expression of matrilysin in human gastric cancer. Carcinogenesis. 2004;25:325–332. doi: 10.1093/carcin/bgh011. [DOI] [PubMed] [Google Scholar]
- 30.Firlej V., Ladam F., Brysbaert G., Dumont P., Fuks F., de Launoit Y., Benecke A., Chotteau-Lelievre A. Reduced tumorigenesis in mouse mammary cancer cells following inhibition of Pea3- or Erm-dependent transcription. J Cell Sci. 2008;121:3393–3402. doi: 10.1242/jcs.027201. [DOI] [PubMed] [Google Scholar]
- 31.Chotteau-Lelievre A., Revillion F., Lhotellier V., Hornez L., Desbiens X., Cabaret V., de Launoit Y., Peyrat J.P. Prognostic value of ERM gene expression in human primary breast cancers. Clin Cancer Res. 2004;10:7297–7303. doi: 10.1158/1078-0432.CCR-04-0593. [DOI] [PubMed] [Google Scholar]
- 32.Monge M., Colas E., Doll A., Gonzalez M., Gil-Moreno A., Planaguma J., Quiles M., Arbos M.A., Garcia A., Castellvi J., Llaurado M., Rigau M., Alazzouzi H., Xercavins J., Alameda F., Reventos J., Abal M. ERM/ETV5 up-regulation plays a role during myometrial infiltration through matrix metalloproteinase-2 activation in endometrial cancer. Cancer Res. 2007;67:6753–6759. doi: 10.1158/0008-5472.CAN-06-4487. [DOI] [PubMed] [Google Scholar]
- 33.Planaguma J., Abal M., Gil-Moreno A., Diaz-Fuertes M., Monge M., Garcia A., Baro T., Xercavins J., Reventos J., Alameda F. Up-regulation of ERM/ETV5 correlates with the degree of myometrial infiltration in endometrioid endometrial carcinoma. J Pathol. 2005;207:422–429. doi: 10.1002/path.1853. [DOI] [PubMed] [Google Scholar]
- 34.Monge M., Colas E., Doll A., Gil-Moreno A., Castellvi J., Diaz B., Gonzalez M., Lopez-Lopez R., Xercavins J., Carreras R., Alameda F., Canals F., Gabrielli F., Reventos J., Abal M. Proteomic approach to ETV5 during endometrial carcinoma invasion reveals a link to oxidative stress. Carcinogenesis. 2009;30:1288–1297. doi: 10.1093/carcin/bgp119. [DOI] [PubMed] [Google Scholar]
- 35.Yuen H.F., Chan Y.P., Law S., Srivastava G., El-Tanani M., Mak T.W., Chan K.W. DJ-1 could predict worse prognosis in esophageal squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2008;17:3593–3602. doi: 10.1158/1055-9965.EPI-08-0214. [DOI] [PubMed] [Google Scholar]
- 36.Yuen H.F., Chiu Y.T., Chan K.K., Chan Y.P., Chua C.W., McCrudden C.M., Tang K.H., El-Tanani M., Wong Y.C., Wang X., Chan K.W. Prostate cancer cells modulate osteoblast mineralisation and osteoclast differentiation through Id-1. Br J Cancer. 2010;102:332–341. doi: 10.1038/sj.bjc.6605480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuen H.F., Kwok W.K., Chan K.K., Chua C.W., Chan Y.P., Chu Y.Y., Wong Y.C., Wang X., Chan K.W. TWIST modulates prostate cancer cell-mediated bone cell activity and is upregulated by osteogenic induction. Carcinogenesis. 2008;29:1509–1518. doi: 10.1093/carcin/bgn105. [DOI] [PubMed] [Google Scholar]
- 38.Sakai N., Kajiyama Y., Iwanuma Y., Tomita N., Amano T., Isayama F., Ouchi K., Tsurumaru M. Study of abnormal chromosome regions in esophageal squamous cell carcinoma by comparative genomic hybridization: relationship of lymph node metastasis and distant metastasis to selected abnormal regions. Dis Esophagus. 2010;23:415–421. doi: 10.1111/j.1442-2050.2009.01026.x. [DOI] [PubMed] [Google Scholar]
- 39.Freije J.M., Balbin M., Pendas A.M., Sanchez L.M., Puente X.S., Lopez-Otin C. Matrix metalloproteinases and tumor progression. Adv Exp Med Biol. 2003;532:91–107. doi: 10.1007/978-1-4615-0081-0_9. [DOI] [PubMed] [Google Scholar]
- 40.Zong Y., Zhang S.T., Zhu S.T. Nicotine enhances migration and invasion of human esophageal squamous carcinoma cells which is inhibited by nimesulide. World J Gastroenterol. 2009;15:2500–2505. doi: 10.3748/wjg.15.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zimmermann K.C., Sarbia M., Weber A.A., Borchard F., Gabbert H.E., Schror K. Cyclooxygenase-2 expression in human esophageal carcinoma. Cancer Res. 1999;59:198–204. [PubMed] [Google Scholar]
- 42.Fong L.Y., Zhang L., Jiang Y., Farber J.L. Dietary zinc modulation of COX-2 expression and lingual and esophageal carcinogenesis in rats. J Natl Cancer Inst. 2005;97:40–50. doi: 10.1093/jnci/dji006. [DOI] [PubMed] [Google Scholar]
- 43.Liu J.F., Zhang S.W., Jamieson G.G., Zhu G.J., Wu T.C., Zhu T.N., Shan B.E., Drew P.A. The effects of a COX-2 inhibitor meloxicam on squamous cell carcinoma of the esophagus in vivo. Int J Cancer. 2008;122:1639–1644. doi: 10.1002/ijc.23288. [DOI] [PubMed] [Google Scholar]
- 44.Liu J.F., Zhu G.J., Jamieson G.G., Wu T.C., Zhu T.N., Shan B.E., Drew P.A. NS-398 induces apoptosis in human esophageal cancer cells through inhibition of NF-kappaB downstream regulation of cyclooxygenase-2. Cancer Invest. 2009;27:17–23. doi: 10.1080/07357900801992913. [DOI] [PubMed] [Google Scholar]
- 45.Hildebrandt M.A., Yang H., Hung M.C., Izzo J.G., Huang M., Lin J., Ajani J.A., Wu X. Genetic variations in the PI3K/PTEN/AKT/mTOR pathway are associated with clinical outcomes in esophageal cancer patients treated with chemoradiotherapy. J Clin Oncol. 2009;27:857–871. doi: 10.1200/JCO.2008.17.6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tachibana M., Shibakita M., Ohno S., Kinugasa S., Yoshimura H., Ueda S., Fujii T., Rahman M.A., Dhar D.K., Nagasue N. Expression and prognostic significance of PTEN product protein in patients with esophageal squamous cell carcinoma. Cancer. 2002;94:1955–1960. doi: 10.1002/cncr.0678. [DOI] [PubMed] [Google Scholar]
- 47.Blanco-Aparicio C., Renner O., Leal J.F., Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28:1379–1386. doi: 10.1093/carcin/bgm052. [DOI] [PubMed] [Google Scholar]
- 48.Uddin S., Ahmed M., Hussain A., Assad L., Al-Dayel F., Bavi P., Al-Kuraya K.S., Munkarah A. Cyclooxygenase-2 inhibition inhibits PI3K/AKT kinase activity in epithelial ovarian cancer. Int J Cancer. 2010;126:382–394. doi: 10.1002/ijc.24757. [DOI] [PubMed] [Google Scholar]
- 49.Hsu A.L., Ching T.T., Wang D.S., Song X., Rangnekar V.M., Chen C.S. The cyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking Akt activation in human prostate cancer cells independently of Bcl-2. J Biol Chem. 2000;275:11397–11403. doi: 10.1074/jbc.275.15.11397. [DOI] [PubMed] [Google Scholar]
- 50.Che S.M., Zhang X.Z., Hou L., Song T.B. Cyclooxygenase-2 inhibitor NS398 enhances radiosensitivity of radioresistant esophageal cancer cells by inhibiting AKT activation and inducing apoptosis. Cancer Invest. 2010;28:679–688. doi: 10.3109/07357907.2010.483504. [DOI] [PubMed] [Google Scholar]
- 51.Macleod K., Mullen P., Sewell J., Rabiasz G., Lawrie S., Miller E., Smyth J.F., Langdon S.P. Altered ErbB receptor signaling and gene expression in cisplatin-resistant ovarian cancer. Cancer Res. 2005;65:6789–6800. doi: 10.1158/0008-5472.CAN-04-2684. [DOI] [PubMed] [Google Scholar]
- 52.Andoh T., Chiueh C.C., Chock P.B. Cyclic GMP-dependent protein kinase regulates the expression of thioredoxin and thioredoxin peroxidase-1 during hormesis in response to oxidative stress-induced apoptosis. J Biol Chem. 2003;278:885–890. doi: 10.1074/jbc.M209914200. [DOI] [PubMed] [Google Scholar]
- 53.Menendez J.A., Vellon L., Colomer R., Lupu R. Effect of gamma-linolenic acid on the transcriptional activity of the Her-2/neu (erbB-2) oncogene. J Natl Cancer Inst. 2005;97:1611–1615. doi: 10.1093/jnci/dji343. [DOI] [PubMed] [Google Scholar]
- 54.Wei Y., Liu D., Ge Y., Zhou F., Xu J., Chen H., Gu J., Jiang J. Identification of E1AF as a target gene of E2F1-induced apoptosis in response to DNA damage. J Biochem. 2008;144:539–546. doi: 10.1093/jb/mvn098. [DOI] [PubMed] [Google Scholar]
- 55.Honda M., Miura A., Izumi Y., Kato T., Ryotokuji T., Monma K., Fujiwara J., Egashira H., Nemoto T. Doxorubicin, cisplatin, and fluorouracil combination therapy for metastatic esophageal squamous cell carcinoma. Dis Esophagus. 2010;23:641–645. doi: 10.1111/j.1442-2050.2010.01070.x. [DOI] [PubMed] [Google Scholar]
- 56.Yu L., Wu W.K., Li Z.J., Liu Q.C., Li H.T., Wu Y.C., Cho C.H. Enhancement of doxorubicin cytotoxicity on human esophageal squamous cell carcinoma cells by indomethacin and 4-[5-(4-chlorophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonami de (SC236) via inhibiting P-glycoprotein activity. Mol Pharmacol. 2009;75:1364–1373. doi: 10.1124/mol.108.053546. [DOI] [PubMed] [Google Scholar]
- 57.Polee M.B., Sparreboom A., Eskens F.A., Hoekstra R., van de Schaaf J., Verweij J., Stoter G., van der Gaast A. A phase I and pharmacokinetic study of weekly paclitaxel and carboplatin in patients with metastatic esophageal cancer. Clin Cancer Res. 2004;10:1928–1934. doi: 10.1158/1078-0432.ccr-03-0319. [DOI] [PubMed] [Google Scholar]
- 58.Su Z.Z., Sarkar D., Emdad L., Duigou G.J., Young C.S., Ware J., Randolph A., Valerie K., Fisher P.B. Targeting gene expression selectively in cancer cells by using the progression-elevated gene-3 promoter. Proc Natl Acad Sci U S A. 2005;102:1059–1064. doi: 10.1073/pnas.0409141102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Su Z., Shi Y., Friedman R., Qiao L., McKinstry R., Hinman D., Dent P., Fisher P.B. PEA3 sites within the progression elevated gene-3 (PEG-3) promoter and mitogen-activated protein kinase contribute to differential PEG-3 expression in Ha-ras and v-raf oncogene transformed rat embryo cells. Nucleic Acids Res. 2001;29:1661–1671. doi: 10.1093/nar/29.8.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Su Z., Shi Y., Fisher P.B. Cooperation between AP1 and PEA3 sites within the progression elevated gene-3 (PEG-3) promoter regulate basal and differential expression of PEG-3 during progression of the oncogenic phenotype in transformed rat embryo cells. Oncogene. 2000;19:3411–3421. doi: 10.1038/sj.onc.1203666. [DOI] [PubMed] [Google Scholar]