

Abstract
The prokaryotic V-ATPase of Enterococcus hirae, closely related to the eukaryotic enzymes, provides a unique opportunity to study the ion-translocation mechanism because it transports Na+, which can be detected by radioisotope (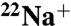 ) experiments and X-ray crystallography. In this study, we demonstrated that the binding affinity of the rotor ring (K ring) for
) experiments and X-ray crystallography. In this study, we demonstrated that the binding affinity of the rotor ring (K ring) for 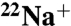 decreased approximately 30-fold by reaction with N,N′-dicyclohexylcarbodiimide (DCCD), and determined the crystal structures of Na+-bound and Na+-unbound K rings modified with DCCD at 2.4- and 3.1-Å resolutions, respectively. Overall these structures were similar, indicating that there is no global conformational change associated with release of Na+ from the DCCD-K ring. A conserved glutamate residue (E139) within all 10 ion-binding pockets of the K ring was neutralized by modification with DCCD, and formed an “open” conformation by losing hydrogen bonds with the Y68 and T64 side chains, resulting in low affinity for Na+. This open conformation is likely to be comparable to that of neutralized E139 forming a salt bridge with the conserved arginine of the stator during the ion-translocation process. Based on these findings, we proposed the ion-translocation model that the binding affinity for Na+ decreases due to the neutralization of E139, thus releasing bound Na+, and that the structures of Na+-bound and Na+-unbound DCCD-K rings are corresponding to intermediate states before and after release of Na+ during rotational catalysis of V-ATPase, respectively.
decreased approximately 30-fold by reaction with N,N′-dicyclohexylcarbodiimide (DCCD), and determined the crystal structures of Na+-bound and Na+-unbound K rings modified with DCCD at 2.4- and 3.1-Å resolutions, respectively. Overall these structures were similar, indicating that there is no global conformational change associated with release of Na+ from the DCCD-K ring. A conserved glutamate residue (E139) within all 10 ion-binding pockets of the K ring was neutralized by modification with DCCD, and formed an “open” conformation by losing hydrogen bonds with the Y68 and T64 side chains, resulting in low affinity for Na+. This open conformation is likely to be comparable to that of neutralized E139 forming a salt bridge with the conserved arginine of the stator during the ion-translocation process. Based on these findings, we proposed the ion-translocation model that the binding affinity for Na+ decreases due to the neutralization of E139, thus releasing bound Na+, and that the structures of Na+-bound and Na+-unbound DCCD-K rings are corresponding to intermediate states before and after release of Na+ during rotational catalysis of V-ATPase, respectively.
Keywords: V1/Vo-ATPase, inhibitor, sodium binding, rotary motor
Vacuolar ATPase (V-ATPase), which resembles ATP synthase (F-ATPase), functions as a proton pump in the acidic organelles and plasma membranes of eukaryotic cells and bacteria (1). Like F-ATPases, V-ATPases have a globular catalytic domain, V1 (equivalent to F1) which hydrolyzes ATP. This V1 domain is attached by central and peripheral stalks to an intrinsic membrane domain, Vo (equivalent to Fo) that pumps H+ (or Na+) across the membrane. In both F- and V-ATPases, ATP hydrolysis drives rotation of the central stalk and the membrane ring attached to it, which is composed of hydrophobic c subunits. H+ (or Na+) is pumped at the interface between the rotating ring and a static membrane component (the “a” subunit) (1). It is widely believed that electrostatic interaction at the glutamate (asparatate) residues within the H+ (or Na+) binding sites of the rotor ring and the conserved arginine residue of the a subunit is crucial for the ion-transport mechanism (2–4). However, the precise mechanism of ion translocation by F- and V-ATPases is still not clear because of the lack of the atomic structure of the a subunit, although high-resolution crystal structures of Na+- and H+-coupled rotor rings have been obtained (5–7).
N,N′-dicyclohexylcarbodiimide (DCCD) specifically inhibits ATPase activity and proton translocation by reacting with the conserved Glu (or Asp) residues of the rotor ring of both F- and V-ATPases (8–10). The exact mechanism by which DCCD reacts with the carboxyl groups of the acidic residues is unclear, but it is widely assumed that the reaction occurs with the protonated form of the carboxyl group. The reaction takes place in two steps as follows (Fig. 1): (i) The carboxylic acid forms a hydrogen bond to one of the nitrogen atoms in DCCD. The electron rearrangement in DCCD leads to formation of an unstable dicyclohexyl-O-acylurea intermediate. (ii) Further rearrangement of the molecule leads to formation of the stable dicyclohexyl-N-acylurea (DCNU) (11). Hence the ion-binding site accommodating the DCNU structure should have no bound H+ (or Na+) available for transport because of neutralization of the conserved carboxylic groups. DCCD has been proposed to influence the ring structure in a way that mimics the arginine residue in the a subunit (7, 9). Recently, Pogoryelov et al. reported a crystal structure of the c ring modified with DCCD in the F-type H+-ATPase from Spirulina platensis, even though the structure was obtained from native c-ring crystals after soaking with 5 mM DCCD and the occupancy of DCCD was low because of partial reaction (modified 27% of the c subunits) (12). The structure of the c ring revealed a small conformational change caused by side-chain movement of E62 modified with DCCD breaking off its hydrogen-bond interactions with Q29 and F60. They proposed that the open conformation of the DCCD-modified E62 corresponds to the neutralized conformation of E62 by forming the salt bridge with the conserved arginine of the a subunit (12).
Fig. 1.
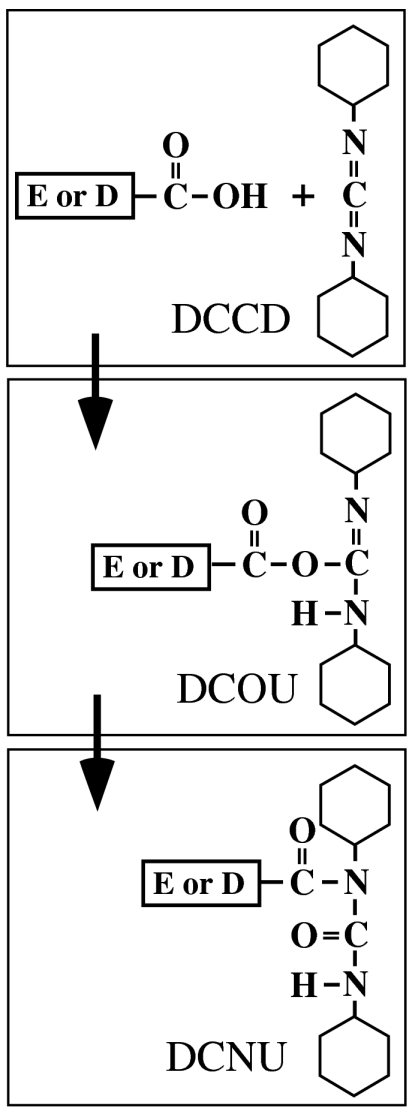
Proposed model of the DCCD-reaction mechanism at the ion-binding sites of the rotor ring. DCCD reacts with protonated forms of the conserved Glu or Asp residues at the ion-binding site in the rotor-ring subunit forming a dicyclohexyl-O-acylurea (DCOU) intermediate with subsequent acyl migration to form the stable DCNU.
We have identified a variant of V-ATPase in the fermentative bacterium Enterococcus hirae, which physiologically transports Na+ and Li+ (13). The enzyme is encoded by nine ntp genes (ntpFIKECGABD) organized within the ntp operon (14). Amino acid sequences of NtpF, -I, -K, -E, -C, -G, -A, -B, and -D are homologous to those of subunits G, a, c, E, d, F, A, B, and D of eukaryotic V-ATPases, respectively (15). The E. hirae V-ATPase possesses a high-binding affinity for the substrate Na+. This property enabled the measurement of Na+ binding to this ATPase using radioisotope (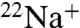 ) (16). We previously proposed an ion-transport mechanism of V-ATPase based on the Na+ binding properties of the rotor ring (K ring) and the crystal structure of the Na+-bound K ring (5, 17). To elucidate the precise ion-transport mechanism of V-ATPase, it is essential to understand the structure of Na+-unbound K ring after releasing bound substrate Na+. Here, we examined the inhibition mechanism of K ring by DCCD and found that the binding affinity for Na+ decreased by the DCCD reaction. Subsequently, we succeeded in obtaining the structures of Na+-bound and Na+-unbound rotor rings modified with DCCD. On the basis of these findings, we present an ion-transport mechanism involving the K ring during rotational catalysis of V-ATPase.
) (16). We previously proposed an ion-transport mechanism of V-ATPase based on the Na+ binding properties of the rotor ring (K ring) and the crystal structure of the Na+-bound K ring (5, 17). To elucidate the precise ion-transport mechanism of V-ATPase, it is essential to understand the structure of Na+-unbound K ring after releasing bound substrate Na+. Here, we examined the inhibition mechanism of K ring by DCCD and found that the binding affinity for Na+ decreased by the DCCD reaction. Subsequently, we succeeded in obtaining the structures of Na+-bound and Na+-unbound rotor rings modified with DCCD. On the basis of these findings, we present an ion-transport mechanism involving the K ring during rotational catalysis of V-ATPase.
Results
Inhibition of 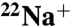 Binding to the Purified K Ring by DCCD.
Binding to the Purified K Ring by DCCD.
DCCD is believed to inhibit 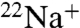 binding and ATPase activities of E. hirae V1Vo-ATPase, by attacking the conserved glutamic acid residue (E139) at the Na+-binding pocket of the NtpK proteolipid (16) (Fig. S1). In the present study, we examined the inhibitory effects of DCCD on
binding and ATPase activities of E. hirae V1Vo-ATPase, by attacking the conserved glutamic acid residue (E139) at the Na+-binding pocket of the NtpK proteolipid (16) (Fig. S1). In the present study, we examined the inhibitory effects of DCCD on 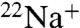 binding to the purified K ring and measured the kinetics of
binding to the purified K ring and measured the kinetics of 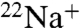 -binding inhibition by 0.2 mM DCCD at pH 6.0 in the absence of Na+ in the reaction buffer (Fig. 2A). Following 1 h of incubation with DCCD at room temperature, approximately 90% reduction in the
-binding inhibition by 0.2 mM DCCD at pH 6.0 in the absence of Na+ in the reaction buffer (Fig. 2A). Following 1 h of incubation with DCCD at room temperature, approximately 90% reduction in the 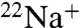 -binding ability was observed. The inhibition curve followed pseudo-first-order kinetics, suggesting that each NtpK monomer in the 10-mer K ring independently reacts with DCCD. The slope of the linear part of the data plotted as log
-binding ability was observed. The inhibition curve followed pseudo-first-order kinetics, suggesting that each NtpK monomer in the 10-mer K ring independently reacts with DCCD. The slope of the linear part of the data plotted as log 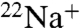 -binding versus incubation time gave an inhibition rate constant (kDCCD = 0.8 × 10-3 ± 1 × 10-4 [S-1]) (see Fig. 2A, Inset) similar to that for V1Vo-ATPase inhibition (Fig. S1).
-binding versus incubation time gave an inhibition rate constant (kDCCD = 0.8 × 10-3 ± 1 × 10-4 [S-1]) (see Fig. 2A, Inset) similar to that for V1Vo-ATPase inhibition (Fig. S1).
Fig. 2.

DCCD inhibition of 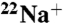 -binding to the purified K ring. (A) Kinetics of
-binding to the purified K ring. (A) Kinetics of 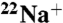 -binding inhibition by DCCD. The time course was initiated by addition of 0.2 mM DCCD. The inhibition rate constant (kDCCD) was estimated from a semilogarithmic plot (Inset). (B) Protective effect of Na+ on the DCCD inhibition of
-binding inhibition by DCCD. The time course was initiated by addition of 0.2 mM DCCD. The inhibition rate constant (kDCCD) was estimated from a semilogarithmic plot (Inset). (B) Protective effect of Na+ on the DCCD inhibition of 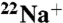 binding. (C) pH dependence of DCCD inhibition. (D) Na+-concentration dependence of
binding. (C) pH dependence of DCCD inhibition. (D) Na+-concentration dependence of 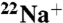 binding to the K ring (▴) and DCCD-modified K ring (●). θ is defined as the number of moles of bound Na+ per mole of K subunit but not K ring. (Inset) Scatchard plot of the specific binding of Na+.
binding to the K ring (▴) and DCCD-modified K ring (●). θ is defined as the number of moles of bound Na+ per mole of K subunit but not K ring. (Inset) Scatchard plot of the specific binding of Na+.
Inhibition of 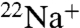 -binding and ATPase activities of the whole complex by DCCD is specifically prevented by the presence of Na+ (16, 18). The protective effects of Na+ against DCCD inhibition have also been observed in bacterial F-type Na+-ATPase (19). In order to examine the protective properties of Na+ against DCCD inhibition, we assessed the ability of DCCD to block
-binding and ATPase activities of the whole complex by DCCD is specifically prevented by the presence of Na+ (16, 18). The protective effects of Na+ against DCCD inhibition have also been observed in bacterial F-type Na+-ATPase (19). In order to examine the protective properties of Na+ against DCCD inhibition, we assessed the ability of DCCD to block 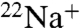 binding to the K ring in the presence of a range of Na+ concentrations (Fig. 2B). Absence of Na+ led to approximately 90% inhibition of Na+ binding by 0.2 mM DCCD. The protective effect of Na+ against DCCD inhibition increased depending on the increment of Na+ concentrations, and the residual activity was retained up to 75% in the presence of more than 1 mM NaCl. Thus, Na+ bound to the K ring probably prevents DCCD binding and inhibition. We previously reported that the K ring binds Na+ and H+ competitively (17). DCCD binding and inhibition were prevented by alkaline pH (> 7.5) (Fig. 2C) in the absence of Na+ where the E139 residue (pKa = 5.5) were deprotonated (17). Thus, DCCD reacts with the protonated E139 in the K ring but not with the deprotonated E139 that binds Na+, consistent with the reaction model described in Fig. 1 whereby DCCD reacts with protonated forms of acidic residues.
binding to the K ring in the presence of a range of Na+ concentrations (Fig. 2B). Absence of Na+ led to approximately 90% inhibition of Na+ binding by 0.2 mM DCCD. The protective effect of Na+ against DCCD inhibition increased depending on the increment of Na+ concentrations, and the residual activity was retained up to 75% in the presence of more than 1 mM NaCl. Thus, Na+ bound to the K ring probably prevents DCCD binding and inhibition. We previously reported that the K ring binds Na+ and H+ competitively (17). DCCD binding and inhibition were prevented by alkaline pH (> 7.5) (Fig. 2C) in the absence of Na+ where the E139 residue (pKa = 5.5) were deprotonated (17). Thus, DCCD reacts with the protonated E139 in the K ring but not with the deprotonated E139 that binds Na+, consistent with the reaction model described in Fig. 1 whereby DCCD reacts with protonated forms of acidic residues.
Binding of 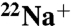 to the Purified K Ring Modified with DCCD.
to the Purified K Ring Modified with DCCD.
Functional analysis has shown that the purified K ring binds one Na+ per K monomer with high affinity (Kd(Na+) = 15 ± 4 μM) (Fig. 2D, filled triangles) as described previously (17), consistent with the crystal structure of the Na+-bound K ring (5). The Na+-binding capacity of the K ring, measured in the presence of 15 μM 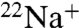 (Kd(Na+) of the purified K ring) decreased after preincubation with DCCD as described in the previous section. Fig. 2D (filled circles) shows that
(Kd(Na+) of the purified K ring) decreased after preincubation with DCCD as described in the previous section. Fig. 2D (filled circles) shows that 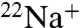 binding of the DCCD-K ring was also dependent on NaCl concentration, although higher concentrations of NaCl were necessary to stimulate binding. The Scatchard plot (Fig. 2D, Inset; filled circles) of this data gave an intercept on the abscissa of approximately one, indicating that one DCCD-K subunit binds one Na+ (SD 0.91 ± 0.12). The slope of the Scatchard plot indicates that the dissociation constant (Kd(Na+)) is 0.5 mM (SD 0.5 ± 0.04 mM), suggesting that the DCCD-K ring can bind Na+ with low affinity.
binding of the DCCD-K ring was also dependent on NaCl concentration, although higher concentrations of NaCl were necessary to stimulate binding. The Scatchard plot (Fig. 2D, Inset; filled circles) of this data gave an intercept on the abscissa of approximately one, indicating that one DCCD-K subunit binds one Na+ (SD 0.91 ± 0.12). The slope of the Scatchard plot indicates that the dissociation constant (Kd(Na+)) is 0.5 mM (SD 0.5 ± 0.04 mM), suggesting that the DCCD-K ring can bind Na+ with low affinity.
Crystal Structure of the DCCD-Modified K Ring.
The purified K ring was incubated with 0.2 mM DCCD in the absence of Na+ at pH 6.0 for 5 h before crystallization. Under these conditions, DCCD reacts with most of the Na+-binding sites in the K ring as shown in Fig. 2A. Crystals were obtained in the presence of 240 mM sodium citrate (pH 7.5), which should allow binding of Na+ to the DCCD-K ring (low affinity for Na+). The structural data are summarized in Table S1. The overall structure of the DCCD-modified K ring, composed of 10 K subunits, was similar (rmsd for main chain; 0.428 Å) to that of the wild-type K ring [Protein Data Bank (PDB) ID 2BL2] (5) except for the ion-binding pocket in each individual K subunit (Fig. 3 A and B). Fig. 3C shows the electron-density map at the ion-binding pocket of the K ring modified with DCCD, calculated using the model structure omitting the side chain of E139 and Na+. The positive electron-density peak (shown in red) around E139 was interpreted as DCNU (see Fig. 1), which was found associated with E139 at all 10 Na+-binding sites in the K-ring structure. No other region that could correspond to additional modifications by DCCD was present in the electron-density map. A strong density peak in the middle of the Na+-binding site was observed (Fig. 3C), similar to that of the wild-type K-ring structure (5) (Fig. 3F). We interpreted this result as Na+ based on the geometry of the coordination and the Na+ (Kd(Na+) = 0.5 mM) affinity of the DCCD-K ring (Fig. 3 C–E; shown in light-blue sphere). Each Na+ in the DCCD-K ring is surrounded by five oxygen atoms, four of them contributed by the side chains of T64, Q65, Q110, and E139 (covalently modified as DCNU), and the fifth contributed by the main-chain carbonyl of L61 (Fig. 3 D and E). Residues in helix 2, 3, and 4 comprise the Na+-binding pocket as seen in the wild-type K-ring structure (Fig. 3 G and H). The distances between Na+ and oxygen atoms of the DCCD-K ring (2.5–3.0 Å; Fig. 3 D and E) are longer than those of the wild-type K ring (2.2–2.3 Å; Fig. 3 G and H). The metal-oxygen distances and the Na+-binding affinity decreased as the result of the neutralization of E139 by DCCD modification.
Fig. 3.

Structure of the DCCD-modified K ring. Helices 0 (residues 1–8), 1 (11–46), 2 (51–79), 3 (85–124), and 4 (127–156), and loops (9–10, 47–50, 80–84, and 125–126) are colored in violet, blue, green, orange, yellow, and red in ribbon representations, respectively. Residues involved in ion binding are shown in stick representation. Oxygen and nitrogen atoms are in red and dark blue, respectively. (A) A side view of the K ring. The DCNU is shown in space-filling representation. The red box indicates the location of the ion-binding pocket. (B) A top view from the cytoplasmic side. (C–K) Ion-binding pocket of the K ring as follows: (C–E) DCCD-modified K ring in the presence of Na+; (F–H) wild-type K ring in the presence of Na+ (PDB ID 2BL2) (5); (I–K) DCCD-modified K ring in the absence of Na+. Na+ are shown as light-blue spheres. (C, F, and I) Omit maps and the coordinates calculated using the model structure omitting the side chain of E139 and Na+ at the binding pocket contoured at 1.5σ. The positive peaks of the |Fo| - |Fc| electron-density map contoured at 3.5σ (C and I) and 5.0 sigma (F) are shown in red. (D, G, and J) Final structure models. The 2|Fo| - |Fc| maps of E139 with and without DCNU were shown in blue. Black lines are Na+-O bonds with distances. E, H, and K are top views from the cytoplasmic side of D, G, and J, respectively.
The DCCD-K ring was also crystallized with reservoir solution in the absence of Na+. In this condition, the DCCD-K ring (low affinity for Na+) should not bind Na+ in the binding pocket. The structural data are summarized in Table S1. The overall structure of the DCCD-K ring was similar (rmsd for main chain; 0.434 Å) to that of the Na+-bound DCCD-K ring crystallized at high-Na+ concentration. Fig. 3I shows the omit map of the ion-binding pocket calculated using the model structure after removing the E139 side chain and Na+ at the binding pocket. The positive electron-density peak (shown in red) around E139 was similar to that of the DCCD-K ring crystallized at high-Na+ concentration, although the density for Na+ in the middle of the Na+-binding pocket was much weaker (Fig. 3I). Based on biochemical analysis indicating low affinity of the DCCD-K ring for Na+ (Fig. 2D) and the crystallographic data obtained, we concluded that the structure obtained in the absence of Na+ corresponds to the Na+-free form of the K ring (Fig. 3 J and K). Similarity between Na+-bound and Na+-unbound structures indicates that there is no global conformational change associated with release of Na+ from the binding pocket modified with DCCD.
Discussion
Mechanism of DCCD Inhibition.
V-ATPases have been implicated in a number of diseases and are known as important drug targets in diseases such as osteoporosis and cancer (1). Therefore, inhibitors of V-ATPase have potential as drugs for these diseases, and it is important to understand the precise inhibition mechanism of V-ATPase in order to develop drugs targeting the V-ATPases. In this study, we examined the inhibition mechanism of DCCD in the ion-binding pocket of the K ring by using a 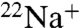 -binding inhibition assay and X-ray crystallography. DCCD inhibited
-binding inhibition assay and X-ray crystallography. DCCD inhibited 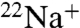 binding at acidic pH in the absence of Na+ (Fig. 2 A and B), suggesting that DCCD reacted with the protonated form of the carboxyl group (E139) located in the binding pocket. These findings correspond with the studies of F-type Na+-ATPase and H+-ATPases (8, 9). The electron density corresponding to DCCD fits well to the DCNU structure, consistent with the proposed model in Fig. 1. The DCCD-K ring still retained Na+-binding ability but the affinity for Na+ was much lower than that of the wild-type K ring (Fig. 2D). Previous results suggested that any Na+ bound to the DCCD K ring is not transported in the whole complex, because ATPase activity and
binding at acidic pH in the absence of Na+ (Fig. 2 A and B), suggesting that DCCD reacted with the protonated form of the carboxyl group (E139) located in the binding pocket. These findings correspond with the studies of F-type Na+-ATPase and H+-ATPases (8, 9). The electron density corresponding to DCCD fits well to the DCNU structure, consistent with the proposed model in Fig. 1. The DCCD-K ring still retained Na+-binding ability but the affinity for Na+ was much lower than that of the wild-type K ring (Fig. 2D). Previous results suggested that any Na+ bound to the DCCD K ring is not transported in the whole complex, because ATPase activity and 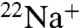 -transport activity were completely inhibited by DCCD (18). We demonstrated that the rate of inhibition by DCCD for ATPase activity of the whole complex was approximately 6.5 times faster than that of
-transport activity were completely inhibited by DCCD (18). We demonstrated that the rate of inhibition by DCCD for ATPase activity of the whole complex was approximately 6.5 times faster than that of 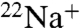 -binding (Fig. S1). Under these inhibition conditions, DCCD did not inhibit the V1-ATPase. These findings suggest that modification of one or two NtpK subunits in the 10-mer K ring of the whole complex is sufficient for complete inhibition of the enzymatic activity, which is also reported in previous studies of F- and V-ATPases (9, 10). The additional bulk modification with DCCD can possibly cause steric hindrance at the interface between the K ring and NtpI (corresponding to the a subunit of other V-ATPases).
-binding (Fig. S1). Under these inhibition conditions, DCCD did not inhibit the V1-ATPase. These findings suggest that modification of one or two NtpK subunits in the 10-mer K ring of the whole complex is sufficient for complete inhibition of the enzymatic activity, which is also reported in previous studies of F- and V-ATPases (9, 10). The additional bulk modification with DCCD can possibly cause steric hindrance at the interface between the K ring and NtpI (corresponding to the a subunit of other V-ATPases).
Ion-Binding/Releasing Mechanism of the K Ring.
We have previously proposed an ion-binding/releasing mechanism of the K ring based on the crystal structures of the ion-bound K ring, and the ion-binding properties and selectivity of the K ring (5, 17) as follows: the bound ion is occluded by E139 and its carbonyl oxygen atom position is stabilized by hydrogen bonds with Y68 and T64 side chains (closed form) (Fig. 3G; shown as thick line). The bound ion can be accessed by changing the torsion angles of the E139 side chain by losing the hydrogen bonds (open form). Binding/releasing of the ion can then be easily achieved through the open space. In the DCCD-K-ring structures obtained, the hydrogen bonds with Y68 and T64 working for stabilization of the “closed” form are disrupted by modification with DCCD (Fig. 3 D and J). Thus, the ion-binding site modified with DCCD probably corresponds to the open conformation, which is consistent with the low binding affinity for Na+ of the DCCD-K ring (Fig. 2D). A high-resolution structure of the c ring from F-type H+-ATPase modified with DCCD has also been shown in a similar open conformation (Fig. S2), which was verified by the molecular dynamic simulations of the c ring (12).
Ion-Transport Mechanism of V-ATPase.
We have previously proposed an ion-transport mechanism (5, 14, 17) as follows: clockwise rotation of the K ring driven by ATP-hydrolysis energy in V1-ATPase, as viewed from the cytoplasm, brings an occupied Na+-binding site into the K ring–NtpI interface (Fig. 4A). The proximity of the Na+ site to the essential R573 residue of NtpI produces an electrostatic interaction between R573 and E139, losing the hydrogen-bound network in the Na+-binding pocket of the K ring, resulting in the release of Na+ into the periplasm via a half-channel in NtpI as indicated by the red arrow in Fig. 4B. Further rotation caused by ATP-hydrolysis energy in V1-ATPase, which may disrupt the Arg-Glu interaction, results in the binding of a cytoplasmic Na+ as indicated by the blue arrow in Fig. 4D.
Fig. 4.

A model for the ion-transport mechanism of Na+-transporting V-ATPase. The model is based on the crystal structures of the K ring with and without DCCD. The views are from the cytoplasm and show the K-ring–NtpI interface at the level of the Na+-binding sites. Residues involved in Na+ binding are shown in stick representation. Colors correspond to Fig. 3. NtpI shown in green is in close proximity to the K ring with the essential R573 (blue stick representation), which has two half-channels connecting to the periplasm and cytoplasm, respectively. The Na+-transport mechanism of V-ATPase is represented by four intermediate states (A–D). See text and Movie S1 for details.
In this study, we found that the binding affinity of the K ring for Na+ decreased approximately 30-fold by reaction with DCCD, and we obtained the structures of the DCCD-K ring with and without the substrate ion Na+. Binding of DCCD effectively neutralized the negative charge of the conserved E139 (Fig. 3 C–E and I–K), resulting in lower affinity for Na+ in the binding pocket (Fig. 2D). Similarity of the Na+-bound and Na+-unbound DCCD-K ring structures to that of the wild-type K ring indicates that there is no global conformational change associated with neutralization of E139 and with release of Na+ from the binding pocket modified with DCCD. We believe that E139 neutralized by DCCD adopts the open conformation corresponding to the similar open conformation through formation of an electrostatic interaction with R573 during the ion-transport process. A similar interpretation was proposed for the rotational model of F-type H+-ATPase (12). The structure of the Na+-bound K ring modified with DCCD (Fig. 3 C–E) seems to fit to the intermediate state prior to the release of Na+ into the periplasm, as hypothesized in Fig. 4B. Neutralization of E139 in the binding site reduces the affinity for the bound Na+, which is readily released into the periplasm (Fig. 4B, indicated by the red arrow). The structure of the Na+-unbound DCCD-K ring (Fig. 3 I–K) is likely to correspond to another intermediate state after release of Na+ into the periplasm, as shown in Fig. 4C. Thus, we propose an ion-transport mechanism of the V-ATPase whereby ion translocation can be accomplished through an affinity change of the Na+ binding sites by neutralization of E139 due to formation of a salt bridge with R573 of the stator (see Movie S1).
Materials and Methods
Protein Preparation.
V1Vo-ATPase was purified from E. hirae cells (18). The K ring was released from the isolated V1Vo-ATPase by treatment with 10% isopropanol and then purified by anion exchange and gel filtration chromatography (20).
DCCD Inhibition Kinetics of Na+ Binding to the Purified K Ring.
The K ring is comprised of ten 16 kDa K subunits. One milligram of purified K ring corresponds to 60 nmol K subunit (6 nmol K ring). The reaction mixture that contained 6 μM purified K subunit (0.6 μM K ring) and 15 μM 22NaCl (3 cpm/nL) in Buffer A (20 mM MES-Tris, 20% glycerol, 0.05% n-dodecyl β-d-maltoside; pH 6.0) was incubated for 2 h at room temperature, which was sufficient to saturate Na+ binding to the K ring. The time-course experiment of DCCD inhibition was initiated by addition of 0.2 mM DCCD to the incubation mixture at room temperature. Free 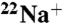 was rapidly separated using a Dowex-50 method at various time intervals (16). The inhibition curve followed pseudo-first-order kinetics. The rate constants (kDCCD) for
was rapidly separated using a Dowex-50 method at various time intervals (16). The inhibition curve followed pseudo-first-order kinetics. The rate constants (kDCCD) for 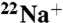 -binding inhibition by DCCD were calculated by fitting the linear part of the data plotted as
-binding inhibition by DCCD were calculated by fitting the linear part of the data plotted as 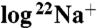 binding versus incubation time. The slope of the line is the negative of the rate constant for inhibition. All
binding versus incubation time. The slope of the line is the negative of the rate constant for inhibition. All 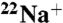 -binding experiments were repeated three times, the data were averaged, and the standard deviations were calculated.
-binding experiments were repeated three times, the data were averaged, and the standard deviations were calculated.
Protection Effect of Na+ from 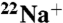 -Binding Inhibition by DCCD.
-Binding Inhibition by DCCD.
The purified K ring in Buffer A was incubated with 0.2 mM DCCD in the presence of several concentrations of NaCl for 2 h at room temperature, which was sufficient to saturate the inhibition of Na+ binding by DCCD. Free DCCD and NaCl in the mixture were removed by quick spin column (Sephadex G-50) equilibrated with Buffer A. The eluate (6 μM purified K subunit) was incubated with 15 μM 22NaCl (3 cpm/nL) for 2 h. The Na+-binding capacity was measured as described in the previous subsection.
pH Dependence of 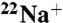 Binding Inhibition by DCCD.
Binding Inhibition by DCCD.
The purified K ring was incubated with 0.2 mM DCCD at varying pH (pH 4–5.5, citrate-Tris; pH 5.5–7, Bistris; pH 7–9, Tris·HCl). Free DCCD and pH buffer were removed by a quick spin column (Sephadex G-50) equilibrated with Buffer A. The Na+-binding capacity was measured as described in the previous subsection.
Na+-Concentration Dependence of 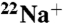 Binding to the DCCD-Modified K Ring.
Binding to the DCCD-Modified K Ring.
DCCD-modified K ring was obtained by incubating with 0.2 mM DCCD for 5 h at 20 °C. The reaction mixture that contained 6 pmol DCCD-bound K ring (or DCCD-unbound K ring) and various concentrations of 22NaCl (30,000 cpm) in 10 μL of Buffer A was incubated for 2 h at room temperature, which was sufficient to saturate Na+ binding to the K ring. Na+-binding capacity was measured as described in the previous subsection.
Crystallization of DCCD-Modified K Ring in the Presence of Na+.
The purified K ring (2.5 mg/mL) in buffer containing 10 mM MES-HCl (pH 6.0), 10% glycerol, 0.32 mM n-dodecyl β-d-maltoside, and 1.2 mM undecyl maltoside was incubated with 0.2 mM DCCD for 5 h at 20 °C. The protein sample (1 μL) was then mixed with reservoir solution (1 μL) consisting of 100 mM Tris·HCl (pH 7.5), 4% glycerol, 240 mM sodium citrate, and 32% PEG 400. Crystals grew at 20 °C in sitting drops by vapor diffusion. The crystals appeared after 5 d and grew to maximum dimensions (200 × 200 × 80 μm) in 1 mo, and were plunged into liquid nitrogen and stored at 100 K.
Crystallization of DCCD-Modified K Ring in the Absence of Na+.
The purified K ring (2.5 mg/mL; 1 μL) modified with DCCD as described in the previous subsection was mixed with reservoir solution (1 μL) consisting of 100 mM Tris·HCl (pH 7.5), 4% glycerol, 240 mM potassium citrate, and 35% PEG 400. Crystals grew at 20 °C in sitting drops by vapor diffusion. The crystals appeared after 10 d and grew to maximum dimensions (150 × 150 × 40 μm) in 1 mo, and were plunged into liquid nitrogen and stored at 100 K.
Data Collection, Structure Determination, and Refinement.
Diffraction data were collected from a single crystal at cryogenic temperature (100 K) on beamline BL26B1 and BL41XU at SPring-8. The data were indexed and integrated with MOSFLM (21) and processed further with the CCP4 programs (22). The structures were solved by molecular replacement with AMoRe (23) using the Na+-bound K-ring structure as a search model (PDB ID code 2BL2) in all subsequent refinement steps, 5% of the data was set aside for calculation of the free R factor. The atomic model was built using the program O (24) and refined using REFMAC5 (25). The coordinates for N-5-cyclohexyl-N-5-[(cyclohexylamino) carbonyl] glutamine were obtained from the PDB file (1E79) of bovine F1-ATPase inhibited with DCCD (26). Tight noncrystallographic symmetry (NCS) restraints (sigma 0.05 Å) were applied to the 10 K protomers (excluding regions in lattice contacts). Translation, libration, and screw-rotation refinement (TLS), with one TLS group per protomer, was carried out in the final stages without NCS restraints. The refined structures were validated with PROCHECK (27). Figures were generated with PyMOL (28).
Supplementary Material
Acknowledgments.
We thank Dr. Bernadette Byrne for critical reading of the manuscript. We also thank the beam-line staff at BL26B1 and BL41XU of SPring-8 for their help during data collection. This work was supported by Targeted Proteins Research Program (S.I. and T.M.), grant-in-aid (18074003), and Special Coordination Funds for Promoting Science and Technology from the Ministry of Education, Culture, Sports, Science, and Technology of the Japanese government, and partially supported by the RIKEN Structural Genomics/Proteomics Initiative (S.Y.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2DB4 and 3AOU).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1103287108/-/DCSupplemental.
References
- 1.Forgac M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 2.Jiang W, Fillingame RH. Interacting helical faces of subunits a and c in the F1Fo ATPsynthase of Escherichia coli defined by disulfide cross-linking. Proc Natl Acad Sci USA. 1998;95:6607–6612. doi: 10.1073/pnas.95.12.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vorburger T, et al. Arginine-induced conformational change in the c-ring/a-subunit interface of ATP synthase. FEBS J. 2008;275:2137–2150. doi: 10.1111/j.1742-4658.2008.06368.x. [DOI] [PubMed] [Google Scholar]
- 4.Kawano M, Igarashi K, Yamato I, Kakinuma Y. Arginine residue at position 573 in Enterococcus hirae vacuolar-type ATPase NtpI subunit plays a crucial role in Na+ translocation. J Biol Chem. 2002;277:24405–24410. doi: 10.1074/jbc.M200973200. [DOI] [PubMed] [Google Scholar]
- 5.Murata T, et al. Structure of the rotor of the V-Type Na+-ATPase from Enterococcus hirae. Science. 2005;308:654–659. doi: 10.1126/science.1110064. [DOI] [PubMed] [Google Scholar]
- 6.Meier T, et al. Structure of the rotor ring of F-Type Na+-ATPase from Ilyobacter tartaricus. Science. 2005;308:659–662. doi: 10.1126/science.1111199. [DOI] [PubMed] [Google Scholar]
- 7.Pogoryelov D, Yildiz O, Faraldo-Gomez JD, Meier T. High-resolution structure of the rotor ring of a proton-dependent ATP synthase. Nat Struct Mol Biol. 2009;16:1068–1073. doi: 10.1038/nsmb.1678. [DOI] [PubMed] [Google Scholar]
- 8.Hermolin J, Fillingame RH. H+-ATPase activity of Escherichia coli F1F0 is blocked after reaction of dicyclohexylcarbodiimide with a single proteolipid (subunit c) of the F0 complex. J Biol Chem. 1989;264:3896–3903. [PubMed] [Google Scholar]
- 9.Kluge C, Dimroth P. Kinetics of inactivation of the F1Fo ATPase of Propionigenium modestum by dicyclohexylcarbodiimide in relationship to H+ and Na+ concentration: Probing the binding site for the coupling ions. Biochemistry. 1993;32:10378–10386. doi: 10.1021/bi00090a013. [DOI] [PubMed] [Google Scholar]
- 10.Arai H, Berne M, Forgac M. Inhibition of the coated vesicle proton pump and labeling of a 17,000-dalton polypeptide by N, N′-dicyclohexylcarbodiimide. J Biol Chem. 1987;262:11006–11011. [PubMed] [Google Scholar]
- 11.Azzi A, Casey RP, Nalecz MJ. The effect of N,N′-dicyclohexylcarbodiimide on enzymes of bioenergetic relevance. Biochim Biophys Acta. 1984;768:209–226. doi: 10.1016/0304-4173(84)90017-x. [DOI] [PubMed] [Google Scholar]
- 12.Pogoryelov D, et al. Microscopic rotary mechanism of ion translocation in the Fo complex of ATP synthases. Nat Chem Biol. 2010;6:891–899. doi: 10.1038/nchembio.457. [DOI] [PubMed] [Google Scholar]
- 13.Murata T, et al. Catalytic properties of Na(+)-translocating V-ATPase in Enterococcus hirae. Biochim Biophys Acta. 2001;1505:75–81. doi: 10.1016/s0005-2728(00)00278-4. [DOI] [PubMed] [Google Scholar]
- 14.Murata T, Yamato I, Kakinuma Y. Structure and mechanism of vacuolar Na+-translocating ATPase from Enterococcus hirae. J Bioenerg Biomembr. 2005;37:411–413. doi: 10.1007/s10863-005-9481-0. [DOI] [PubMed] [Google Scholar]
- 15.Kakinuma Y, Yamato I, Murata T. Structure and function of vacuolar Na+-translocating ATPase in Enterococcus hirae. J Bioenerg Biomembr. 1999;31:7–14. doi: 10.1023/a:1005499126939. [DOI] [PubMed] [Google Scholar]
- 16.Murata T, Igarashi K, Kakinuma Y, Yamato I. Na+ binding of V-type Na+-ATPase in Enterococcus hirae. J Biol Chem. 2000;275:13415–13419. doi: 10.1074/jbc.275.18.13415. [DOI] [PubMed] [Google Scholar]
- 17.Murata T, et al. Ion binding and selectivity of the rotor ring of the Na+-transporting V-ATPase. Proc Natl Acad Sci USA. 2008;105:8607–8612. doi: 10.1073/pnas.0800992105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murata T, et al. Properties of the V0V1 Na+-ATPase from Enterococcus hirae and its V0 moiety. J Biochem. 1999;125:414–421. doi: 10.1093/oxfordjournals.jbchem.a022302. [DOI] [PubMed] [Google Scholar]
- 19.Kluge C, Dimroth P. Specific protection by Na+ or Li+ of the F1F0-ATPase of Propionigenium modestum from the reaction with dicyclohexylcarbodiimide. J Biol Chem. 1993;268:14557–14560. [PubMed] [Google Scholar]
- 20.Murata T, et al. The membrane domain of the Na+-motive V-ATPase from Enterococcus hirae contains a heptameric rotor. J Biol Chem. 2003;278:21162–21167. doi: 10.1074/jbc.M301620200. [DOI] [PubMed] [Google Scholar]
- 21.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992;26:22–23. [Google Scholar]
- 22.CCP4 (Collaborative Computational Project N) The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 23.Navaza J. Implementation of molecular replacement in AMoRe. Acta Crystallogr D Biol Crystallogr. 2001;57:1367–1372. doi: 10.1107/s0907444901012422. [DOI] [PubMed] [Google Scholar]
- 24.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 25.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 26.Gibbons C, Montgomery MG, Leslie AG, Walker JE. The structure of the central stalk in bovine F(1)-ATPase at 2.4 A resolution. Nat Struct Biol. 2000;7:1055–1061. doi: 10.1038/80981. [DOI] [PubMed] [Google Scholar]
- 27.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 28.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.