

Abstract
Type 1 diabetes mellitus (T1D) is an autoimmune disease caused by the destruction of pancreatic insulin-producing β cells by autoreactive T cells early in life. Despite daily insulin injections, patients typically develop cardiovascular and other complications; and intensive efforts are being directed toward identifying therapeutic targets to prevent the disease without directly impinging on the host defense. Fas ligand (FasL) is one potential target. Fas-FasL interactions primarily regulate T-cell homeostasis, not activation. Nevertheless, spontaneous gene mutation of Fas (called lpr mutation) or FasL (called the gld mutation) prevents autoimmune diabetes in nonobese diabetic (NOD) mice, the widely used model for T1D. Furthermore, although homozygous gld mutations cause age-dependent lymphoproliferation, limiting the gld mutation to one allele (NOD-gld/+) or treating NOD–wild-type mice with FasL-neutralizing monoclonal antibody completely prevents the disease development without causing lymphoproliferation or immune suppression. Herein, we show that the heterozygous gld mutation inhibits the accumulation of diabetogenic T cells in the pancreas, without interfering with their proliferation and expansion in the draining pancreatic lymph nodes. Pancreata from NOD-gld/+ mice contained B cells that expressed CD5 and produced IL-10, which was critical for maintenance of the disease resistance because its neutralization with an IL-10 receptor–blocking monoclonal antibody allowed accumulation of CD4 T cells in the pancreas and led to insulitis development. The results provide novel insights into the pathogenesis of T1D that could have important therapeutic implications.
The Fas pathway regulates immune homeostasis.1–6 Engagement of Fas by its ligand initiates an apoptotic cascade that leads to activation of caspase 3, chromatin condensation, DNA fragmentation, and T-cell death.7 Mice bearing spontaneous loss-of-function mutations in Fas (the lpr mutation) or Fas ligand (FasL; the gld mutation) develop T-cell lymphoproliferation, splenomegaly, and lupuslike autoimmune disease.8,9 In humans, defects in the Fas pathway cause an autoimmune lymphoproliferative syndrome that is similar to the disease in mutant mice.10 The lymphoproliferation is largely because of slow accumulation of chronically activated T cells, in which an α/β T-cell subset that lacks both CD4 and CD8 coreceptors predominates, which is referred to as double-negative T cells.9,11,12 The contraction of expanded T cells after a short-term adaptive immune response is mainly mediated by the proapoptotic molecule, Bim.13–15 Thus, there is reportedly minor impact of Fas or FasL deficiency on expansion and contraction of T cells after immunization with model antigens or viral infections.13–17 T cells from lymphocytic choriomeningitis virus–infected lpr and gld mice showed normal expansion and cytolytic function, and their number and cytolytic activity decreased to normal postinfection levels.16
Paradoxically, despite systemic T-cell lymphoproliferation, the lpr and gld mutations prevent many organ-specific T-cell autoimmune diseases, including type 1 diabetes mellitus (T1D) and multiple sclerosis, in animal models.18–20 The expression of homozygous lpr or gld mutations in nonobese diabetic (NOD) mice confers complete protection from autoimmune diabetes,18,19,21–23 leading to the idea that the protection was because of abrogation of Fas-mediated death of β cells.18 However, subsequent studies19,21–23 found that the role of the Fas pathway in the death of β cells is dispensable and that the precise nature of the protective mechanism remained unclear. In the absence of alternative mechanistic explanations, the view prevailed that disease resistance is somehow a product of the extensive distortion of the immune system associated with generalized lymphoproliferation; and interest in the therapeutic potential of targeting the Fas pathway waned.
Recently, we began to revisit this phenomenon after the observations by Su et al24 that NOD mice bearing a heterozygote gld mutation (NOD-gld/+) are protected from T1D without developing lymphoproliferation. The heterozygous gld mutation reduces FasL activity by approximately 85% because FasL functions as a homotrimer and mixing of wild-type (wt) and mutant FasL chains produces nonfunctional signaling complexes and dominant-negative interference.25 Analysis of the NOD-gld/+ mice by our group26 confirmed the original findings by Su et al.24 Since then, we have adopted the NOD-gld/+ mouse as a model to analyze the mechanisms of protection in the absence of lymphoproliferation. More important, we found that antibody blockade of FasL protects NOD-wt mice from T1D without causing lymphoproliferation, thereby directly linking the protection to FasL blockade.26,27 In addition, disease-resistant NOD-gld/+ mice harbor diabetogenic T cells that cause disease in NOD–severe combined immunodeficiency adoptive hosts.26
We hypothesize that inactivation of FasL leads to augmentation of an immunoregulatory mechanism that keeps the diabetogenic T cells in check. In support of this hypothesis, we show that the genetic and antibody blockade of FasL was associated with accumulation of IL-10–producing CD5+ B cells in the pancreata of NOD and that blockade of IL-10 impaired control of diabetogenic T cells and led to insulitis development. The results provide a valuable mechanistic step in understanding how inactivation of FasL protects from autoimmune diabetes. Potential mechanisms by which FasL blockade promoted CD5+ B-cell accumulation in the pancreata of NOD mice and the implications of the results for developing useful therapeutic interventions to T1D are discussed.
Materials and Methods
Mice
The NOD-wt and NOD-gld/+ mice used in this study were littermates produced by intercrossing of NOD-gld/+ mice. The original parental NOD-gld/+ mice were genotyped for polymorphic microsatellites linked to the IDDM susceptibility (Idd) genes, as previously described: Idd1, Idd3, Idd5, Idd10, and Idd16.24 Only female NOD mice between the ages of 8 and 12 weeks were used, unless otherwise indicated. BDC2.5 Thy1.1+ T-cell receptor (TCR) transgenic female NOD mice were from the Juvenile Diabetes Research Foundation (JDRF) Center on immunological tolerance in Type 1 diabetes (Harvard Medical School, Boston, MA) and were used between the ages of 3 and 5 weeks. All mice were bred and maintained at the Animal Care Facility of Johns Hopkins University School of Medicine (Baltimore, MD), and all of the experimental procedures have been approved by the Animal Use and Care Committee of The Johns Hopkins University.
Reagents
All fluorochrome-conjugated monoclonal antibodies were obtained from BD PharMingen (San Jose, CA) or eBioscience (San Diego, CA), unless otherwise indicated. The antibodies used were as follows: anti-B220 (RA3-6B2), anti-CD1d (1B1), anti-CD4 (GK1.5), anti-CD5 (53-7.3), anti-CD11c (N418), anti-CD19 (1D3), anti-CD40 (1C10), anti-CD44 (IM7), anti-CD80 (16-10A1), anti-CD86 (GL1), anti-FasL (MFL3), anti-IL-10 (JES5-16E3), anti-major histocompatibility complex II (AMS-32.1), anti-TCR (H57-597), and anti-Thy1.1 (HIS51). The anti-FasL monoclonal antibody (mAb; MFL4) was produced as previously described and has been used in many studies.26,28,29
Preparation of Hemopoietic Cells from Secondary Lymphoid Tissue and Pancreas
Spleens, pancreatic lymph nodes (PLNs), and inguinal lymph nodes were disrupted mechanically and passed through a 70-μm cell strainer; and red blood cells were removed with ACK lysising buffer (Quality Biological, Inc., Gaithersburg, MD). Pancreata were rapidly excised and transferred into a 60-mm dish with ice-cold washing buffer. After thorough washing and blood removal, pancreata were cut into small pieces and digested with collagenase D (1 mg/mL in 10 mL of PBS-containing calcium) for 30 minutes in a 37°C water bath in 50-mL conical tubes. Digestion was discontinued by the addition of washing buffer and filtration through a 40-μm cell strainer. Samples were washed twice, treated with ACK lysis buffer, resuspended in RPMI 1640 medium containing 2% fetal calf serum, and kept on ice for further testing.
Cell Enrichment and Adoptive Transfers
Thy1.1+ BDC2.5 T cells were isolated from combined spleens and lymph nodes of 3- to 5-week-old female BDC2.5 NOD mice using a Dynal mouse T-cell negative isolation kit (Invitrogen, Carlsbad, CA). Dendritic cells (DCs) were purified from splenocytes using CD11c microbeads from Miltenyi Biotec (Bergisch Gladbach, Germany). Purity was >95% for T cells and >92% for DCs. Freshly purified BDC2.5 CD4 T cells were labeled with carboxyfluorescein diacetate succinyl ester (CFSE) and injected i.v. (1.5 × 106 to 2 × 106 in 100 μL of PBS) into 10-week-old Thy1.2+ NOD-wt or NOD-gld/+ female littermates. The sample size was six to eight mice per genotype. Three independent experiments were conducted.
FACS Analysis of Surface Molecules and Intracellular IL-10
Single-cell suspensions on ice were stained with predetermined optimal concentrations of indicated antibodies and isotype-matched control Ig (BD Biosciences, San Diego, CA, or eBioscience, San Diego, CA). Surface-labeled cells were analyzed using an FACSCalibur flow cytometer (BD Biosciences) and FCS Express software (Becton Dickinson, San Diego, CA). For assessing intracellular IL-10, single-cell suspensions were cultured with lipopolysaccharide (10 μg/mL), phorbol myristate acetate (PMA) (50 ng/mL; Sigma-Aldrich, St. Louis, MO), ionomycin (500 ng/mL; Sigma-Aldrich), and Golgi-Plug (2 mmol/L; eBioscience) for 5 hours, as described.30 Cells were stained for surface markers, fixed, and permeabilized with the Cytofix/Cytoperm kit (BD PharMingen), according to the manufacturer's instructions. Permeabilized cells were stained with phycoerythrin-conjugated IL-10 mAb (JES5-16E3; BD PharMingen) or isotype-matched irrelevant antibody (eBioscience).
In Vivo Blockade of FasL or IL-10 Receptor by Neutralizing mAbs
The FasL-neutralizing mAb (MFL4) was previously described.26,28,29 For IL-10 receptor (IL-10R) blockade, 7-week-old NOD-gld/+ female or NOD-wt mice were injected i.p. with IL-10R–neutralizing mAb (1B1.3A) with an initial dose of 20 mg/kg body weight, followed by 10 mg/kg body weight every 4 days for 4 consecutive weeks. Littermate controls were treated in parallel with rat isotype-matched IgG1. The sample size was six to eight animals per group for NOD-gld/+ mice and four to five mice per group for NOD-wt mice. Two independent experiments were conducted.
Histological Features and Scoring of Insulitis
For analysis of insulitis, pancreata were fixed in 10% neutral-buffered formalin, dehydrated by graded ethanol series, cleared in xylene, and embedded in paraffin blocks. The blocks (5-μm sections) were stained with H&E by following standard protocols. Nonsequential sections were evaluated by light microscopy. Damage to the islets was scored in a blinded fashion and graded as follows: 0, no visible infiltration; 1, peri-insulitis; 2, insulitis with <50% islet infiltration; and 3, insulitis with >50% islet infiltration. At least 50 islets in each group of five animals were scored.
Statistical Analysis
Mean proportions were compared by Student's t-test after square-root transformations of the data and F-test to compare variances among the groups. Mean cell numbers were compared by Student's t-test.
Results
Diabetogenic T Cells Expand in PLNs but Fail to Accumulate in the Pancreata of NOD-gld/+ Mice
Priming of diabetogenic T cells usually occurs in PLNs, followed by infiltration of pancreatic islets, resulting in the development of insulitis.31 To investigate the mechanisms by which NOD-gld/+ mice control diabetogenic T cells, we examined the ability of clonotypic T cells isolated from islet-specific BDC2.5 TCR-transgenic donors32 to proliferate and expand in PLNs of NOD-gld/+ or NOD-wt littermates. We labeled BDC2.5 T cells with CFSE and examined their proliferation/expansion and islet accumulation 4 days after adoptive transfer into recipients. Donor BDC2.5 T cells, identified with the Thy1.1 allelic marker, were analyzed for proliferation by assessing CFSE dilution and for expansion by counting their absolute numbers in different organs. BDC2.5 T cells proliferated vigorously in PLNs of both NOD-gld/+ and NOD-wt recipients but not in irrelevant lymph nodes (Figure 1; see also Supplemental Figure S1 at http://ajp.amjpathol.org). BDC2.5 T cells found in the pancreata of NOD-gld/+ mice also diluted CFSE to levels comparable to those in the pancreata of NOD-wt littermates. Nevertheless, in contrast to NOD-wt recipients, BDC2.5 T cells did not accumulate in the pancreata of NOD-gld/+ mice. Only a few endogenous diabetogenic T cells were found in the pancreata of NOD-gld/+ mice compared with their NOD-wt counterparts (see Supplemental Figure S2 at http://ajp.amjpathol.org). Together, these results show that, despite normal proliferation and expansion in the draining PLNs, diabetogenic T cells failed to accumulate efficiently in pancreatic islets of NOD-gld/+ mice.
Figure 1.

Diabetogenic T cells expand in PLNs of gld mice but fail to accumulate in pancreatic islets. A: Normal expansion of Thy1.1+ BDC2.5 cells in PLNs of gld/+ mice. Dot plots show CFSE dilution of donor Thy1.1+ BDC2.5 cells in PLNs of 10-week-old NOD-gld/+ and NOD-wt intercross littermates on day 4 after adoptive transfer. The bar graph shows absolute numbers of Thy1.1+ BDC2.5 T cells in PLNs. B: Failure of Thy1.1+ BDC2.5 cells to accumulate in the pancreata of NOD-gld/+ mice. Dot plots show CFSE dilution of donor Thy1.1+ BDC2.5 cells in the pancreata of the described mice on day 4 after adoptive transfer. The bar graph shows absolute numbers of Thy1.1+ BDC2.5 T cells in the pancreata on day 4 after transfer. Results are presented as mean ± SD, with six to eight mice per genotype per group from three independent experiments. *P = 0.002.
CD5+ B Cells Constitute a Significant Proportion of B Cells in the Pancreata of NOD-gld/+ Mice
Because BDC2.5 T cells were from FasL-sufficient wt donors, their failure to accumulate efficiently in pancreatic islets of NOD-gld/+ mice indicated that the gld mutation regulates diabetogenic T cells by an extrinsic mechanism. Previous data26 showed that the heterozygous gld mutation does not alter the frequency of regulatory T cells in NOD mice. Therefore, we assessed the impact of the gld mutation on DCs and B cells that are usually crucial players in disease pathogenesis. We found no detectable change in the frequency and phenotype of DCs in PLNs and pancreata of NOD-gld/+ mice compared with DCs from NOD-wt littermates (see Supplemental Figure S3 at http://ajp.amjpathol.org). On the other hand, there was a significant pancreatic tissue–specific increase in the frequency of CD5-expressing B cells that was not observed in PLNs or spleens of NOD-gld/+ mice (Figure 2, A and B). B cells in the pancreata of NOD-gld/+ and NOD-wt littermates expressed comparable levels of CD1d, a marker that is characteristic of B10 B cells.30 Because most CD5+ B cells are found in the peritoneal cavity, we compared the frequency of CD5+ B cells in the peritoneum of NOD-gld/+ with NOD-wt littermates but found no significant differences (see Supplemental Figure S4 at http://ajp.amjpathol.org). Moreover, the frequency of CD5+ B cells in the blood was not altered by the gld mutation (see Supplemental Figure S4 at http://ajp.amjpathol.org).
Figure 2.

Prevalence of CD5+ B cells in the pancreata of NOD-gld/+ mice. A: Increased frequency of CD5-expressing B cells in the pancreata of NOD-gld/+ mice. CD19+B220+ cells from pancreas (Panc), spleen (SP), and PLNs of 9- to 10-week-old NOD-gld/+ and NOD-wt littermate females were gated and analyzed for CD5, CD1d, and major histocompatibility complex class II (MHCII) expression. B: Percentages of CD5+ B cells in the indicated organs of NOD-gld/+ and NOD-wt littermates. Results are presented as mean ± SD, with five to six mice per genotype per group from three independent experiments. *P = 0.002.
To confirm that the increased frequency of pancreatic CD5+ B cells was due to impaired FasL function, we injected NOD-wt mice with FasL-neutralizing or control IgG and assessed the impact on the frequency of CD5+ B cells. Treatment of NOD-wt mice with FasL-neutralizing mAb led to a significant and pancreatic tissue–specific increase in the frequency of CD5+ B cells in NOD-wt mice (Figure 3A). In contrast, treatment with the FasL-neutralizing mAb did not affect the frequency of CD5+ B cells in NOD-gld/gld mice with completely inactivated FasL, excluding a nonspecific effect (data not shown), which is consistent with previous results.26,28,29 Thus, both genetic and pharmacological results directly related the higher presence of CD5+ B cells in the pancreata of NOD mice to inhibition of FasL activity. In addition, consistent with previous results,27 treatment with FasL-neutralizing mAb halted progression of insulitis in NOD-wt mice compared with control mice (Figure 3B).
Figure 3.
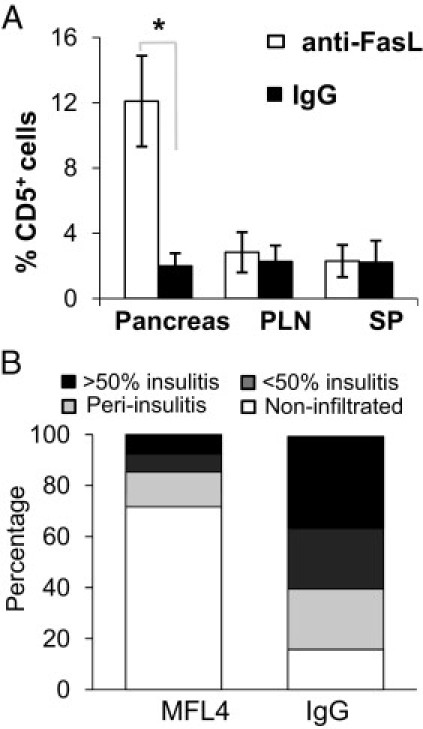
Accumulation of CD5+ B cells in the pancreas of NOD-wt mice after antibody blockade of FasL. Seven-week-old NOD-wt female littermates were injected i.p. with FasL-neutralizing mAb (MFL4) or IgG (20 mg/kg initially, followed by 10 mg/kg injections) every 4 days for 4 consecutive weeks. A: Frequency of CD5+ B cells in the indicated organs at the end of treatment. Data are given as the mean ± SD from four to five mice per group. SP indicates spleen. B: The severity of insulitis in the pancreata of mice from the two groups (n = 4 to 5 per group), scored as described in Materials and Methods. *P = 0.002.
An important feature of pancreatic CD5+ B cells is their enhanced ability to produce IL-10 after stimulation. Most pancreatic CD5+ B cells (90%) expressed IL-10 after in vitro stimulation with PMA, ionomycin, and lipopolysaccharide, compared with <20% of CD5- B cells (Figure 4). Together, these results show that the gld mutation is associated with increased presence of IL-10–producing CD5+ B cells in the pancreas, correlating with protection of the pancreas from diabetogenic T cells.
Figure 4.

Pancreatic CD5+ B cells express IL-10 anti-inflammatory cytokine. A: FACS analyses of IL-10 production by B cells. Single cells from spleens (SPs), PLNs, and pancreata of 10-week-old NOD-wt or NOD-gld/+ female littermates were cultured with lipopolysaccharide, PMA, and ionomycin for 5 hours. CD19+ cells were gated and analyzed for intracellular IL-10. The bar graph shows the frequencies of IL-10–producing B cells in NOD-gld/+ or NOD-wt littermates in the pancreas (Panc), PLNs, and irrelevant lymph node (iLN). Results are given as the mean ± SD from five to six mice per group from three independent experiments. B: Most IL-10–producing B cells belong to the CD5+ subset. Pancreatic cells were stimulated and stained as previously described and then CD19+ cells were gated cells and analyzed for intracellular IL-10 (left), followed by analysis of CD5 expression on each subset (right). The data are from one of three different experiments with similar results. *P = 0.007.
IL-10 Is a Key Regulator of Diabetogenic T Cells in NOD-gld/+ Mice
The IL-10 cytokine plays a key role in down-regulating the onset or initiation of many autoimmune diseases, including experimental autoimmune encephalomyelitis,21 collagen-induced arthritis,33 and inflammatory bowel disease,34 in murine models.35 However, the role of IL-10 in autoimmune diabetes has been paradoxical. Several studies have shown that deficiency of IL-10 fails to accelerate spontaneous autoimmune diabetes. Nevertheless, in other studies, recombinant IL-10 has inhibited insulitis development in NOD-wt mice36,37 and infusion of B-cell receptor–activated B cells has prevented autoimmune diabetes by an IL-10–dependent mechanism.38 Given that NOD-gld/+ mice harbor diabetogenic T cells26 that remain effectively restrained and that more B cells in the pancreata of NOD-gld/+ mice expressed IL-10, we thought that IL-10 might be involved in the gld mutation–mediated protection of pancreatic islets from diabetogenic T cells. Therefore, we determined whether IL-10R blockade with a neutralizing mAb (clone JES5-16E3) would break the control of diabetogenic T cells in NOD-gld/+ mice. The results of the intervention confirmed this notion. We found that blockade of IL-10R did, indeed, result in significant infiltration of islet cells of NOD-gld/+ mice by mainly CD4 T cells, with a minimal impact on CD8 T cells (Figure 5A and data not shown). Consistent with the fluorescence-activated cell sorter (FACS) data, a histological examination of pancreatic tissue demonstrated that treatment with the IL-10R–neutralizing mAb also increased the severity of insulitis (Figure 5B). Similarly, IL-10R blockade increased the frequency of adoptively transferred BDC2.5 T cells (see Supplemental Figure S5 at http://ajp.amjpathol.org). Thus, it appeared that an IL-10–dependent mechanism restricted diabetogenic CD4 T cells from accumulating in the pancreata of NOD-gld/+ mice. In contrast, and in concordance with previous results,36,37 blockade of the IL-10R had a minimal impact on the diabetogenic process in NOD-wt mice. There was comparable islet infiltration by CD4 T cells in NOD-wt mice that received IL-10R–neutralizing mAb or those that received control IgG and minimal impact on insulitis progression (Figure 5, C and D). These results establish a key role for IL-10 in controlling diabetogenic CD4 T cells in NOD-gld/+, but not NOD-wt, mice.
Figure 5.
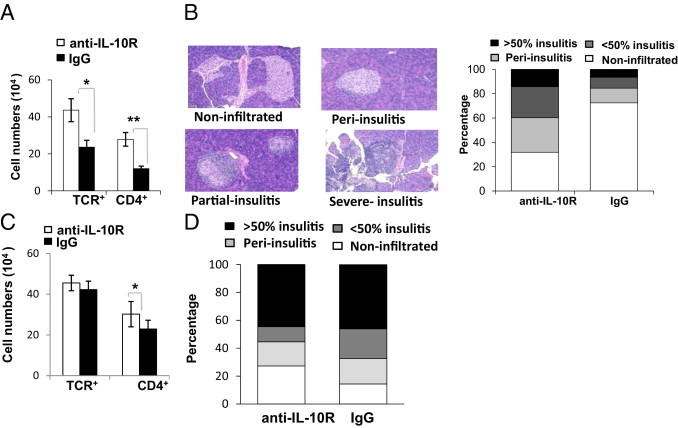
IL-10 is a key regulator of gld-mediated resistance to insulitis. Seven-week-old NOD-gld/+ females were injected i.p. with IL-10R–neutralizing mAb or control IgG for 4 consecutive weeks (initial dose of 20 mg/kg, followed by 10 mg/kg injections). A: Absolute numbers of TCR+ and CD4+ in the pancreata of NOD-gld/+ mice treated with anti-IL-10R or IgG isotype control. *P = 0.09 and **P = 0.007. B: Scores of insulitis after mice were treated with Abs. Results are from two independent experiments (n = 6 to 8 mice per group). Treatment of NOD-wt mice with IL-10R–blocking mAb has minimal impact on islet infiltration. C: Absolute numbers of TCR+ and CD4+ in the pancreata of NOD-wt mice treated with anti-IL-10R or IgG isotype control (n = 4 to 5 per group). *P > 0.05. D: Scores of insulitis after treatment termination.
In summary, the results of this study show that gld mutation of FasL leads to protection from diabetogenic T cells by an IL-10–regulated mechanism that is associated with increased local frequency of IL-10–producing CD5+ B cells in the pancreas.
Discussion
The inhibition of FasL, whether genetic or pharmacological, completely prevents autoimmune diabetes in NOD mice and, thus, offers a rare example of a molecule that is not essential for T-cell activation; however, its modulation leads to restraint of diabetogenic T cells. The therapeutic potential of this phenomenon is yet to be seriously considered. A major discouraging factor was the tight association of homozygous gld mutations with an age-dependent lymphoproliferation that led to the prevailing view that the two phenomena were inseparable. In addition, the lymphoproliferation made it difficult to examine the underlying mechanism. Recently, however, researchers11,26 have shown that FasL-mediated protection from autoimmune diabetes is dissociable from the lymphoproliferation. Dissociation of the two phenomena is demonstrated genetically by expression of a single gld allele in the NOD (NOD-gld/+) mice and pharmacologically by treatment of NOD-wt mice with FasL-neutralizing mAb.11,26 This key finding raises the possibility that FasL could be harnessed into a safer immunotherapeutic target. Since then, we have adopted the NOD-gld/+ model together with the pharmacological approach to investigate the underlying mechanism. Previous work26 shows that NOD-gld/+ mice harbor diabetogenic T cells that remain continuously controlled; however, the underlying mechanism remained unclear.
In the present study, we showed that the IL-10 cytokine played a critical role in controlling diabetogenic T cells in NOD-gld/+ mice. We detected a significant presence of CD5+ B cells in the pancreata of NOD-gld/+ mice but not NOD-wt littermates, most of which expressed IL-10 after lipopolysaccharide-PMA-ionomycin stimulation. A functional role of IL-10 in islet protection in mutant mice was confirmed by accumulation of CD4 T cells in the pancreata of NOD-gld/+ mice and development of insulitis after neutralization of IL-10 by using IL-10R–blocking mAb. In contrast, this mechanism is weak in NOD-wt mice because IL-10R blockade did not significantly alter diabetogenic T-cell responses or progression of insulitis. The latter results are in agreement with the previously reported dispensable role for endogenous IL-10 in disease pathogenesis in NOD-wt mice.37,39 Thus, the inactivation of FasL unmasked a protective role for IL-10 in controlling diabetogenic T cells in this widely used model of the disease. However, it is unclear whether blockade of FasL potentiated a regulatory mechanism that is otherwise compromised in NOD-wt mice or whether the IL-10 protective effect was an epiphenomenon related to FasL blockade in an autoimmune microenvironment. In either case, given the potent effect of FasL blockade on disease pathogenesis in this widely used model of the disease, this finding could have important implications for developing a therapeutic strategy for T1D.
IL-10 has a short range and half-life, and the tissue localization of IL-10–producing cells at the site of inflammation significantly affects its effectiveness.36 In experimental autoimmune encephalomyelitis, local delivery of IL-10 effectively ameliorates the disease40–42; systemic administration partially inhibits the disease, lacks efficacy, or even causes adverse effects.43,44 Therefore, it was of interest that IL-10–producing CD5+ B cells localized predominantly in the pancreata, the target of autoimmunity in T1D. This was seen both genetically in NOD-gld/+ mice and pharmacologically in NOD-wt mice treated with FasL-neutralizing mAb compared with unmanipulated NOD-wt controls. Preferential localization of IL-10–producing B cells in the target organ of inflammation has previously been reported in a colitis model of inflammatory bowel disease.34 It is postulated that the inflammatory milieu recruits IL-10–producing B cells into the intestinal mucosa.34 In the case of autoimmune diabetes, IL-10–producing B cells were detected at a low frequency in the pancreata of NOD-wt mice but significantly increased when FasL was genetically or pharmacologically blocked. Given the primary role of FasL-Fas interactions in mediating apoptosis, blockade of FasL may increase the frequency of IL-10–producing CD5+ B cells by promoting survival. Alternatively, but not mutually exclusive, it is possible that FasL blockade enhances IL-10–producing CD5+ B-cell recruitment into the pancreas. Therefore, distinguishing between these possibilities and understanding the mechanistic relationship between accumulation of IL-10–producing B cells in the pancreas and blockade of FasL will require further analysis.
Although IL-10 is a broadly expressed cytokine,39 we were unable to identify additional IL-10 sources at the diabetogenic sites that were differentially regulated by the gld mutation but cannot definitively exclude this possibility. However, our unpublished data, consistent with previous studies,40 show no changes in helper polarization of CD4 T cells in NOD-gld/+ mice. In addition, T cells isolated from the pancreata showed signs of activation; and islet-reactive BDC2.5 T cells injected into NOD-gld/+ mice showed evidence of comparable or increased proliferation (Figure 1; see also Supplemental Figure S6 at http://ajp.amjpathol.org). Future studies should examine the cellular and molecular mechanisms by which gld mutation of FasL regulates the tolerogenic role of IL-10 in autoimmune diabetes.
It will also be important to examine why diabetogenic T cells fail to cause overt disease after infiltrating pancreatic islets of anti-IL-10R–treated NOD-gld/+ mice, even after several weeks of treatment (data not shown). A simple explanation is insufficient blockade of IL-10 activity. Alternatively, it is possible that transition from insulitis to the destructive phase is regulated by an additional or independent mechanism in NOD-gld/+ mice. In line with the latter possibility, previous work26 shows that the few diabetogenic T cells that are found in pancreata of NOD-gld/+ mice are controlled by an IL-2–dependent mechanism. This is because the mild peri-islet infiltration that occurs in NOD-gld/+ mice rapidly progresses to severe insulitis and overt diabetes after treatment of the mice with IL-2–neutralizing mAb.26 Therefore, we speculate that diabetogenic T cells in NOD-gld/+ mice are controlled by at least two mechanisms: a primary IL-10–dependent mechanism that regulates initial T-cell accumulation in pancreatic islets and a secondary IL-2–regulated mechanism that controls the ability of islet-infiltrating T cells to destroy β cells. Previous work26 shows that neutralization of IL-2 or blocking of cytotoxic T-lymphocyte antigen-4–negative signals abrogates disease resistance in NOD-gld/+ mice, suggesting that Foxp3+ Treg cells, which depend on the two pathways for survival and function, are involved in controlling diabetogenic T cells that infiltrate gld/+ pancreata. In line with this idea, deficiency of Foxp3+ Treg cells does not affect initial diabetogenic T-cell activation or accelerate the onset of T-cell infiltration; instead, it makes insulitis immediately destructive.41 A better understanding of these protective mechanisms and investigation of independent or overlapping roles of regulatory T and B cells in gld-mediated tolerance to autoimmune diabetes will be investigated in future studies. This will include generation of IL-10 and FasL double-knockout NOD mice (both genes are localized on chromosome 1 but are 13 centimorgans apart).
In conclusion, patients with T1D typically develop severe complications, including retinopathy, neuropathy, cardiovascular disease, and nephropathy, despite daily insulin injections.42,43 A significant effort is ongoing to develop immunotherapies that effectively prevent and/or treat T1D in the clinic. Although the blockade of many stimulatory molecules and soluble inflammatory mediators is an effective approach to control autoimmune diseases, most of these therapies are inherently associated with a general systemic immunomodulatory effect.44,45 The findings of this study provide a valuable mechanistic step in dispelling the notion that gld-mediated resistance to autoimmune diabetes is because of extensive distortion of the immune system. Given that the Fas pathway is not essential for host defense, developing a new therapy based on targeting the Fas pathway may significantly reduce the risk of immunosuppression and offer a strategy that can be combined with current approaches46 to eventually protect high–risk children47 from developing T1D.
Acknowledgment
We thank Pamela Talalay for reviewing and editing the manuscript.
Footnotes
Supported by the Juvenile Diabetes Research Foundation (grant JDRF-1-2006-676), the NIH (grant 1R56AI083444-01A2), and the American Heart Association (10GRNT4200003).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.04.016.
Supplementary data
Comparable numbers of T cells in PLNs but not in the pancreata of the NOD-gld/+ and NOD-wt mice. The bar graphs show absolute numbers of TCRb+ T cells in the PLNs and pancreata on day 4 after transfer. The results are presented as the mean ± SD, with six to eight mice per genotype per group from three independent experiments. *P = 0.003.
No proliferation of diabetogenic T cells in irrelevant lymph. The dot plots show CFSE staining of Thy1.1+ BDC2.5 T cells in inguinal lymph nodes of recipients on day 4 after transfer. Similar results are observed in six to eight mice per genotype per group from three independent experiments.
Phenotype of DCs from NOD-wt and gld/+ mice. Single-cell suspensions of 9- to10-week-old NOD-wt and gld/+ littermate females from pancreata, PLNs, and spleens (SPs) were analyzed by FACS. CD11c+ cells were gated and analyzed for expression of indicated molecules. The average frequency of CD11c+ cells in PLNs of wt was 6.6% compared with 5.9% in mutant mice; in the pancreas (Panc), their frequency was 3.8% versus 3.6%, respectively. SSC-H, side scattering;; MHCII, major histocompatibility complex II.
No statistically significant differences in the frequency of CD5+ B cells in the peritoneum and blood of NOD-gld/+ and NOD-wt mice. Peritoneal lavage and blood mononuclear cells from 8-week-old NOD-wt and NOD-gld/+ mice were analyzed for expression of CD19 and CD5 by FACS, as described in Figure 2. Results are given as the mean ± SD from five mice per group.
Anti-IL-10 treatment facilitates accumulation of adoptively transferred BDC2.5 CD4 T cells in the pancreata of NOD-gld/+ mice. Ten-week-old NOD-gld/+ littermates were treated with anti-IL-10 or istoype control IgG (20 mg/kg, followed by 10 mg i.p. every 4 days). On day 10, mice in both groups were injected i.v. with 3 × 106 or 4 × 106 BDC2.5 T cells (total, n = 4 per group in two independent experiments). Four days later, T cells were isolated from the pancreata; and the numbers of CFSE+ BDC2.5 CD4 T cells were determined. The graph shows the fold increase in the number of BDC2.5 T cells isolated from the pancreata of anti-IL-10–treated mice relative to the number of BDC2.5 T cells isolated from the pancreata of IgG controls. Data are shown as the mean ± SEM.
Reduced accumulation of BDC2.5 T cells in the pancreata of NOD-gld/+ mice is detected 18 hours after transfer. NOD-gld/+ or NOD-wt mice (10 weeks old) were injected with CFSE-labeled BDC2.5 T cells (10 × 106 per mouse); 18 hours later, single-cell suspensions of pooled pancreata from two mice in each group were analyzed for accumulation of the donor CFSE+ BDC2.5 T cells. The dot plots show the percentage of CFSE+ T cells in the pancreata in NOD-gld/+ (left) and NOD-wt (right). The normalized number of CFSE+ T cells per pancreas was 4 × 103 and 13 × 104 in the gld and wt pancreata, respectively. Left: TCR expression by gated CFSE cells in the gld and wt pancreata. Right: Comparison of CFSE dilution by gated donor T cells in the gld and wt pancreata (the numbers in parenthesis indicate the percentages in the M1 gate).
References
- 1.Ashany D., Savir A., Bhardwaj N., Elkon K.B. Dendritic cells are resistant to apoptosis through the Fas (CD95/APO-1) pathway. J Immunol. 1999;163:5303–5311. [PubMed] [Google Scholar]
- 2.Guo Z., Zhang M., An H., Chen W., Liu S., Guo J., Yu Y., Cao X. Fas ligation induces IL-1beta-dependent maturation and IL-1beta-independent survival of dendritic cells: different roles of ERK and NF-kappaB signaling pathways. Blood. 2003;102:4441–4447. doi: 10.1182/blood-2002-11-3420. [DOI] [PubMed] [Google Scholar]
- 3.Guo Z., Zhang M., Tang H., Cao X. Fas signal links innate and adaptive immunity by promoting dendritic-cell secretion of CC and CXC chemokines. Blood. 2005;106:2033–2041. doi: 10.1182/blood-2004-12-4831. [DOI] [PubMed] [Google Scholar]
- 4.Pinkoski M.J., Green D.R. Fas ligand, death gene. Cell Death Differ. 1999;6:1174–1181. doi: 10.1038/sj.cdd.4400611. [DOI] [PubMed] [Google Scholar]
- 5.Rescigno M., Piguet V., Valzasina B., Lens S., Zubler R., French L., Kindler V., Tschopp J., Ricciardi-Castagnoli P. Fas engagement induces the maturation of dendritic cells (DCs), the release of interleukin (IL)-1beta, and the production of interferon gamma in the absence of IL-12 during DC-T cell cognate interaction: a new role for Fas ligand in inflammatory responses. J Exp Med. 2000;192:1661–1668. doi: 10.1084/jem.192.11.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tada Y., O-Wang J., Takiguchi Y., Tatsumi K., Kuriyama T., Okada S., Tokuhisa T., Sakiyama S., Tagawa M. Cutting edge: a novel role for Fas ligand in facilitating antigen acquisition by dendritic cells. J Immunol. 2002;169:2241–2245. doi: 10.4049/jimmunol.169.5.2241. [DOI] [PubMed] [Google Scholar]
- 7.Lenardo M., Chan F., Hornug F., McFarland H., Siegel R., Wang J., Zheng L. Mature T lymphocytes apoptosis-immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi T., Tanaka M., Brannan C.I., Jenkins N.A., Copeland N.G., Suda T., Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe-Fukunaga R., Brannan C., Copeland N., Jenkins N., Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 10.Fisher G.H., Rosenberg F.J., Straus S.E., Dale J.K., Middelton L.A., Lin A.Y., Strober W., Lenardo M.J., Puck J.M. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 11.Hamad A.R. Analysis of gene profile, steady state proliferation and apoptosis of double-negative T cells in the periphery and gut epithelium provides new insights into the biological functions of the Fas pathway. Immunol Res. 2010;47:134–142. doi: 10.1007/s12026-009-8144-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagata S., Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today. 1995;16:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 13.Hildeman D.A., Zhu Y., Mitchell T.C., Bouillet P., Strasser A., Kappler J., Marrack P. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002;16:759–767. doi: 10.1016/s1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- 14.Snow A.L., Oliveira J.B., Zheng L., Dale J.K., Fleisher T.A., Lenardo M.J. Critical role for BIM in T cell receptor restimulation-induced death. Biol Direct. 2008;3:34. doi: 10.1186/1745-6150-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stranges P.B., Watson J., Cooper C.J., Choisy-Rossi C.M., Stonebraker A.C., Beighton R.A., Hartig H., Sundberg J.P., Servick S., Kaufmann G., Fink P.J., Chervonsky A.V. Elimination of antigen-presenting cells and autoreactive T cells by fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lohman B.L., Razvi E.S., Welsh R.M. T-lymphocyte downregulation after acute viral infection is not dependent on CD95 (Fas) receptor-ligand interactions. J Virol. 1996;70:8199–8203. doi: 10.1128/jvi.70.11.8199-8203.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohamood A.S., Bargatze D., Xiao Z., Jie C., Yagita H., Ruben D., Watson J., Chakravarti S., Schneck J.P., Hamad A.R. Fas-mediated apoptosis regulates the composition of peripheral alphabeta T cell repertoire by constitutively purging out double negative T cells. PLoS One. 2008;3:e3465. doi: 10.1371/journal.pone.0003465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chervonsky A.V., Wang Y., Wong F.S., Visintin I., Flavell R.A., Janeway C.A., Jr, Matis L.A. The role of Fas in autoimmune diabetes. Cell. 1997;89:17–24. doi: 10.1016/s0092-8674(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 19.Kim S., Kim K.A., Hwang D.Y., Lee T.H., Kayagaki N., Yagita H., Lee M.S. Inhibition of autoimmune diabetes by Fas ligand: the paradox is solved. J Immunol. 2000;164:2931–2936. doi: 10.4049/jimmunol.164.6.2931. [DOI] [PubMed] [Google Scholar]
- 20.Thomas H.E., Darwiche R., Corbett J.A., Kay T.W. Evidence that beta cell death in the nonobese diabetic mouse is Fas independent. J Immunol. 1999;163:1562–1569. [PubMed] [Google Scholar]
- 21.Allison J., Strasser A. Mechanisms of beta cell death in diabetes: a minor role for CD95. Proc Natl Acad Sci U S A. 1998;95:13818–13822. doi: 10.1073/pnas.95.23.13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Apostolou I., Hao Z., Rajewsky K., von Boehmer H. Effective destruction of Fas-deficient insulin-producing beta cells in type 1 diabetes. J Exp Med. 2003;198:1103–1106. doi: 10.1084/jem.20030698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savinov A.Y., Tcherepanov A., Green E.A., Flavell R.A., Chervonsky A.V. Contribution of Fas to diabetes development. Proc Natl Acad Sci U S A. 2003;100:628–632. doi: 10.1073/pnas.0237359100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su X., Hu Q., Kristan J.M., Costa C., Shen Y., Gero D., Matis L.A., Wang Y. Significant role for Fas in the pathogenesis of autoimmune diabetes. J Immunol. 2000;164:2523–2532. doi: 10.4049/jimmunol.164.5.2523. [DOI] [PubMed] [Google Scholar]
- 25.Siegel R.M., Frederiksen J.K., Zacharias D.A., Chan F.K., Johnson M., Lynch D., Tsien R.Y., Lenardo M.J. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 2000;288:2354–2357. doi: 10.1126/science.288.5475.2354. [DOI] [PubMed] [Google Scholar]
- 26.Mohamood A.S., Guler M.L., Xiao Z., Zheng D., Hess A., Wang Y., Yagita H., Schneck J.P., Hamad A.R. Protection from autoimmune diabetes and T-cell lymphoproliferation induced by FasL mutation are differentially regulated and can be uncoupled pharmacologically. Am J Pathol. 2007;171:97–106. doi: 10.2353/ajpath.2007.070148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakayama M., Nagata M., Yasuda H., Arisawa K., Kotani R., Yamada K., Chowdhury S.A., Chakrabarty S., Jin Z.Z., Yagita H., Yokono K., Kasuga M. Fas/Fas ligand interactions play an essential role in the initiation of murine autoimmune diabetes. Diabetes. 2002;51:1391–1397. doi: 10.2337/diabetes.51.5.1391. [DOI] [PubMed] [Google Scholar]
- 28.Hao Z., Hampel B., Yagita H., Rajewsky K. T cell-specific ablation of Fas leads to Fas ligand-mediated lymphocyte depletion and inflammatory pulmonary fibrosis. J Exp Med. 2004;199:1355–1365. doi: 10.1084/jem.20032196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kayagaki N., Yamaguchi N., Nagao F., Matsuo S., Maeda H., Okumura K., Yagita H. Polymorphism of murine Fas ligand that affects the biological activity. Proc Natl Acad Sci U S A. 1997;94:3914–3919. doi: 10.1073/pnas.94.8.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsushita T., Yanaba K., Bouaziz J.D., Fujimoto M., Tedder T.F. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoglund P., Mintern J., Waltzinger C., Heath W., Benoist C., Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J Exp Med. 1999;189:331–339. doi: 10.1084/jem.189.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katz J.D., Wang B., Haskins K., Benoist C., Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 33.Mauri C., Gray D., Mushtaq N., Londei M. Prevention of arthritis by interleukin 10-producing B cells. J Exp Med. 2003;197:489–501. doi: 10.1084/jem.20021293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizoguchi A., Mizoguchi E., Takedatsu H., Blumberg R.S., Bhan A.K. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 2002;16:219–230. doi: 10.1016/s1074-7613(02)00274-1. [DOI] [PubMed] [Google Scholar]
- 35.O'Garra A., Barrat F.J., Castro A.G., Vicari A., Hawrylowicz C. Strategies for use of IL-10 or its antagonists in human disease. Immunol Rev. 2008;223:114–131. doi: 10.1111/j.1600-065X.2008.00635.x. [DOI] [PubMed] [Google Scholar]
- 36.Balasa B., Van Gunst K., Jung N., Katz J.D., Sarvetnick N. IL-10 deficiency does not inhibit insulitis and accelerates cyclophosphamide-induced diabetes in the nonobese diabetic mouse. Cell Immunol. 2000;202:97–102. doi: 10.1006/cimm.2000.1658. [DOI] [PubMed] [Google Scholar]
- 37.Pennline K.J., Roque-Gaffney E., Monahan M. Recombinant human IL-10 prevents the onset of diabetes in the nonobese diabetic mouse. Clin Immunol Immunopathol. 1994;71:169–175. doi: 10.1006/clin.1994.1068. [DOI] [PubMed] [Google Scholar]
- 38.Hussain S., Delovitch T.L. Intravenous transfusion of BCR-activated B cells protects NOD mice from type 1 diabetes in an IL-10-dependent manner. J Immunol. 2007;179:7225–7232. doi: 10.4049/jimmunol.179.11.7225. [DOI] [PubMed] [Google Scholar]
- 39.Moore K.W., de Waal Malefyt R., Coffman R.L., O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 40.Serreze D.V., Chapman H.D., Post C.M., Johnson E.A., Suarez-Pinzon W.L., Rabinovitch A. Th1 to Th2 cytokine shifts in nonobese diabetic mice: sometimes an outcome, rather than the cause, of diabetes resistance elicited by immunostimulation. J Immunol. 2001;166:1352–1359. doi: 10.4049/jimmunol.166.2.1352. [DOI] [PubMed] [Google Scholar]
- 41.Chen Z., Herman A.E., Matos M., Mathis D., Benoist C. Where CD4+CD25+ T reg cells impinge on autoimmune diabetes. J Exp Med. 2005;202:1387–1397. doi: 10.1084/jem.20051409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodriguez B.L., Fujimoto W.Y., Mayer-Davis E.J., Imperatore G., Williams D.E., Bell R.A., Wadwa R.P., Palla S.L., Liu L.L., Kershnar A., Daniels S.R., Linder B. Prevalence of cardiovascular disease risk factors in U.S. children and adolescents with diabetes: the SEARCH for diabetes in youth study. Diabetes Care. 2006;29:1891–1896. doi: 10.2337/dc06-0310. [DOI] [PubMed] [Google Scholar]
- 43.Wadwa R.P. Cardiovascular disease risk in youth with diabetes mellitus. Rev Endocr Metab Disord. 2006;7:197–204. doi: 10.1007/s11154-006-9016-y. [DOI] [PubMed] [Google Scholar]
- 44.Ejrnaes M., von Herrath M.G., Christen U. Cure of chronic viral infection and virus-induced type 1 diabetes by neutralizing antibodies. Clin Dev Immunol. 2006;13:337–347. doi: 10.1080/17402520600800721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haller M.J., Atkinson M.A., Schatz D.A. The road not taken: a path to curing type 1 diabetes. Eur J Immunol. 2009;39:2054–2058. doi: 10.1002/eji.200939517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von Herrath M. Combination therapies for type 1 diabetes: why not now. Immunotherapy. 2010;2:289–291. doi: 10.2217/imt.10.23. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L., Gianani R., Nakayama M., Liu E., Kobayashi M., Baschal E., Yu L., Babu S., Dawson A., Johnson K., Jahromi M., Aly T., Fain P., Barker J., Rewers M., Eisenbarth G.S. Type 1 diabetes: chronic progressive autoimmune disease. Novartis Found Symp. 2008;292:85–94. doi: 10.1002/9780470697405.ch7. [DOI] [PubMed] [Google Scholar]; discussion, 94–98, 122–129, 202–203
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparable numbers of T cells in PLNs but not in the pancreata of the NOD-gld/+ and NOD-wt mice. The bar graphs show absolute numbers of TCRb+ T cells in the PLNs and pancreata on day 4 after transfer. The results are presented as the mean ± SD, with six to eight mice per genotype per group from three independent experiments. *P = 0.003.
No proliferation of diabetogenic T cells in irrelevant lymph. The dot plots show CFSE staining of Thy1.1+ BDC2.5 T cells in inguinal lymph nodes of recipients on day 4 after transfer. Similar results are observed in six to eight mice per genotype per group from three independent experiments.
Phenotype of DCs from NOD-wt and gld/+ mice. Single-cell suspensions of 9- to10-week-old NOD-wt and gld/+ littermate females from pancreata, PLNs, and spleens (SPs) were analyzed by FACS. CD11c+ cells were gated and analyzed for expression of indicated molecules. The average frequency of CD11c+ cells in PLNs of wt was 6.6% compared with 5.9% in mutant mice; in the pancreas (Panc), their frequency was 3.8% versus 3.6%, respectively. SSC-H, side scattering;; MHCII, major histocompatibility complex II.
No statistically significant differences in the frequency of CD5+ B cells in the peritoneum and blood of NOD-gld/+ and NOD-wt mice. Peritoneal lavage and blood mononuclear cells from 8-week-old NOD-wt and NOD-gld/+ mice were analyzed for expression of CD19 and CD5 by FACS, as described in Figure 2. Results are given as the mean ± SD from five mice per group.
Anti-IL-10 treatment facilitates accumulation of adoptively transferred BDC2.5 CD4 T cells in the pancreata of NOD-gld/+ mice. Ten-week-old NOD-gld/+ littermates were treated with anti-IL-10 or istoype control IgG (20 mg/kg, followed by 10 mg i.p. every 4 days). On day 10, mice in both groups were injected i.v. with 3 × 106 or 4 × 106 BDC2.5 T cells (total, n = 4 per group in two independent experiments). Four days later, T cells were isolated from the pancreata; and the numbers of CFSE+ BDC2.5 CD4 T cells were determined. The graph shows the fold increase in the number of BDC2.5 T cells isolated from the pancreata of anti-IL-10–treated mice relative to the number of BDC2.5 T cells isolated from the pancreata of IgG controls. Data are shown as the mean ± SEM.
Reduced accumulation of BDC2.5 T cells in the pancreata of NOD-gld/+ mice is detected 18 hours after transfer. NOD-gld/+ or NOD-wt mice (10 weeks old) were injected with CFSE-labeled BDC2.5 T cells (10 × 106 per mouse); 18 hours later, single-cell suspensions of pooled pancreata from two mice in each group were analyzed for accumulation of the donor CFSE+ BDC2.5 T cells. The dot plots show the percentage of CFSE+ T cells in the pancreata in NOD-gld/+ (left) and NOD-wt (right). The normalized number of CFSE+ T cells per pancreas was 4 × 103 and 13 × 104 in the gld and wt pancreata, respectively. Left: TCR expression by gated CFSE cells in the gld and wt pancreata. Right: Comparison of CFSE dilution by gated donor T cells in the gld and wt pancreata (the numbers in parenthesis indicate the percentages in the M1 gate).