

Abstract
Objectives
To investigate the role that oxidative stress plays in the development of diabetic cystopathy.
Materials and methods
Comparative gene expression in the bladder of non-diabetic and streptozotocin (STZ)-induced 2-month-old diabetic rats was carried out using microarray analysis. Evidence of oxidative stress was investigated in the bladder by analyzing glutathione S-transferase activity, lipid peroxidation, and carbonylation and nitrosylation of proteins. The activity of protein degradation pathways was assessed using western blot analysis.
Results
Analysis of global gene expression showed that detrusor smooth muscle tissue of STZ-induced diabetes undergoes significant enrichment in targets involved in the production or regulation of reactive oxygen species (P = 1.27 × 10−10). The microarray analysis was confirmed by showing that markers of oxidative stress were all significantly increased in the diabetic bladder. It was hypothesized that the sequelae to oxidative stress would be increased protein damage and apoptosis. This was confirmed by showing that two key proteins involved in protein degradation (Nedd4 and LC3B) were greatly up-regulated in diabetic bladders compared to controls by 12.2 ± 0.76 and 4.4 ± 1.0-fold, respectively, and the apoptosis inducing protein, BAX, was up-regulated by 6.76 ± 0.76-fold.
Conclusions
Overall, the findings obtained in the present study add to the growing body of evidence showing that diabetic cystopathy is associated with oxidative damage of smooth muscle cells, and results in protein damage and activation of apoptotic pathways that may contribute to a deterioration in bladder function.
Keywords: diabetic cystopathy, oxidative stress, protein degradation
Introduction
Diabetes mellitus (DM) is rapidly increasing in prevalence in the USA, resulting in a corresponding increase in urological disorders [1,2]. Bladder pathology, including detrusor overactivity and cystopathy, is one of the most common complications of DM and has a profound negative effect on the quality of life in those diabetic patients [3]. Both neurogenic and myogenic effects have been implicated in DM-associated cystopathy, such as changes in the physiology of detrusor smooth-muscle cells, changes in the innervation or functioning of the neuronal component, and urothelial dysfunction [4]. Diabetes-related alterations of smooth-muscle cells are attributed to several mechanisms: changes in cellular excitability or intercellular communication; changes in receptor density, distribution and function; and alterations to intracellular signal transduction as well as molecular or genetic changes.
There is increasing evidence suggesting that oxidative stress plays a significant role in the development of diabetic complications [5–7]. Preliminary studies have also indicated that oxidative stress plays a role in bladder pathology in rat models of diabetes. For example, in the streptozotocin (STZ)-induced diabetic rat bladder, there is a reduction in chloramphenicol acetyl transferase-like activity, increased thiobarbituric acid reactive substances levels, increased inducible nitric oxide synthase protein expression, and an increased number of apoptotic cells in the diabetic bladder [8]. In alloxan induced diabetes in rabbits, a decrease in detrusor smooth muscle force generation was found to be associated with an increase in lipid peroxide (LPO) and sorbitol levels concomitant with over-expression of aldose reductase and polyol pathway activation [9]. Recently, genomic and proteomic approaches have been applied to study global changes in the urogenital system accompanying diabetes. A previous proteomic study on the effects of STZ-induced diabetes on the bladder suggested that the development of diabetes related complications in this model involves the down-regulation of structural and extracellular matrix proteins in smooth muscle that are essential for normal muscle contraction and relaxation [10]. In the same study, diabetes also induced changes in proteins that are associated with cell proliferation and inflammation. Preliminary results from a microarray study on the effects on gene expression in the rat bladder after 1 week of STZ-induced diabetes are also available [11]. These studies showed that there are changes in genes involved in cell proliferation, metabolism, actin cytoskeleton and myosin, as well as decreases in gene expression involved in cell motility and regulation of muscle contraction, suggesting that, at a very early stage in diabetes, there is protein remodelling of the bladder tissue. In a study on a different urogenital tissue (the rat cavernosum) in rats after 10 weeks of STZ-induced diabetes, microarray analysis revealed that there was a decrease in numerous extracellular matrix genes (e.g. collagen and elastin related) and an increase in oxidative stress-associated genes [12].
In the present study, the effect of STZ-diabetes on the global expression of genes in the rat bladder was investigated using microarray analysis. Significant changes were observed in genes that are involved in the modulation of oxidative stress and activation of protein degradation pathways. The impact of these genes on the biochemistry of the diabetic bladder tissue was then confirmed. The observations of the present study suggest that diabetic cystopathy may involve activation of oxidative stress pathways followed by degradation of damaged proteins, which could explain several of the manifestations of diabetic cystopathy.
Materials and methods
Animals
Diabetes was induced in eight male F-344 rats (Taconic Farms, Germantown, NY, USA; 8–10 weeks-old and weighing 200–240 g) by one i.p. injection of STZ (35 mg/kg, dissolved in citrate buffer, 0.6 M citric acid/0.08 M Na2HPO4, pH 4.6). Eight age-matched control rats were injected with vehicle. Diabetes was confirmed by the presence of blood glucose levels of > 250 mg/dL for three consecutive days. From these animals, four were given a daily injection of 2 units of long-acting insulin (Lantus, 100 units/mL, Sanofi-Aventis, Bridgewater, NJ, USA) beginning at 1 week after the STZ injection; these rats were referred to as the insulin-treated group. After confirming the diabetic state, rats were killed 2 months later in a CO2 asphyxiation chamber, and the bladder and corpora were excised. The bladder was quickly denuded of the urothelial and suburothelial layers and flash-frozen in liquid nitrogen and stored at −70 °C until RNA preparation. All experimental protocols were approved by the Animal Institute Committee of the Albert Einstein College of Medicine.
Microarray studies
For the GeneChip studies, the RG-U34A rat GeneChip (Affymetrix Inc., Sunnyvale, CA, USA) containing ≈8799 genes was used. RNA was prepared from the bladder of age-matched control and STZ-diabetic rats using a Mixer Miller (MM3; Qiagen, CA, USA). To determine that the isolated RNA was not degraded, RNA was run on a Bioanalyzer. After confirming its quality using a bioanalyzer, the isolated RNA was reverse transcribed to cDNA, labelled with biotin and fragmented according to Affymetrix protocols (http://www.affymetrix.com). Subsequent hybridization to microarray chips was carried out at the Albert Einstein College of Medicine microarray core facilities using an Affymetrix Fluidics Station. Test chips were used to screen for the quality of the labelled cRNA, before proceeding to the RG-U34A chip. Data was analyzed from four age-matched control and four STZ-induced diabetic bladders.
Quality control
Quality-control measures were applied to raw data by using two Bioconductor packages (simpleaffy and AffyQCReport; http://www.bioconductor.org/) and in-house R code [13]. Quality-control results include signal intensity box-plots and density-plots, β-actin and glyceraldehyde 3-phosphate dehydrogenase Quality-control statistics, positive and negative border element plots, as well a correlation matrix. Array data containing flaws or biases (or of which RNA was strongly degraded) were excluded from the subsequent data analysis.
Differential expression analysis
The gene expression measures were normalized by the robust multi-array analysis approach in the Bioconductor affy package [14]. Ranking of genes by degree of differential expression was performed using significance analysis of microarrays [15] (SAM; http://www-stat.stanford.edu/~tibs/SAM/). Selection of significantly different gene expression profiles between the two classes unpaired condition was based on the false discovery rate permutation statistic [16]. All data was permutated by 100 cycles. Significant genes were identified by 0.05 ≤ median false discover rate ≤ 0.15, corresponding to type 1 error rates of 5–15%, and a fold-difference in mean expression ≥|1.5|. For each comparison group, the results from SAM are compared to identify consistency. Concordance in selection of differentially expressed genes across the four time points was determined by Venn diagrams.
Glutathione S-transferase assay
To perform assays for the activity of glutathione S-transferase (GST) bladder tissue homogenates were prepared in 100 mM phosphate buffer (pH 6.5) containing 2 mM EDTA at 4 °C. The supernatants were separated by centrifugation using a Sorvall Centrifuge (Thermo Fisher Scientific, Waltham, MA, USA) at 15 000 g for 30 min. The protein concentration was determined in the tissues extracts by the Bio-Rad DC protein assay method (Bio-Rad Laboratories, Hercules, CA, USA). The GST assay was carried out in accordance with the method of Habig et al. [17]. To 900 μl of phosphate buffer (pH 6.5), 33.3 μl of 75 mM reduced glutathione (Sigma, St Louis, MO, USA), 33.3 μl of 30 mM 1-chlroro-2,4-dinitrobenzene (Sigma) in ethanol and 33.3 μl of tissue homogenates were added. The coloured product formed was read at 340 nm using a Beckman Spectophotometer (Beckman Coulter, Fullerton, CA, USA) for 5 min. The blank reaction was carried out without the enzyme. The GST activity was expressed as nmoles/milligram protein.
Lipid peroxidation
To determine lipid peroxidation, bladder tissue homogenates were prepared in 1.15% potassium chloride. The supernatants were separated by centrifugation at 15 000 g for 30 min using an Sorvall centrifuge (Thermo Fisher Scientific). The lipid peroxidation assay was carried out by the method of Ohkawa et al. [18]. 1,1,3,3,-Tetramethoxypropane (Sigma) was used as an external standard, ranging from 2–10 nM for the standard curve. To the standards and 0.5 ml of rat tissue homogenate, 200 μl of 8% SDS, 1.5 ml of 0.8% thiobarbituric acid (Sigma) and 20% glacial acetic acid (pH 3.5) was added and boiled at 95 °C for 1 h, the samples were cooled and the LPOs were extracted in 15:1 ratio of n-butnaol and pyridine solvent (Sigma) by centrifugation at 4000 g for 15 min. The LPO levels were measured at 532 nm using a Beckman Spectophotometer. The LPOs were expressed as malondialdehyde (MDA) formed (nM/g tissue).
Preparation of protein extracts and western blot analysis
The expression of proteins in bladder tissue was analyzed using western blots. Protein extractions were carried at 4 °C in accordance with the method of Kanika et al. [18]. Briefly, bladder tissues were homogenized using a polytron homogenizer in 50 mM Tris-HCl buffer (pH 7.4) containing 10 mM EDTA, 30 mM sucrose and 10 μl of mammalian protease inhibitor cocktail (Sigma) and the supernatants were separated by centrifugation at 15 000 g for 30 min. Protein concentrations were determined in the samples by the Bio-Rad DC protein assay method. The proteins were electrophoresed on Nu PAGE® 10% Bis-Tris gels (Invitrogen, Carlsbad, CA, USA) and then transferred to a poly(vinylidene fluoride) membrane (Immun-Blot™ PVDF Membrane; Bio-Rad Laboratories) by semi-dry electroblotting for 1 h. The membranes were blocked with 5% milk in Tris-buffered saline containing 0.05% Tween-20 for 1 h, the membranes were probed with anti-LC3B antibody (dilution 1 : 1000), anti-Nedd-4 (dilution 1 : 5000), anti-MDM2 (dilution 1 : 6000) (Millipore, Upstate, NY, USA), anti-nitrotyrosine (NOY-7A5; Alexis Biochemicals, San Diego, CA, USA), anti-glyceraldehyde 3-phosphate dehydrogenase (dilution 1 : 20,000; Abcam, Cambridge, MA, USA), and anti-α-actin and anti-BCL2 (dilution 1 : 500) antibodies (BD Transduction Laboratories; Becton-Dickinson Biosciences, Franklin Lakes, NJ, USA) for 1 h at room temperature. The bound antibodies were detected by probing with HRP-labelled anti-mouse or anti-rabbit secondary antibody (dilution 1 : 10000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h at room temperature. Enhanced chemiluminescence was performed with Pierce® ECL Western blotting substrate (Pierce, Rockford, IL, USA) and bands were quantified by densitometry using Quantity One® software (Bio-Rad Laboratories).
Oxyblot analysis
The protein oxidation levels were determined using Oxyblot™ Protein Oxidation Detection Kit (Chemicon International Inc., Temecula, CA, USA) in accordance with the manufacturer’s instructions. Bladder tissue extracts were prepared in protein extraction buffer containing 50 mM dithiothreitol at 4 °C, the supernatants were separated by centrifugation at 15 000 g for 30 min. Briefly, to 5 μl (15 μg) of protein, 5 μl of 12% SDS and 10 μl of control or dinitrophenylhydrazine derivatizing agents were added and incubated at room temperature for 15 min. The reaction was terminated by the addition of 7.5 μl of the neutralization buffer supplied with the kit. The oxyblot standard was loaded with 2.5 μl of SDS-sample buffer. The carbonyl derivatized samples were loaded on Nu PAGE® 10% Bis-Tris gels (Invitrogen) and then transferred on to a PVDF membrane (Immun-Blot™ PVDF Membrane; Bio-Rad Laboratories) by semi-dry electroblotting for 1 h. After the transfer, membranes were blocked with 1% BSA in Tris-buffered saline containing 0.05% Tween-20 for 1 h at room temperature. The membranes were probed with primary antibody (dilution 1 : 150) for 1 h and with secondary antibody (dilution 1 : 300) for 1 h at room temperature. Enhanced chemiluminiscence was performed with Pierce® ECL Western Blotting Substrate (Pierce) and bands were quantified by densitometry using Quantity One® software (Bio-Rad Laboratories). Bands were quantified by densitometry using Quantity One® software (Bio-Rad Laboratories).
Statistical analysis
Student’s t-test was used to determine the significant difference in expression between experimental and control groups. P < 0.05 was considered statistically significant.
Results
Changes in mitochondrial function and level of oxidative stress in diabetic bladders
To gain insight into the molecular mechanisms that may result in the bladder pathophysiology after 2 months of STZ-diabetes, a GeneChip microarray (the affymetrix RGU34a chip) was used to look for significant changes in gene expression. The 2-month time point was chosen because previous studies indicate that this represents a duration of diabetes resulting in significant changes in the physiology of the rat bladder [19]. Using a false discovery rate of 9.7%, and a significant change in expression of > 1.5-fold, out of the approximately 8799 genes represented on the RGU34a chip, 1046 genes were down-regulated in expression and 598 genes were up-regulated in expression in STZ-diabetic rats compared to age-matched controls (AMC). Table 1 shows the 30 most up- and down-regulated genes. Although the specific genes may warrant individual study, the approach employed was that, with such a large number of gene changes, the study should focus on investigating the impact that an ontological group may have on the biochemistry of the diabetic bladder. Ingenuity pathway analysis showed that genes changed in bladder of STZ-induced diabetes compared to AMC are significantly enriched in targets involved in production or regulation of reactive oxygen species (ROS; P = 1.27 × 10−10). Some of the genes involved in oxidative stress pathways that were significantly changed in the genomic analysis are shown in Table 2, and their distribution on a pre-existing mitochondrial dysfunction pathway is depicted in Fig. 1. Our published proteomic studies when subjected to similar analysis also showed significant changes in the expression of genes that generate or respond to the production of ROS (P = 1.37 × 10−3) [10].
Table 1.
The top 30 genes that were down- or up-regulated in expression compared to age-matched controls at 2 months.
| 2 Month Up Regulated Genes | 2 Month Down Regulated Genes | ||||
|---|---|---|---|---|---|
| Gene Name | Gene Symbol | Fold Change | Gene Name | Gene Symbol | Fold Change |
| spermine binding protein | Sbp | 58.84158218 | nuclear receptor subfamily 4, group A, member 3 | Nr4a3 | 0.140948809 |
| prostatic steroid binding protein 1 | Psbp1 | 22.35410061 | cysteine rich protein 61 | Cyr61 | 0.239881357 |
| matrix metalloproteinase 7 | Mmp7 | 20.10072299 | immediate early gene transcription factor NGFI-B | Nr4a1 | 0.252981521 |
| probasin | LOC54193 | 14.4324318 | butyrophilin-like 2 (MHC class II associated) | Btnl2 | 0.278787027 |
| androgen regulated 20 kDa protein | Andpro | 8.32406535 | small inducible cytokine subfamily A11 | Scya11 | 0.28040929 |
| probasin | LOC54193 | 8.246853733 | alpha-2-macroglobulin | A2m | 0.281696275 |
| extracellular proteinase inhibitor | Expi | 6.125447792 | B-cell translocation gene 2, anti-proliferative | Btg2 | 0.294026367 |
| chemokine (C-X-C motif) ligand 2 | Cxcl2 | 5.149889936 | early growth response 1 | Egr1 | 0.295198118 |
| chemokine (C-C motif) ligand 3 | Ccl3 | 4.003739622 | insulin-like growth factor binding protein 3 | Igfbp3 | 0.301123929 |
| complement component 3 | C3 | 3.825484305 | immediate early gene transcription factor NGFI-B | Nr4a1 | 0.305269856 |
| claudin 3 | Cldn3 | 3.752516668 | Fibroblast growth factor receptor 1 | Fgfr1 | 0.312227857 |
| superoxide dismutase 2 | Sod2 | 3.669789563 | early growth response 2 | Egr2 | 0.316472591 |
| S100 calcium-binding protein A8 (calgranulin A) | S100a8 | 3.552234263 | early growth response 1 | Egr1 | 0.330379792 |
| gro | Gro1 | 3.51098163 | cAMP responsive element modulator | Crem | 0.340710177 |
| orosomucoid 1 | Orm1 | 3.097160132 | biglycan | Bgn | 0.362080385 |
| ornithine decarboxylase 1 | Odc1 | 3.045201121 | apolipoprotein D | Apod | 0.366552013 |
| claudin 3 | Cldn3 | 2.876686229 | insulin-like growth factor 1 | Igf1 | 0.368770881 |
| lipocalin 2 | Lcn2 | 2.864320127 | amphiregulin | Areg | 0.375508894 |
| interleukin 1 alpha | II1a | 2.729642093 | thymus cell antigen 1, theta | Thy1 | 0.375675629 |
| spongiotrophoblast specific protein | Tpbp | 2.581845227 | guanosine diphosphate dissociation inhibitor 1 | Gdi1 | 0.37739635 |
| chemokine (C-X3-C motif) ligand 1 | Cx3cl1 | 2.574015066 | adipsin | Adn | 0.377744894 |
| arginosuccinate synthetase | Ass | 2.538114204 | nuclear factor I/X | Nfix | 0.382101757 |
| prolactin receptor | Prlr | 2.476002977 | lipoprotein lipase | Lpl | 0.387506366 |
| cytochrome P450, 4a12 | Cyp4a12 | 2.370902632 | Fibroblast growth factor receptor 1 | Fgfr1 | 0.389871085 |
| small inducible cytokine subfamily A20 | Scya20 | 2.326088414 | lysyl oxidase | Lox | 0.393805833 |
| ATPase Na+/K+ transporting beta 1 polypeptide | Atp1b1 | 2.323959651 | caveolin | Cav | 0.395313842 |
| ceruloplasmin | Cp | 2.310252708 | myosin phosphatase, target subunit 1 | Mypt1 | 0.397582176 |
| tumor necrosis factor superfamily, member 2 | Tnf | 2.296100096 | protein tyrosine phosphatase, non-receptor type 16 | Ptpn16 | 0.397993828 |
| haptoglobin | Hp | 2.26686919 | mitogen-responsive phosphoprotein | Dab2 | 0.400719731 |
| peroxisome proliferator activated receptor delta | Ppard | 2.255560289 | B-cell translocation gene 2, anti-proliferative | Btg2 | 0.400859782 |
| colony stimulating factor 3 | Csf3 | 2.229054724 | collagen, type V, alpha 2 | Col5a2 | 0.402249658 |
Only identifiable genes are included in the list.
Table 2.
Targets and their fold changes involved in oxidative stress and mitochondrial dysfunction.
| ID | Name | Description | Fold |
|---|---|---|---|
| 116686 | GSR | glutathione reductase | 1.71 |
| 24248 | CAT | catalase | 1.61 |
| 24787 | SOD2 | superoxidedismutase2, mitochondrial | 3.67 |
| 25035 | CYB5R3 | cytochrome b5 reductase3 | −2.15 |
| 298376 | GPX7 | glutathione peroxidase7 | −1.88 |
| 29253 | MAOA | monoamine oxidase A | −2.01 |
| 497811 | XDH | xanthine dehydrogenase | −1.64 |
| 54226 | APP | amyloid beta (A4) precursor protein | −2.04 |
| 25756 | CPT1B | carnitine palmitoyltransferase1 B (muscle) | 2.02 |
| 691001 | NDUFA3 | NADH dehydrogenase1 alpha subcomplex 3, | 1.51 |
| 315167 | NDUFA6 | NADH dehydrogenase(ubiquinone) 1 alpha | 2.64 |
| 293991 | NDUFB8 | NADH dehydrogenase (ubiquinone) 1 beta | 1.65 |
| 84683 | COX4I2 | cytochrome c oxidasesubunit IVisoform2 (lung) | 1.62 |
| 25278 | COX6A2 | cytochrome c oxidasesubunit VIa polypeptide 2 | 1.83 |
| 171335 | COX8A | cytochrome c oxidasesubunit 8A (ubiquitous) | 2.23 |
| 157074 | SDHA | Succinate dehydrogenasecomplex, subunit A * | −1.79 |
Targets identified from proteomics. The fold column indicates fold changes in diabetic animals compared to age-matched controls.
Fig. 1.

Canonical pathway of mitochondrial dysfunction in an ingenuity pathway analysis knowledge base overlaid with targets changed in 2-month diabetic bladder compared to age-matched controls. Green indicates down-regulation and red indicates up-regulation in diabetes.
Abundance changes for genes that are involved in ROS generation and removal, as well as up-regulation of gene expression of components of respiratory complex I and III, are highlighted in Fig. 1. These changes indicate increased oxidative stress resulting in oxidative damage of mitochondrial proteins and the cellular efforts to reduce oxidative stress. Genomic mitochondrial targets involved in the generation of ROS (monoamine oxidase A, xanthine dehydrogenase, amyloid b precursor protein and cytochrome b5 reductase 3) are down-regulated and mitochondrial targets involved in antioxidant (superoxide dismutase 2, chloramphenicol acetyl transferase and glutathione reductase) are up-regulated in the bladder of diabetic animals. Antioxidant enzymes are frequently up-regulated with increased oxidative stress. In addition, most of the changed targets involved in oxidative phosphorylation (subunits of respiratory complex I and III; NADH dehydrogenase 1 α subcomplex 3; NADH dehydrogenase, ubiquinone, 1 α; NADH dehydrogenase, ubiquinone, 1 β; cytochrome c oxidase subunit IV isoform 2; cytochrome c oxidase subunit VIa polypeptide 2; and cytochrome c oxidase subunit 8A, ubiquitous) are up-regulated. The down-regulation of targets involved in ROS generation and up-regulation in antioxidant enzymes in the mitochondria may be working synergistically in an attempt to reduce oxidative stress in mitochondria.
Markers of oxidative stress are up-regulated in the diabetic bladder
To determine oxidative stress levels in the bladder of STZ-diabetic animals, the level of lipid peroxidation and GST activity was measured compared to AMC. As shown in Fig. 2A, lipid peroxidation, as determined by MDA levels in the bladder tissue, significantly increased (by an average of 2.52-fold) in STZ-diabetic animals compared to AMC. Treating the diabetic animals with insulin significantly reversed MDA levels; however, they did not return to the levels in AMC. Insulin therefore partially reversed the effects of diabetes on MDA levels in the bladder. GST activity is also an indicator of oxidatives stress, with its activity being reduced in the presence of ROS [20]. GST activity in the STZ-diabetic bladder was significantly reduced in diabetic animals (by an average of 18%) and was reversed in animals treated with insulin (Fig. 2B).
Fig. 2.

(A) Lipid peroxidation was measured by determining malondialdehyde (MDA) in bladder tissues of four age-matched control rats, four rats with streptozotocin (STZ)-induced diabetes for 2 months, and four rats with STZ-induced diabetes treated with insulin. (B) Glutathione S-transferase (GST) activity was also measured in the same animals. *Student’s t-test was used to determine a significant difference from control (P < 0.05).
Diabetes results in increased oxidative damage of bladder proteins
Proteins are one of the major targets of ROS. Oxidation of proteins introduces carbonyl groups at lysine, arginine, threonine or proline residues in a site-specific manner. Therefore, whether oxidatively damaged proteins are increased in the bladder of STZ-diabetic animals was analyzed compared to AMC by detecting and quantifying the carbonyl groups introduced into proteins by ROS. Figure 3A shows a representative western blot using an antibody against carbonyl residues in proteins extracted from the bladder of AMC, diabetic and insulin-treated animals. Densitometric analysis from a total of four animals in each group showed that the levels of oxidized proteins are increased in the diabetic bladder compared to AMC (1.43 ± 0.13-fold; P < 0.05). In STZ-diabetic animals treated with insulin, the levels of carbonyl residues in the bladder were not significantly different from AMC.
Fig. 3.

(A) Levels of oxidatively damaged proteins were determined in the bladder of four age-matched control (AMC), four streptozotocin-diabetic and four insulin-treated) were detected by Oxyblot. A control reaction (‘con’) was run in which proteins were prepared in the absence of dinitrophenylhydrazine derivatizing agents and appear blank relative to reactions where the derivitization reagent was present (‘exp’). A representative blot of two animals from each of these groups is shown. (B) Nitrosylation of proteins in four AMC, diabetic and insulin-treated diabetic animals. A representative blot is shown for three animals from each group.
Another potential indicator of oxidative stress in the bladder is the level of nitrotyrosylated proteins. The peroxynitrite radical (ONOO−), the reaction product of nitric oxide and superoxide, is implicated as the key oxidative species in several pathologies and results in S-nitrosylation or nitrotyrosylation of proteins. The level of nitrotyrosylated proteins in bladder tissue from AMC, 2 month STZ-diabetic and insulin-treated animals was measured (Fig. 3B). Densitometric analysis from a total of four animals in each group showed that the mean level of nitrotyrosylation was significantly increased in the diabetic animals (4.3 ± 0.13-fold). The level of protein nitrotrosylation was partly reversed in the bladder of insulin-treated animals (2.38 ± 0.011-fold compared to the AMC).
Diabetes results in the activation of proteins resulting in protein degradation
Increased oxidative damage of proteins would be expected to result in changes in the expression of genes involved in pathways resulting in protein degradation and removal. Therefore, using western blot analysis, specific proteins were investigated in the bladder that are involved in pathways leading to degradation of oxidatively damaged proteins in AMC, STZ-diabetic and insulin-treated animals. Figure 4 shows a representative blot for two out of the four animals in each group probed with antibodies for LC3B, UHLC3, Nedd4, MDM2 and BAX. Densitometic analysis for expression levels are presented in Table 3.
Fig. 4.
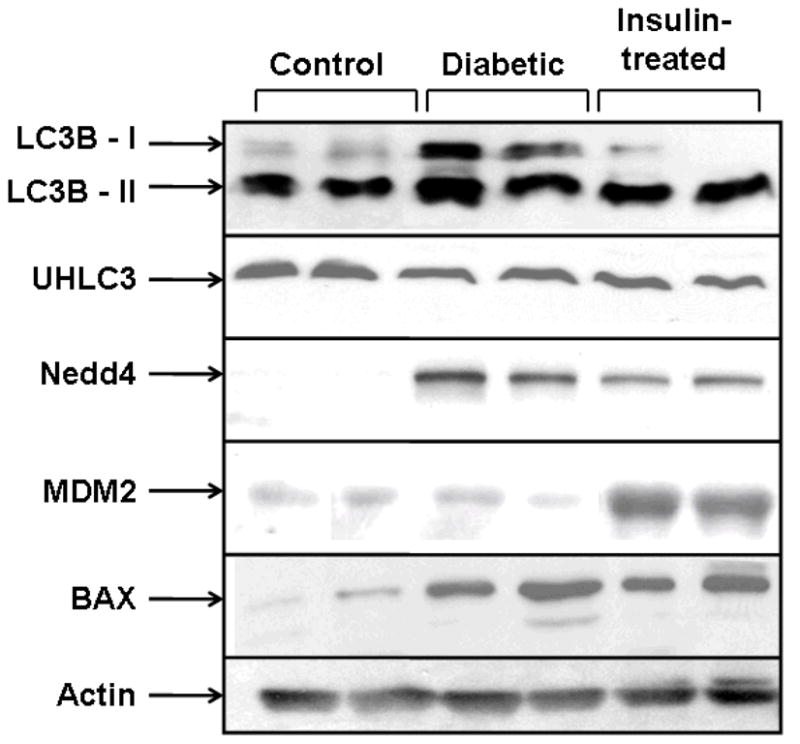
Changes in the expression of proteins involved in ubiquitination and in the lysosomal autophage pathway. A representative western blot is shown for protein extract from the bladder of two control, two STZ-diabetic and two insulin-treated animals probed with LC3.
Table 3.
Densitometric analysis of proteins involved in ubiquitination and in the lysosomal autophage pathway (significantly different expression compared to non-diabetic control.
| Control | Diabetic | Insulin | |
|---|---|---|---|
| LC3B-I | 1.0 ±0.02 | 4.4 ± 1.0* | 0.75 ± 0.015* |
| LC3B-II | 1.0 ±0.5 | 1.5 ± 0.74 | 1.54 ± 0.52 |
| UHLC3 | 1.0 ±0.08 | 0.75 ± 0.05* | 0.84 ± 0.07 |
| Nedd -4 | 1.0 ±0.6 | 12.2 ± 0.78* | 8.3 ± 0.28* |
| MDM2 | 1.0 ±0.24 | 0.54 ± 0.05* | 2.17 ± 0.25* |
| BAX | 1.0 ±0.005 | 6.76 ± 0.8* | 5.86 ± 0.18* |
Student’s t-test, P < 0.05.
The lysosomal autophage pathway for protein degradation was analyzed by determining the expression of the short and long form of LC3 (Fig. 4 and Table 3). The expression of LC3 can serve as a marker for autophagy [21,22]. Densitometric analysis of both the short form (LC3-I) and long form (LC3-II) of four replicates (four control, four diabetic and four insulin-treated) revealed increases in the bladder of diabetic animals compared to AMC (Table 3). When diabetic animals were treated with insulin, the changes were reversed.
As shown in Fig. 4, the levels of several proteins involved in ubiquitination (MDM2, Nedd4 and UCHL3) were also analyzed in age-matched control, STZ-diabetic and insulin-treated diabetic bladder. Densitometric analysis showed that the average level of MDM2 and UCHL3 were slightly down-regulated in the bladder of diabetic animals compared to AMC, whereas Nedd4 was markedly up-regulated. There was a partial reversal of the effect of expression on MDM2 and UCHL3 in the insulin-treated animals. However, insulin treatment resulted in increased MDM2 expression relative to the AMC. Expression of BAX was also analyzed, which exerts a pro-apoptotic effect on cell. Its expression was increased in the diabetic animals, and this effect was partly reversed by insulin treatment.
Discussion
The STZ-induced diabetic rat is a well-established model for studies on the physiological and molecular consequences of experimental diabetes. STZ has been shown to have specific cytotoxic effects on pancreatic islet B cells and therefore STZ-treated rats are one of the commonest animal models of type 1 diabetes [23]. An in-depth study of the changes in bladder function that occur in this animal model was recently presented [19]. Measurements of bladder dynamics after 2 months of diabetes indicated an increase in bladder capacity in STZ-animals, which was reflected by an increase in urinary volume and bladder weight. In addition to bladder capacity, micturition pressure and volume in the diabetic animals was significantly greater than in the AMC group. Bladder compliance was also increased significantly at 2 months and voiding frequency was significantly lower for diabetic animals.
The genomic analysis presented here, as well as the previously reported proteomic analysis [10], show that 2 months of STZ-induced diabetes affects expression of a large number of genes. One approach to understanding the effect of diabetes on bladder physiology would be to investigate the impact of each of the genes changed in expression individually. However, the present study took the approach of determining the physiological impact of changes in an ontological group of genes, specifically, those related to oxidative stress. Ingenuity pathway analysis of our published proteomic data [10] and the genomic data reported in the present study here show that, in the bladder of STZ-induced diabetic rats, there is significant enrichment in targets involved in the production or regulation of ROS (genomic analysis; P = 1.27 × 10−10 proteomic analysis P = 1.37 × 10−3). Therefore, there could be both a direct effect on the levels of oxidative stress in the bladder tissues, and a compensatory increase in cellular mechanisms that could protect the cell.
Similar to observations made by Beshay et al. [8], in the present study, several cellular indicators of oxidative stress are shown to be up-regulated in the diabetic bladder. Beshay et al. [8] measured different characteristics of oxidative stress to those performed in our own studies, observing a reduction in the catalase-like activity and increase in thiobarbituric acid reactive substances in the bladders from diabetic animals. Our studies show that levels of lipid peroxidation were elevated and the activity of GST reduced in diabetic animals. In addition, the environment of increased oxidative stress results in damage to bladder proteins. There is an increase in oxidatively damaged proteins as observed using an oxyblot and an increase in nitrotyrosylated proteins. It is possible that an increased oxidative environment combined with a previously reported increase in the number of inducible nitric oxide synthase-positive cells in the smooth muscle cell layers of the diabetic rat bladder [8] contributes to increases nitrosylation of amino acids in proteins.
It is considered that increased damage of proteins as a result of oxidative stress would lead to activation of protein degradation pathways. ROS are known to be required to trigger autophagy probably because they oxidize a critical cysteine residue in Atg4 [24,25]. Atg4 is involved in the regulation of LC3 [26] and it was shown that, in diabetic bladder, there is an up-regulation of the protein LC3 (LC3B-I), which would suggest an increase in autophagy [21,22]. The up-regulation of LC3 is reversed when diabetic animals are treated with insulin.
The levels of several proteins involved in ubiquitination (MDM2, Nedd4 and UCHL3) were also analyzed in AMC, STZ-diabetic and insulin-treated diabetic bladder. Densitometric analysis showed that the average level of MDM2 and UCHL3 were only slightly down-regulated in the bladder of diabetic animals compared to AMC. However, Nedd4 was markedly up-regulated in the diabetic bladder. The up-regulation of Nedd4 with the onset of diabetes has not been reported previously; however, its up-regulation in bladder cancer suggests that it plays an important role in maintaining homeostasis of signal mechanisms in the bladder [27].
Diabetes in the STZ-rat animal model is also associated with increased apoptosis in the bladder [8]. With regard to this effect of diabetes, a significant increase was noted in the expression of BAX in the diabetic bladders. Accumulation of active BAX at mitochondria will result in apoptosis through a mechanism that involves the release of cytochrome c, which in turn triggers activation of the caspase cascade and induces apoptotic cell death [28–30]. It has previously been suggested that enhanced apoptosis in detrusor smooth muscle cells as part of a remodelling response during compensatory hyperplasia and hypertrophy occurring in response to urinary outflow obstruction in the fetal ovine bladder is mediated through up-regulation of BAX [31]. The reduction of the detrusor smooth muscle cell mass that occurs with apoptosis will impair the contractile capacity of the bladder associated with dysfunction.
The present study shows that, after 2 months of experimental diabetes, there is up-regulation of markers of oxidative stress in the bladder, accompanied by an increase in the activity of pathways involved in protein degradation. Previous studies have shown that, after 2 months of experimental diabetes, there is a significant deterioration in bladder function [19]. Although, there is an association between diabetes, bladder dysfunction and markers of oxidative stress, these studies are unable to distinguish whether changes in oxidative stress are a forerunner or a consequence of bladder dysfunction. To distinguish between these possibilities in future studies, it will be of interest to correlate the time course of oxidative stress with changes in bladder physiology.
Bladder dysfunction is a major source of morbidity in patients with DM. More than two-thirds of diabetic patients present with urodynamically abnormal bladder function [32]. Our research adds to the growing body of evidence that diabetic cystopathy is associated with oxidative damage of smooth muscle cells, and results in protein damage and activation of apoptotic pathways that may contribute to a deterioration in bladder function. These results suggest antioxidant therapy may have therapeutic potential for treating diabetic cystopathy.
Acknowledgments
This work was supported by a grant awarded by the NIH/NIDDK to Kelvin P. Davies (R01DK077665).
Abbreviations
- AMC
age-matched controls
- DM
diabetes mellitus
- LPO
lipid peroxide
- GST
glutathione S-transferase
- MDA
malondialdehyde
- PVDF
poly(vinylidene fluoride)
- ROS
reactive oxygen species
- STZ
streptozotocin
References
- 1.Daneshgari F, Moore C. Diabetic uropathy. Semin Nephrol. 2006;26(2):182–5. doi: 10.1016/j.semnephrol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 2.Goldstraw MA, Kirby MG, Bhardwa J, Kirby RS. Diabetes and the urologist: a growing problem. BJU Int. 2007;99(3):513–7. doi: 10.1111/j.1464-410X.2006.06588.x. [DOI] [PubMed] [Google Scholar]
- 3.Epstein LB, Goldberg RP. The overactive bladder and quality of life. Int J Fertil Womens Med. 2005;50(1):30–6. [PubMed] [Google Scholar]
- 4.Yoshimura N, Chancellor MB, Andersson KE, Christ GJ. Recent advances in understanding the biology of diabetes-associated bladder complications and novel therapy. BJU Int. 2005;95(6):733–8. doi: 10.1111/j.1464-410X.2005.05392.x. [DOI] [PubMed] [Google Scholar]
- 5.Grattagliano I, Palmieri VO, Portincasa P, Moschetta A, Palasciano G. Oxidative stress-induced risk factors associated with the metabolic syndrome: a unifying hypothesis. J Nutr Biochem. 2008;19(8):491–504. doi: 10.1016/j.jnutbio.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Maritim AC, Sanders RA, Watkins JB., 3rd Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol. 2003;17(1):24–38. doi: 10.1002/jbt.10058. [DOI] [PubMed] [Google Scholar]
- 7.Roberts CK, Sindhu KK. Oxidative stress and metabolic syndrome. Life Sci. 2009;84(21–22):705–12. doi: 10.1016/j.lfs.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 8.Beshay E, Carrier S. Oxidative stress plays a role in diabetes-induced bladder dysfunction in a rat model. Urology. 2004;64(5):1062–7. doi: 10.1016/j.urology.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 9.Changolkar AK, Hypolite JA, Disanto M, Oates PJ, Wein AJ, Chacko S. Diabetes induced decrease in detrusor smooth muscle force is associated with oxidative stress and overactivity of aldose reductase. J Urol. 2005;173(1):309–13. doi: 10.1097/01.ju.0000141583.31183.7a. [DOI] [PubMed] [Google Scholar]
- 10.Yohannes E, Chang J, Christ GJ, Davies KP, Chance MR. Proteomics analysis identifies molecular targets related to diabetes mellitus associated bladder dysfunction. Mol Cell Proteomics. 2008 doi: 10.1074/mcp.M700563-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hipp JD, Davies KP, Tar M, Valcic M, Knoll A, Melman A, et al. Using gene chips to identify organ-specific, smooth muscle responses to experimental diabetes: potential applications to urological diseases. BJU Int. 2007;99(2):418–30. doi: 10.1111/j.1464-410X.2007.06676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sullivan CJ, Teal TH, Luttrell IP, Tran KB, Peters MA, Wessells H. Microarray analysis reveals novel gene expression changes associated with erectile dysfunction in diabetic rats. Physiol Genomics. 2005;23(2):192–205. doi: 10.1152/physiolgenomics.00112.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson CL, Miller CJ. Simpleaffy: a BioConductor package for Affymetrix Quality Control and data analysis. Bioinformatics. 2005;21(18):3683–5. doi: 10.1093/bioinformatics/bti605. [DOI] [PubMed] [Google Scholar]
- 14.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98(9):5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Efron B, Tibshirani R. Empirical bayes methods and false discovery rates for microarrays. Genet Epidemiol. 2002;23(1):70–86. doi: 10.1002/gepi.1124. [DOI] [PubMed] [Google Scholar]
- 17.Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974;249(22):7130–9. [PubMed] [Google Scholar]
- 18.Kanika ND, Tar M, Tong Y, Kuppam DS, Melman A, Davies KP. The mechanism of opiorphin-induced experimental priapism in rats involves activation of the polyamine synthetic pathway. Am J Physiol Cell Physiol. 2009 doi: 10.1152/ajpcell.00656.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melman A, Zotova E, Kim M, Arezzo J, Davies K, DiSanto M, et al. Longitudinal studies of time-dependent changes in both bladder and erectile function after streptozotocin-induced diabetes in Fischer 344 male rats. BJU Int. 2009;104(9):1292–300. doi: 10.1111/j.1464-410X.2009.08573.x. [DOI] [PubMed] [Google Scholar]
- 20.Tamai K, Satoh K, Tsuchida S, Hatayama I, Maki T, Sato K. Specific inactivation of glutathione S-transferases in class Pi by SH-modifiers. Biochem Biophys Res Commun. 1990;167(1):331–8. doi: 10.1016/0006-291x(90)91769-o. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 22.Ezaki J, Kominami E. The intracellular location and function of proteins of neuronal ceroid lipofuscinoses. Brain Pathol. 2004;14(1):77–85. doi: 10.1111/j.1750-3639.2004.tb00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohkuwa T, Sato Y, Naoi M. Hydroxyl radical formation in diabetic rats induced by streptozotocin. Life Sci. 1995;56(21):1789–98. doi: 10.1016/0024-3205(95)00150-5. [DOI] [PubMed] [Google Scholar]
- 24.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J. 2007;26(7):1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meijer AJ, Codogno P. Autophagy: a sweet process in diabetes. Cell Metab. 2008;8(4):275–6. doi: 10.1016/j.cmet.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 26.Yoshimura K, Shibata M, Koike M, Gotoh K, Fukaya M, Watanabe M, et al. Effects of RNA interference of Atg4B on the limited proteolysis of LC3 in PC12 cells and expression of Atg4B in various rat tissues. Autophagy. 2006;2(3):200–8. doi: 10.4161/auto.2744. [DOI] [PubMed] [Google Scholar]
- 27.Chen C, Matesic LE. The Nedd4-like family of E3 ubiquitin ligases and cancer. Cancer Metastasis Rev. 2007;26(3–4):587–604. doi: 10.1007/s10555-007-9091-x. [DOI] [PubMed] [Google Scholar]
- 28.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288(5467):870–4. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 29.Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, et al. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol Cell Biol. 2002;22(13):4929–42. doi: 10.1128/MCB.22.13.4929-4942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144(5):891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thiruchelvam N, Nyirady P, Peebles DM, Fry CH, Cuckow PM, Woolf AS. Urinary outflow obstruction increases apoptosis and deregulates Bcl-2 and Bax expression in the fetal ovine bladder. Am J Pathol. 2003;162(4):1271–82. doi: 10.1016/S0002-9440(10)63923-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bilous RW. Diabetic autonomic neuropathy. Bmj. 1990;301(6752):565–7. doi: 10.1136/bmj.301.6752.565. [DOI] [PMC free article] [PubMed] [Google Scholar]