

Abstract
The high levels of hepatitis B virus (HBV) surface antigen (HBsAg)-bearing subviral particles in the serum of chronically infected individuals play an important role in suppressing HBV-specific immune response, and are only mildly affected by the current small molecule therapies. Thus, a therapy that specifically reduces HBsAg serum levels could be used in combination therapy with nucleos(t)ide drugs, or permit therapeutic vaccination for the treatment of HBV infection. Herein, we report the design, synthesis and evaluation of novel triazolo-pyrimidine inhibitors (1, 3 and 4) of HBsAg cellular secretion, with activity against drug-resistant HBV variants. Extensive SAR led to substantial improvements in the EC50 of the parent compound, 5 (HBF-0259), with the best being 3c, with EC50 = 1.4 ± 0.4 μM, SI ≥ 36. The lead candidates, both 1a (PBHBV-001) and 3c (PBHBV-2-15), were well-tolerated in both normal and HBV-transgenic mice, and exhibited acceptable pharmacokinetics and bioavailability in Sprague-Dawley rats.
Introduction
Chronic hepatitis B affects more than 350 million people worldwide, and is associated with as many as 1 million deaths per year, due to the development of hepatocellular carcinoma, cirrhosis, and other complications.1-4 The current treatment options for hepatitis B include immune system modulation with two forms of interferon-α (IFN-α), and direct inhibition of the viral polymerase through five nucleos(t)ide analogues (Figure 1). Different versions of IFN-α show clinical benefit in up to 60% of patients, but result in complete clearance in only 7%.5, 6 However, there are several undesirable aspects to its use that render it far from ideal, chief among these being a high rate of adverse side effects that cause many patients to discontinue therapy.7, 8 The FDA-approved small molecule drugs offer advantages over IFN-α including oral bioavailability, low toxicity, and efficacy in almost all patients, but complete clearance of infection (as measured by complete HBsAg and viremia loss) is rare, and the emergence of resistance remains a problem.9 Further development of effective therapeutics awaits antiviral drugs that target the viral life cycle through mechanisms other than inhibition of the viral polymerase, whether for monotherapy or combination therapy.10
Figure 1.

Nucleos(t)ide Drugs Approved by FDA for the Treatment of Chronic Hepatitis B
One of the classic hallmarks of chronic hepatitis B is the high levels of hepatitis B virus surface antigen (HBsAg) in the serum of patients, which may reach 400 μg/mL (0.4% of total serum protein).11, 12 The antigenemia resulting from production of subviral particles is thought to play an important role in suppressing the HBV-specific immune response. In addition, recent reports have suggested that HBsAg acts directly on dendritic cells to limit cytokine production and adaptive immunity.13, 14 Functionally, the role of HBsAg in eliciting HBV-specific immune tolerance has been demonstrated in woodchucks infected with woodchuck hepatitis virus (WHV), an animal model that closely recapitulates chronic hepatitis B in humans. Reduction of antigenemia with the experimental antiviral clevudine resulted in a partial restoration of virus-specific immune response.15 Thus, inhibitors of HBsAg secretion16, 17 could potentially enable the therapeutic use of the HBV vaccine, or be used as combination therapy with nucleos(t)ide drugs for the treatment of HBV infection.
Our group is advancing the concept that control of HBsAg antigenemia, for which existing therapies have limited value, will be an important arm of future hepatitis B therapy regimens. Recently, we performed high-throughput screening of an in-house small-molecule library, using the HBV-expressing cell line HepG2.2.15, and identified 7-(2-chloro-6-fluorophenyl)-5-(4-chlorophenyl)-4,5,6,7-tetrahydro-tetrazolo[1,5-a]pyrimidine 5 (HBF-0259, Figure 2),18 as an effective inhibitor of HBV surface antigen secretion. Herein, we describe our follow-up lead optimization studies of a series of novel triazolo-pyrimidine inhibitors of HBsAg secretion, including the rational structure design, synthesis and biological evaluation. The structure exploration involves the A, B, C and D rings of 5 as shown in Scheme 1. In addition, biological profiles of selected compounds including activities against HBV mutants (5, 1a and 3c), in vivo toxicity and pharmacokinetic (PK) studies (1a and 3c) are also reported.
Figure 2.
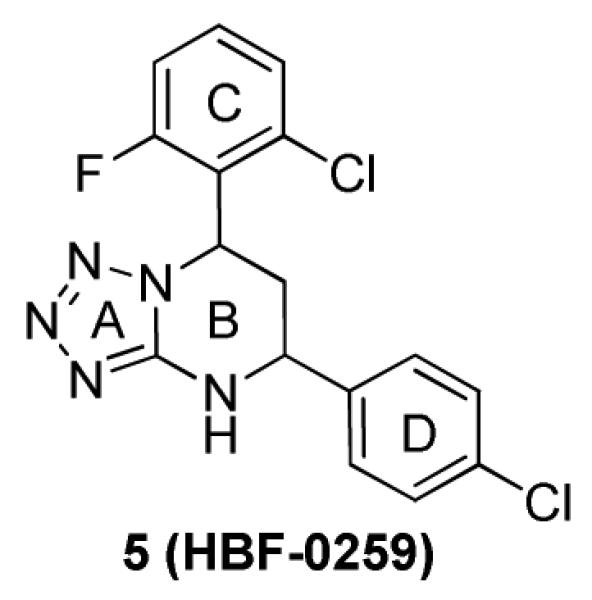
Structure of 7-(2-Chloro-6-fluorophenyl)-5-(4-chlorophenyl)-4,5,6,7-tetrahydro-tetrazolo[1,5-a] pyrimidine 5
Scheme 1.

Structure Exploration on A, B, C and D Rings
Chemistry
Scheme 1 illustrates our strategy for the optimization of the original hit, 5: 1) modification of the A ring to address the potential chemical stability issue of 5; 2) structure exploration at the C and D side chains; and 3) understanding the effects of the chirality of the B ring on activity.
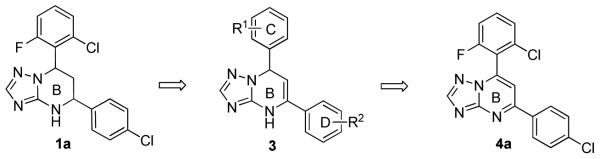
The tetrahydro-tetrazolo-pyrimidine structure of 5 could undergo rearrangement via the open azide form19 to give another isomer, as shown in Scheme 2. To address this potential stability issue, the triazole analogue 1a, by replacing N-2 with a CH group (on the A ring), was designed and synthesized to block the rearrangement process.
Scheme 2.

Ring Opening and Rearrangement of the 4,5,6,7-Tetrahydro-tetrazolo[1,5-a]pyrimidine
The synthetic sequence of 1a and related triazolo-pyrimidine analogues 1, 3 and 4 was described in Scheme 3. Condensation of corresponding substituted benzaldehydes with acetophenones in the presence of sodium hydroxide in methanol, at room temperature, formed chalcones 2.20 Reaction of α,β-unsaturated ketones with 1H-1,2,4-triazol-3-amine in DMF at refluxing temperature gave dihydrotriazolo-pyrimidine analogues 3, which were then reduced by sodium borohydride in methanol to afford tetrahydro-triazolo-pyrimidine analogues 1.21, 22 Analogues 4 were readily prepared through the oxidation of 3 with NBS.21 Although there are two chiral centers present in the molecule, the triazolo-pyrimidine analogues 1 exist only as a pair of thermodynamically more stable cis-enantiomers,23, 24 evidenced by chiral HPLC (Figure 4) and NMR analysis. X-ray crystal structure of 1a (Figure 3) confirmed that the C ring and D ring adopt the cis-conformation, while the tetrahydro-pyrimidine ring (B ring) adopts “half chair” conformation. Furthermore, the resolution of two enantiomers of 1a was successfully carried out through chiral column chromatography (See “Experimental Section”).
Scheme 3.

Synthesis of the Triazole Analogues 1, 3 and 4
(a) NaOH, MeOH, rt; (b) 1H-1,2,4-triazol-3-amine, DMF, reflux; (c) NaBH4, MeOH, reflux; (d) NBS, iPrOH, reflux.
Figure 4.

Chiral Resolution of Analogue 1a: Panel A, HPLC Spectrum of Racemic 1a; Panel B, HPLC Spectrum of cis-enantiomer 1 of 1a; Panel C, HPLC Spectrum of cis-enantiomer 2 of 1a
Figure 3.

X-ray Crystal Structure of 1a
Results and Discussions
Inhibition of HBsAg Secretion
In the HepG2.2.15 cell line, the inhibitory activity (EC50) and the cytotoxicity (CC50) of 1a were measured by the HBsAg ELISA and the MTT assay (Table 1), respectively, as we previously reported.18 The EC50 of 1a for the inhibition of surface antigen secretion was 4.1 ± 2.1 μM, almost three-fold more active than 5 (EC50 = 11.3 ± 3.2 μM). No cytotoxicity was observed for either analogue up to the highest concentration tested (50 μM). Furthermore, inhibitory activity of HBsAg secretion was enantio-selective and was only observed for the enantiomer 1 (Figure 4) of 1a (EC50 = 4.2 ± 1.6 μM), while the enantiomer 2 was largely inactive (EC50 = 35.6 ± 15.3 μM).25 Since neither of them exhibited cytotoxicity up to 50 μM, all the other compounds were tested as a cis racemic mixture in the following study.
Table 1.
SAR Study on A, C and D Rings: EC50s of 1a-v, Compared with 5a
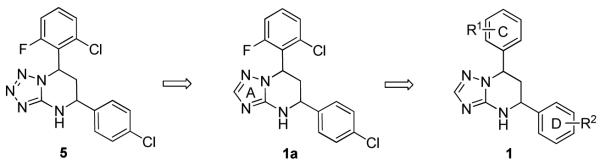 | ||||
|---|---|---|---|---|
| Compounds | C ring | D ring | ELISA, EC50 (μM) | MTT, CC50 (μM) |
| 5 | - | - | 11.3 ± 3.2 | > 50 |
| 1a |
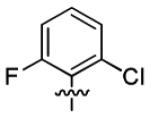
|
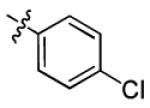
|
4.1 ± 2.1 | > 50 |
| 1b |
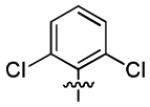
|
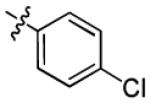
|
4.7 ± 1.9 | > 50 |
| 1c |
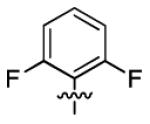
|
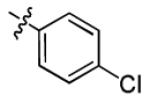
|
2.7 ± 1.0 | > 50 |
| 1d |
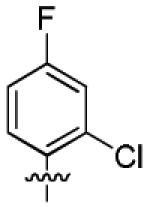
|
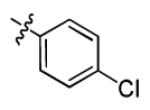
|
2.7 ± 0.7 | > 50 |
| 1e |
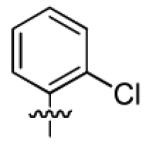
|
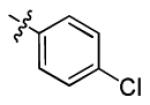
|
3.0 ± 0.7 | > 50 |
| 1f |
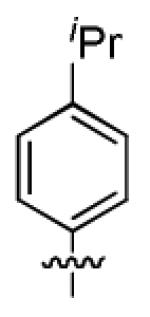
|
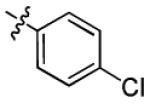
|
8.8 ± 3.2 | > 50 |
| 1g |
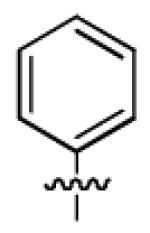
|
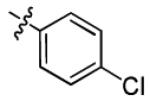
|
34.2 ± 13.6 | > 50 |
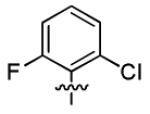
| 1h |
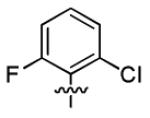
|
|
3.2 ± 1.8 | > 50 |
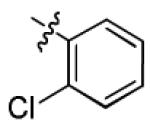
| 1i |
|
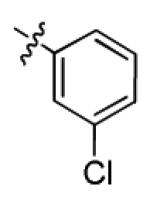
|
12.1 ± 3.6 | > 50 |
| 1j |
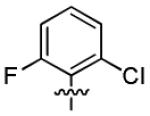
|
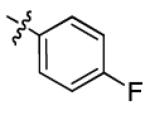
|
5.9 ± 3.3 | > 50 |
| 1k |
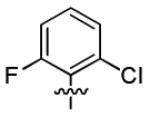
|
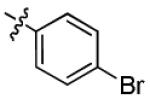
|
2.1 ± 1.0 | > 50 |
| 1l |
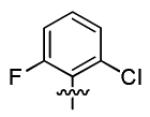
|
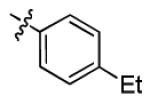
|
7.4 ± 1.7 | > 50 |
| 1m |
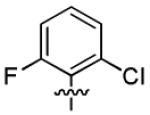
|
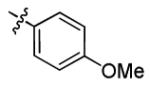
|
46.6 ± 3.2 | > 50 |
| 1n |
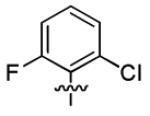
|
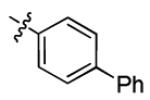
|
3.0 ± 1.3 | > 50 |
| 1o |
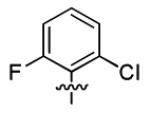
|
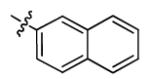
|
2.9 ± 0.7 | > 50 |
| 1p |
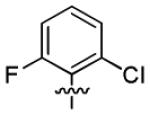
|
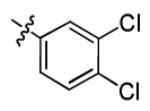
|
5.2 ± 1.9 | > 50 |
| 1q |
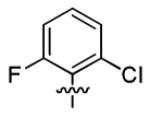
|
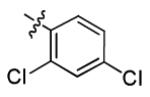
|
5.7 ± 1.9 | > 50 |
| 1r |
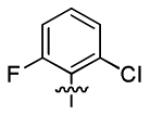
|
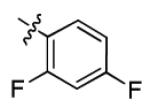
|
11.2 ± 2.2 | > 50 |
| 1s |
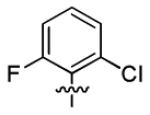
|
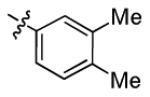
|
2.2 ± 0.5 | > 50 |
| 1t |
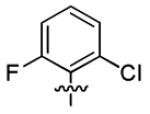
|
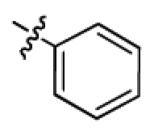
|
40.8 ± 15.9 | > 50 |
| 1u |
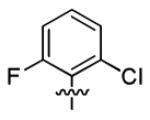
|
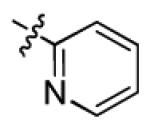
|
> 50 | > 50 |
| 1v |
|
|
> 50 | > 50 |
EC50: 50% effective concentration, measured by the HBsAg ELISA assay; CC50: 50% cytotoxic concentration, measured by the MTT assay; both CC50 and EC50 are the average of at least two independent determinations.
With this encouraging result in hand, we commenced exploration of the structure and activity relationship (SAR) on the side chains (C and D rings), based on triazolo-pyrimidine 1a. Both EC50s and CC50s were investigated in the HepG2.2.15 cell line using the same assay described above. We did not observe cytotoxicity for any of these triazole analogues up to the highest concentration tested (50 μM) (Table 1). The SAR study on the C ring in compounds 1a-1r indicated that chloro/fluoro group at the ortho position (1a-e, compared with 1g) was necessary for the inhibitory activity of HBsAg secretion. The EC50 was improved when the fluoro group was moved from the ortho (1a) to the para (1d) position. Incorporation of an electron donating group (1f) instead of halides on the C ring decreased the potency. Compounds (1a and 1j-k) with halide groups at the para position on the D ring exhibited good EC50s, with 4-bromo group (1k, EC50= 2.1 ± 1.0 μM) being the optimal one. The activity was maintained when the halide group was moved from the para (1a) to the ortho position (1h). However, the activity was decreased, when the 4-halide group was moved to the meta position (1i) or replaced with electron donating groups (1l-m). Di-substitution (1p-r) on the D ring didn’t improve the inhibitory activities, except for the 2,3-dimethyl analogue (1s), which showed an EC50 of 2.2 ± 0.5 μM. Removal of the halide groups on the D ring (1t) significantly decreased the activity. Nevertheless, it is worth noting that biphenyl (1n) and 2-naphthyl (1o) analogues without halide substituents on the side chain exhibited significant potency. The introduction of a nitrogen atom to the D ring as shown in analogues 1u and 1v was associated with a loss of activity. This result could tentatively be attributed to the increased hydrophilicities (ClogP: 1u, 2.37 and 1v, 3.17 vs 1a, 4.58), which may make it difficult for the compounds to pass through the cell membrane; alternatively, it is equally likely that interaction with a target binding site may also be affected.
Next, further structural modification on the core (B ring) of 1a by introducing double bonds led to analogues 3a and 4a. Cell culture studies (Table 2) showed that 3a, the precursor with only one chiral center for the synthesis of 1a (Scheme 3), had a modest, but consistently better EC50 (2.3 ± 2.1 μM) compared with 1a. However, the non-chiral analogue 4a completely lost its activity. To further investigate the scope of this observation, we tested more compounds with one chiral center (3b-d and 3h-s). Compared with their corresponding analogues with two chiral centers, most of them (3b-d, 3i-j, 3l-n and 3p-s) exhibited better EC50 values. In this series, SAR study indicated 2,6-dihalide (3a-3c, compared with 3d) was the optimal substituent on the C ring, and on the D ring para (3a) and meta (3i) chlorination equally improved the activity. Overall, the best compound was the difluoro analogue (3c), which possessed an EC50 of 1.4 ± 0.4 μM, with minimum selectivity index (SI) of 36 (CC50 > 50 μM). This result indicated that the activities of the triazolo-pyrimidine analogues are very sensitive to the conformation of the molecule.
Table 2.
Further SAR Study on B ring: EC50s of 3a-4a Compared with 1a; and EC50s of 3b-d and 3h-sa
 | ||||
|---|---|---|---|---|
| Compounds | C ring | D ring | ELISA, EC50 (μM) | MTT, CC50 (μM) |
| 1a | - | - | 4.1 ± 2.1 | > 50 |
| 3a |
|
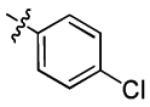
|
2.3 ± 2.1 | > 50 |
| 4a | - | - | > 50 | > 50 |
| 3b |
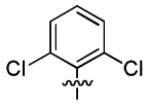
|
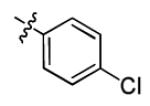
|
1.8 ± 1.2 | > 50 |
| 3c |
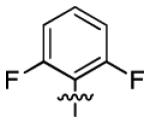
|
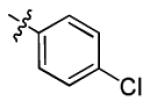
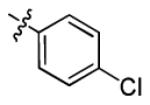
|
1.4 ± 0.4 | > 50 |
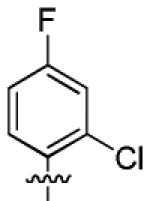
| 3d |
|
|
3.6 ± 2.0 | > 50 |
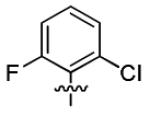
| 3h |
|
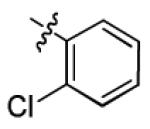
|
7.8 ± 3.2 | > 50 |
| 3i |
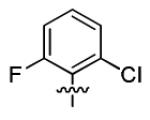
|
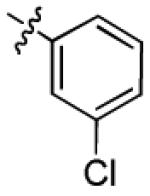
|
2.2 ± 0.9 | > 50 |
| 3j |
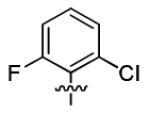
|
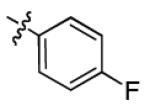
|
2.5 ± 1.2 | > 50 |
| 3k |
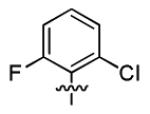
|
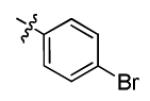
|
3.0 ± 1.6 | > 50 |
| 3l |
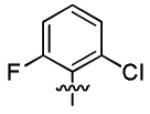
|
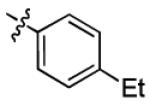
|
3.2 ± 1.2 | > 50 |
| 3m |
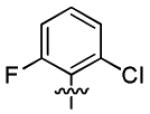
|
|
8.9 ± 3.6 | > 50 |
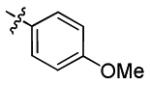
| 3n |
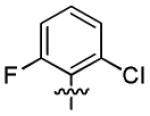
|
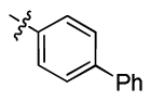
|
2.8 ± 2.1 | > 50 |
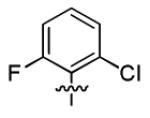
| 3o |
|
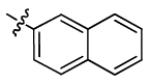
|
3.1 ± 1.4 | > 50 |
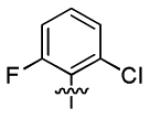
| 3p |
|
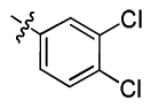
|
2.3 ± 1.3 | > 50 |
| 3q |
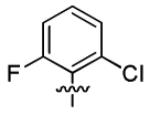
|
|
2.6 ± 0.9 | > 50 |
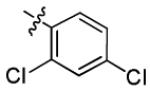
| 3r |
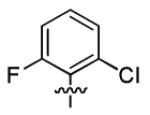
|
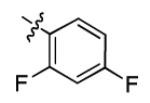
|
2.8 ± 2.0 | > 50 |
| 3s |
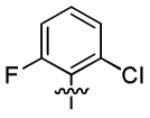
|
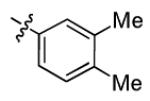
|
1.7 ± 1.1 | > 50 |
EC50: 50% effective concentration, measured by the HBsAg ELISA assay; CC50: 50% cytotoxic concentration, measured by the MTT assay; both CC50 and EC50 are the average of at least two independent determinations.
Assessment of Inhibitory Activity against Drug-resistant HBV Strains
One of the problems associated with the current small molecule antiviral HBV medications is the emergence of drug resistant strains. The rate of resistant infection among the patient population varies with the drugs, ranging as high as 70% after three years of lamivudine/3TC treatment,26 down to 5% after 2 years of entecavir treatment.27, 28 However, lamivudine/3TC treatment predisposes patients to entecavir resistance, such that it occurs at 10% after 2 years,29 and 43% after 4 years.30 This is due to the frequent acquisition of lamivudine/3TC resistance,26 and the subsequent partial cross-resistance to entecavir exhibited by the lamivudine resistant mutants. The newest approved drug, tenofovir, reportedly has had no verifiable resistance, but has been in widespread use for only two years, making firm conclusions on the emergence of resistance premature.
Resistance, which is due to mutations of the the viral polymerase, leads to renewed clinical symptoms. It is thus extremely important that new HBV medications be evaluated for activity against drug resistant HBV strains, as this would be a desirable feature. Although the mechanism of action of 5 and its analogues appears distinct from the HBV polymerase inhibitors in clinical use, because of overlapping open reading frames there are often HBsAg peptide changes that result from the emergence of polymerase drug resistant (DR) mutations,31, 32 for instance, polymerase mutations M552V, M552I, A529V, T532G, and S550I result in the HBsAg alterations I195M, W196S, L173F, L176V, and V194F, respectively. Thus, there is the possibility that the HBsAg of DR mutants may respond differently than the wild type to an inhibitor of HBsAg secretion. Therefore, the ability of the triazolo-pyrimidines to inhibit secretion of HBsAg from wild type and DR-HBV was compared.
To confirm activity against the DR mutants, 5, 1a, and 3c were tested against representatives of the major classes of DR HBV strains, including lamivudine/telbivudine, adefovir, and entecavir resistant mutants. All but two of these mutants carry corresponding changes in HBsAg. For practical reasons, a transient transfection strategy was used rather than establishing stably-transfected cell lines of each virus. The DR point mutations were introduced into a plasmid bearing a 1.3-mer HBV genome, subtype ayw, and the resulting constructs (plus wild type) were transfected into HepG2 cells. Following transfection, the cells were seeded into 96 well plates where they were treated with compounds in concentration from 50 μM to 0.016 μM, in duplicate samples, and colorimetric HBsAg-specific capture ELISA was used to detect levels of secreted surface antigen. Interestingly, the secretion of HBsAg from wild type and DR strains in this transfection assay appears to be less sensitive than in the HepG2.2.15 stable cell line, possibly due to increased levels of HBsAg expression in the transfected cells. However, 5, 1a and 3c all demonstrated significant activity against either wild type or DR mutants, confirming their possible utility in patients who have developed drug resistant strains (Table 3).
Table 3.
Activity of Lead Compounds against HBsAg Secretion from HBV Mutants Bearing Drug Resistant Mutations
| Resistance Phenotype >> | Adefovir | Lamivudine/Telbivudine | Entecavir | |||
|---|---|---|---|---|---|---|
| Genotype >> | Wild Type EC50 (μM) |
A529V EC50 (μM) |
N584T EC50 (μM) |
M552I EC50 (μM) |
L528M/M552V EC50 (μM) |
L528M/M552V/ T532G/S550I EC50 (μM) |
| 5 | 32.0 ± 25.4 | 9.0 ± 2.6 | 5.6 ± 2.2 | 5.5 ± 1.6 | 14.2 ± 5.5 | 6.4 ± 2.5 |
| 1a | 8.4 ± 2.3 | 5.8 ± 1.7 | 5.6 ± 1.8 | 9.1 ± 1.8 | 10.3 ± 8.2 | 8.3 ± 1.3 |
| 3c | 6.9 ± 0.6 | 1.8 ± 0.5 | 6.4 ± 0.4 | 4.0 ± 0.5 | 8.5 ± 1.2 | 8.4 ± 3.7 |
EC50: 50% effective concentration, the average of two independent determinations.
Assessment of Acute and Long Term Toxicity In Vivo
In preparation for in vivo pharmacokinetic and bioavailability studies, and eventual progression to efficacy studies in relevant animal models, we conducted a short term acute toxicity study on 1a and 3c. Both analogues were selected as representatives of two structural classes that were superior to the parent compound 5. CL57BL/6 mice were administered a single intravenous bolus dose of each formulated compound at 25.0 and 7.9 mg/kg of 1a and 3c respectively (maximum possible doses based on solubility limits), or vehicle alone. Over 24 hours, neither compound caused observable morbidity, mortality, or weight loss. Blood cell counts were not affected in a statistically significant manner, although administration of 25 mg/kg of 1a did show a trend towards reduction of platelets and neutrophils, and also an increase in lymphocytes. Serum chemistry was unaffected by either compound (See “Supporting Information”).
To determine longer-term effects of exposure to 1a and 3c, HBV-transgenic mice (strain 1.3.32)33 were used to approximate treatment in a disease-like physiological setting. It should be noted that due to overall low levels of serum HBsAg and highly variable expression, as well as a non-human cellular context, this model is not suitable for efficacy testing. Therefore, future studies will be conducted in a murine model in which chimeric human/mouse livers support HBV infection and high levels of HBsAg secretion in a human cellular context.34 Animals were dosed with the formulated compounds, or vehicle alone, by oral gavage once daily for 14 days. This study was carried after pharmacokinetic profiling and determination of oral bioavailability (described below). Oral dosing was used to simulate typical clinical use. No animals were observed to have died as a result of compound exposure, with some deaths in both vehicle and compound groups attributable to stress from the gavage procedure. No weight loss, morphological or behavioral changes were noted in any treated animals. Treatment with 1a resulted in elevated levels of alanine transaminase (ALT) and bilirubin in a minority of treated animals, indicating some loss of liver function. Both compounds caused a slight rise in total sodium. Treatment did not result in any other obvious and significant dose-dependent serum chemistry changes, with 3c being particularly inert (See “Supporting Information”).
Assessment of Pharmacokinetics In Vivo
After assessment of acute (24 hours) toxicity in mice, the oral bioavailability and pharmacokinetics of 1a and 3c were evaluated in male Sprague-Dawley rats. Each test compound was administered intravenously at 5 mg/kg and orally at 25 mg/kg in fasted rats. Plasma concentrations of each test compound were determined by LC-MS/MS. Following intravenous dosing at 5 mg/kg, 1a had an average plasma half life (t1/2) of 2.17 ± 0.74 hours. Its total body clearance rate (CL) was low, with an average value of 0.70 ± 0.19 L/hr/kg, and its average volume of distribution (Vss) was 1.6 ± 0.3 L/kg. After oral dosing at 25 mg/kg, 1a reached an average maximum plasma concentration (Cmax) of 2437 ± 218 ng/mL, within an average time (Tmax) of 2.3 hours. The average bioavailability (F) was calculated at 31 ± 3% (See “Supporting Information”).
Following intravenous dosing at 5 mg/kg, 3c had an average plasma half life (t1/2) of 2.40 ± 0.38 hours. Its total body clearance rate (CL) was moderate with an average value of 1.0 ± 0.4 L/hr/kg, and its average volume of distribution (Vss) was 2.6 ± 0.4 L/kg. After oral dosing at 25 mg/kg, 3c reached an average maximum plasma concentration (Cmax) of 1937 ± 474 ng/mL, within an average time (Tmax) of 4.0 hours. The average bioavailability (F) was calculated at 42 ± 16% (See “Supporting Information”). For both 1a and 3c, PK characteristics are favorable and suggest that an effective dosing regimen can be developed. In addition, oral bioavailability is likely underestimated due to the fact that a significant amount of the test compounds remained in the systemic circulation at the final collection time point (6 hours) after oral dosing. Future studies will include later time points and/or a lower dosing concentration to allow for the determination of the elimination phase of the test compounds and a better estimation of the bioavailability.
Conclusion
The results of a cell-based phenotypic screen have led to a series of disubstituted triazolo-pyrimidines as novel and specific inhibitors of hepatitis B virus surface antigen (HBsAg) secretion. Although the exact mechanism of action is still under investigation and will be the focus of a future publication, the parent compound was shown to not be an inhibitor of viral genomic replication, but rather a specific inhibitor of HBV envelope secretion. As such, the mechanism is distinct from the current hepatitis B small molecule drugs. In this work, we presented the results of extensive SAR studies, which successfully improved the potency and chemical tractability of the parent compound. These triazolo-pyrimidine derivatives were also active in inhibiting HBsAg secretion of HBV variants that are resistant to current small molecule drugs. Selected lead analogues, both 1a (PBHBV-001) and 3c (PBHBV-2-15), were well tolerated in CL57BL/6 mice and displayed desirable pharmacokinetics profiles in male Sprague-Dawley rats. After 14 days of treatment of HBV transgenic mice, both 1a and 3c were fairly well tolerated, with 3c showing no signs of toxicity through serum chemistry analysis.
Experimental Section
Chemistry
1H NMR spectra were recorded on 500 MHz or 300 MHz INOVA VARIAN (75 MHz for 13C NMR; 282 MHz for 19F NMR) spectrometer. Chemical shifts values are given in ppm and referred as the internal standard to TMS (tetramethylsilane). The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet and dd, doublet of doublets. The coupling constants (J) are reported in Hertz (Hz). Melting points were determined with a national micromelting point apparatus without corrections. Mass Spectra were obtained on an Aligent LC-MS spectrometer (ES-API, Positive). Silica gel column chromatography was performed over silica gel 100-200 mesh, and the eluent was a mixture of ethyl acetate (EtOAc) and Hexanes. All the tested compounds possess a purity of at least 95% as determined by HPLC. Analytical HPLC was run on the Agilent 1100 HPLC instrument, equipped with Phenomenex® C12 column. Eluent system was: A (MeCN, 0.05%TFA) and C (H2O, 0.05%TFA); flow rate = 1 mL/min; Method A: 60%A, 40%C, λ = 219 nm; Method B: 70%A, 30%C, λ = 254 nm; Method C: 80%A, 20%C, λ = 254 nm. Retention times (tR) are given in minutes. Chiral resolution of compound 1a was carried out on the Waters 2695 HPLC instrument, equipped with CHIRALPAK® AD-RH column. Eluent system was: 30%A (H2O), 70%B (EtOH); flow rate = 0.5 mL/min; λ = 219 nm. (See “Supporting Information” for full experimental details and characterization data.)
a. Preparation of 1,3-Diaryl-propenone 2. General Procedure A
To a stirred solution of substituted benzaldehyde (20 mmol) and acetophenone (20 mmol) in methanol (15 mL), was added dropwise a solution of sodium hydroxide (26 mmol) in methanol (15 mL). The resulting mixture was stirred at room temperature for 6 h, then filtered and washed with water to yield the crude product. Pure compound 2 was got by recrystallization from methanol or through silica gel column chromatography (EtOAc/Hexanes 2:98) as a light yellow solid in 38-97% yield.
b. Preparation of 5,7-Diaryl-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine 3. General Procedure B
A solution of 2 (5 mmol) and 3-amino-1,2,4-triazole (7.5 mmol) in DMF (5 mL) was refluxed for 2 h. The reaction mixture was cooled to room temperature, diluted with water (50 mL) and stirred sufficiently. The resulting mixture was filtered and washed with water to give the crude product, which was further purified either by recrystallization from EtOAc or through silica gel column chromatography (EtOAc/Hexanes 30:70) to afford 3 as a white solid in 41-92% yield.
5-(4-Chlorophenyl)-7-(2,6-difluorophenyl)-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine (3c)
The product was obtained according to general procedure B, as a white solid. Yield: 83%. m.p. 223-224 °C. Rf = 0.40 (EtOAc/Hexanes 50:50). MS: MH+ = 345. 1H NMR (300 MHz, DMSO-d6): δ 10.13 (s, 1H, NH), 7.66-7.61 (m, 3H, CHar), 7.49-7.41 (m, 3H, CHar), 7.13-7.07 (m, 2H, CHar), 6.63 (d, J = 3.6 Hz, 1H, CH=C), 5.26-5.25 (m, 1H, CH); 19F NMR (282 MHz, DMSO-d6): δ −118.26. 13C NMR (75 MHz, DMSO-d6): δ 160.6 (dd, JC-F = 247.9, 7.7 Hz, C), 150.5 (C), 150.4 (C), 135.9 (C), 134.3 (C), 133.5 (CH), 131.4 (t, JC-F = 10.7 Hz, C), 129.2 (CH), 128.4 (CH), 117.2 (t, JC-F = 15.2 Hz, C), 112.8 (d, JC-F = 24.9 Hz, CH), 95.1 (CH), 50.8 (CH). HPLC: 98.8% (Method B, tR = 4.96 min).
c. Synthesis of 5,7-Diaryl-4,5,6,7-tetrahydro-[1,2,4]triazolo[1,5-a]pyrimidine 1. General Procedure C
A sample of sodium borohydride (10 mmol) was added to a suspension of 3 (1 mmol) in methanol (5 mL). The reaction mixture was refluxed for 30 min, then cooled to room temperature, diluted with water (50 mL) and stirred sufficiently. The resulting mixture was filtered and washed with water to give the crude product, which was further purified by recrystallization from a mixture of EtOAc and Hexanes to afford 1 as a white solid in 64-94% yield.
7-(2-Chloro-6-fluorophenyl)-5-(4-chlorophenyl)-4,5,6,7-tetrahydro-[1,2,4]triazolo[1,5-a]pyrimidine (1a)
The product was obtained according to general procedure C, as a white solid. Yield: 89%. m.p. 227-228 °C. Rf = 0.16 (EtOAc/Hexanes 33.3:66.7). MS: MH+ = 363. 1H NMR (500 MHz, DMSO-d6): δ 7.54-7.15 (m, 8H, CHar), 5.98-5.94 (m, 1H, CH), 4.82-4.78 (m, 1H, CH), 2.47-2.13 (m, 2H, CH2). HPLC: 99.8% (Method A, tR= 3.67 min).
d. Synthesis of 7-(2-Chloro-6-fluorophenyl)-5-(4-chlorophenyl)-[1,2,4]triazolo [1, 5-a]pyrimidine 4a
A sample of NBS (299 mg, 1.68 mmmol) was added to a solution of 3a (101 mg, 0.28 mmol) in isopropanol (5 mL). The reaction mixture was refluxed for 36 h, then cooled to room temperature, treated with saturated NaHCO3 solution (20 mL) and extracted with EtOAc (15 mL × 3). The combined organic layer was dried over Na2SO4 and evaporated under reduced pressure. The given residue was purified through silica gel column chromatography (EtOAc/Hexanes 30:70) to afford a 42 mg white solid of 4a in 42% yield. m.p. 238-239 °C. Rf = 0.33 (EtOAc/Hexanes 30:70). MS: MH+ = 359. 1H NMR (500 MHz, CDCl3): δ 8.48 (s, 1H, CHar), 8.15 (d, J = 7.5 Hz, 2H, CHar), 7.57-7.39 (m, 5H, CHar), 7.20-7.19 (m, 1H, CHar). HPLC: 97.9 % (Method C, tR = 5.47 min).
Generation and Testing of Drug-resistant HBV Variants
The plasmid pTREHBV, encoding the HBV genome of ayw serotype,35 was used as the background wild type construct. Standard molecular biology procedures employing the GeneTailor site-directed mutagenesis system (Invitrogen, Carlsbad CA) were used to generate single or double point mutations that give rise to the following amino acid changes in the polymerase open reading frame: A529V, N584T, M552I, M552V, L528M/M552V, and L528M/M552V/T532G/S550I.31, 32 To generate the double and quadruple mutants, nucleotide substitutions were introduced sequentially by generating single mutations, confirming the sequence, and introducing additional mutations into the intermediate construct. All constructs were confirmed by sequencing through Integrated DNA technologies (Coralville IA). Primer sequences used for mutagenesis are available upon request.
Transient expression of HBsAg from the mutant and wildtype HBV constructs was accomplished through transient transfection of plasmids into HepG2 cells seeded on 75 cm2 flasks, using Lipofectamine 2000 reagent (Invitrogen) according to directions. 24 hours following transfection, cells were harvested by trypsinization and reseeded into 96-well plates at a density of 5 × 105 cells/well. 24 hours later media in each well was replaced with media containing test compounds in concentrations ranging (in half-log steps) from 0.016 to 50.0 × 10−6 M, in 0.5% DMSO. Control wells contained DMSO alone. Each compound concentration was tested in duplicate. HBsAg levels was determined by ELISA Assay,18 and EC50 values were calculated as described therein. For each mutant HBV genome, compounds were tested at least 5 and up to 10 times, allowing calculation of standard deviation.
Formulation for Intravenous Administration
To produce 2 mg/mL solutions for dosing, 2 mg of compound powders were initially dissolved in a 0.2 ml 1:1 solution (volume : weight) of 1-Methyl-2-pyrrolidinone (NMP, Sigma Aldrich) : Solutol HS15 (BASF) with vortexing. After dissolution, 0.8 ml of sterile saline (0.95% NaCl in H2O) was added slowly while vortexing. Solution was injected into test animals within 10 minutes of preparation.
Assessment of Toxicity In Vivo
The work was done at Utah State University. For the acute toxicity study, female C57BL/6 mice were randomized to the treatment groups, with four animals per group. 1a and 3c were prepared as detailed above for intravenous formulation, and administered by tail vein injection at 25 and 7.9 mg/kg, with one group injected only with vehicle. Animals were observed closely for the initial hour following administration. After 24 hours, animals were sacrificed and necropsied to examine for gross changes of internal organs, and whole blood and serum were collected for analysis of complete blood cell counts (CBCs, specifically red blood cells, white blood cells, platelets, lymphocytes, neutrophils, monocytes, percent eosinophils, percent basophils, hemoglobin, hematoctrit, mean corpuscular volume, mean platelet volume, red cell distribution width, and mean corpuscular hemoglobin concentration, using a Hemavet 950FS, Drew Scientific, Anaheim, CA) and serum chemistry (alanine aminotransferase, blood urea nitrogen, creatinine, total bilirubin, albumin, alkaline phosphatase amylase, globulin, total protein, glucose, Ca++, Na+, K+, and phosphorous through a VetScan® Chemistry, Electrolyte and T4 Analyzer, Abaxis, Union City, CA).
For the 14-day toxicity study, HBV-transgenic mice (strain 1.3.32)33 were randomized across groups with 10 mice per group. 1a and 3c were prepared as detailed above for intravenous formulation, and administered by oral gavage at 25 and 7.9 mg/kg once daily for 14 days, with one group injected only with vehicle. Animals were weighed prior to first administration and after 3 days. Blood was collected from tail vein three hours prior to first administration, at 7 days (three hours after administration), and after necropsis (three hours after last administration) at 14 days. Animals were observed closely for one hour for signs of distress after each administration. Serum chemistry (alanine aminotransferase, blood urea nitrogen, creatinine, total bilirubin, albumin, alkaline phosphatase amylase, globulin, total protein, glucose, Ca++, Na+, K+, and phosphorous through a VetScan® Chemistry, Electrolyte and T4 Analyzer, Abaxis, Union City, CA).
Assessment of Pharmacokinetics and Bioavailability
This analysis was conducted under contract with Absorption Systems (Exton PA). Male Sprague-Dawley rats were randomized as to treatment group, with 3 animals per treatment (12 total). Two days prior to dosing, animals were surgically fitted with jugular vein cannulae for withdrawal of blood samples; for intravenous administration, a second cannula was fitted. At 12 hours prior to dosing, animals were fasted, but supplied with water. 1a and 3c were prepared as detailed above. For intravenous administration, animals were dosed at 5 mg/kg; for oral adminstration, animals were dosed at 25 mg/kg by gavage. Animals were observed for signs of distress throughout the procedure approximately 0.3 ml of whole blood were harvested from the cannulae at predosing time point, 5 minutes, 15 minutes, 30 minutes, 1 hour, 3 hours and 6 hours. Plasma was prepared, and quantitatively analyzed for test compounds by LC/MS after 8-point standard curve was established. Half-life, Cmax, Tmax, mean residence time, and bioavailability were calculated by standard methods.
Supplementary Material
Acknowledgement
This project was supported by the Commonwealth of Pennsylvania, the Hepatitis B Foundation, NIH Grant 1R43AI077123-01A1 and NIH Contract N01-AI50036.
Abbreviations
- HBV
hepatitis B virus
- HBsAg
hepatitis B virus surface antigen
- SAR
structure and activity relationship
- EC50
50% effective concentration
- CC50
50% cytotoxic concentration
- SI
selectivity index
- IFN
interferon
- FDA
food and drug administration
- WHV
woodchuck hepatitis virus
- ELISA
enzyme-linked immunosorbent assay
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PK
pharmacokinetics
- DR
drug resistant
Footnotes
Supporting Information Available: Characterization data and NMR spectra of compounds 1, 3 and 4; X-ray data of 1a; in vivo data of 1a and 3c. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Lee WM. Hepatitis B virus infection. N. Engl. J. Med. 1997;337:1733–1745. doi: 10.1056/NEJM199712113372406. [DOI] [PubMed] [Google Scholar]
- (2).Hoofnagle JH. Therapy of viral hepatitis. Digestion. 1998;59:563–578. doi: 10.1159/000007532. [DOI] [PubMed] [Google Scholar]
- (3).Robinson WS, Klote L, Aoki N. Hepadnaviruses in cirrhotic liver and hepatocellular carcinoma. J. Med. Virol. 1990;31:18–32. doi: 10.1002/jmv.1890310106. [DOI] [PubMed] [Google Scholar]
- (4).Beasley RP. Hepatitis B virus. The major etiology of hepatocellular carcinoma. Cancer. 1988;61:1942–1956. doi: 10.1002/1097-0142(19880515)61:10<1942::aid-cncr2820611003>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- (5).Hollinger FB, The North American Regional Study Group Controlling hepatitis B virus transmission in North America. Vaccine. 1990;(Suppl):S122–128. doi: 10.1016/0264-410x(90)90232-b. discussion: S134-138. [DOI] [PubMed] [Google Scholar]
- (6).Marcellin P, Lau GK, Bonino F, Farci P, Hadziyannis S, Jin R, Lu ZM, Piratvisuth T, Germanidis G, Yurdaydin C, Diago M, Gurel S, Lai MY, Button P, Pluck N. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 2004;351:1206–1217. doi: 10.1056/NEJMoa040431. [DOI] [PubMed] [Google Scholar]
- (7).Hoofnagle JH, di Bisceglie AM. The treatment of chronic viral hepatitis. N. Engl. J. Med. 1997;336:347–356. doi: 10.1056/NEJM199701303360507. [DOI] [PubMed] [Google Scholar]
- (8).Mailliard ME, Gollan JL. Emerging therapeutics for chronic hepatitis B. Annu. Rev. Med. 2006;57:155–166. doi: 10.1146/annurev.med.57.121304.131422. [DOI] [PubMed] [Google Scholar]
- (9).Liaw YF, Chu CM. Hepatitis B virus infection. Lancet. 2009;373:582–592. doi: 10.1016/S0140-6736(09)60207-5. [DOI] [PubMed] [Google Scholar]
- (10).Deres K, Schröder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Krämer T, Niewöhner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RNA, Reimann A, Jaeger R, Groβ R, Beckermann B, Schlemmer K, Haebich D, Rübsamen-Waigmann H. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003;299:893–896. doi: 10.1126/science.1077215. [DOI] [PubMed] [Google Scholar]
- (11).Heermann KH, Goldmann U, Schwartz W, Seyffarth T, Baumgarten H, Gerlich WH. Large surface proteins of hepatitis B virus containing the pre-s sequence. J. Virol. 1984;52:396–402. doi: 10.1128/jvi.52.2.396-402.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hollinger FB, Liang JT. Fields Virology. 4th ed. Lippincott Williams & Wilkins; Philadelphia: 2001. Hepatitis B virus; pp. 2971–3036. [Google Scholar]
- (13).Op den Brouw ML, Binda RS, Geijtenbeek TB, Janssen HL, Woltman AM. The mannose receptor acts as hepatitis B virus surface antigen receptor mediating interaction with intrahepatic dendritic cells. Virology. 2009;393:84–90. doi: 10.1016/j.virol.2009.07.015. [DOI] [PubMed] [Google Scholar]
- (14).Xu Y, Hu Y, Shi B, Zhang X, Wang J, Zhang Z, Shen F, Zhang Q, Sun S, Yuan Z. HBsAg inhibits TLR9-mediated activation and IFN-alpha production in plasmacytoid dendritic cells. Mol. Immunol. 2009;46:2640–2646. doi: 10.1016/j.molimm.2009.04.031. [DOI] [PubMed] [Google Scholar]
- (15).Menne S, Roneker CA, Korba BE, Gerin JL, Tennant BC, Cote PJ. Immunization with surface antigen vaccine alone and after treatment with 1-(2-fluoro-5-methyl-beta-L-arabinofuranosyl)-uracil (L-FMAU) breaks humoral and cell-mediated immune tolerance in chronic woodchuck hepatitis virus infection. J. Virol. 2002;76:5305–5314. doi: 10.1128/JVI.76.11.5305-5314.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Korba BE, Montero AB, Farrar K, Gaye K, Mukerjee S, Ayers MS, Rossignol JF. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Res. 2008;77:56–63. doi: 10.1016/j.antiviral.2007.08.005. [DOI] [PubMed] [Google Scholar]
- (17).Mahtab MA, Bazinet M, Vaillant A. REP 9AC: A potent HBsAg release inhibitor that can rapidly restore immunocompetence in patients with chronic hepatitis B. The Liver Meeting® (AASLD) 2010 #482. [Google Scholar]
- (18).Dougherty AM, Guo H, Westby G, Liu Y, Simsek E, Guo J, Mehta A, Norton PG,B, Block T, Cuconati A. A substituted tetrahydro-tetrazolo-pyrimidine is a specific and novel inhibitor of hepatitis B virus surface antigen secretion. Antimicrob. Agents Chemother. 2007;51:4427–4437. doi: 10.1128/AAC.00541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Desenko SM, Gladkov ES, Komykhov SA, Shishkin OV, Orlov VD. Partially hydrogenated aromatic substituted tetrazolo[1,5-α]pyrimidines. Chem. Heterocycl. Compd. 2001;37:747–754. [Google Scholar]
- (20).Zhao P, Liu C, Huang W, Wang Y, Yang G. Synthesis and fungicidal evaluation of novel chalcone-based strobilurins analogues. J. Agric. Food Chem. 2007;55:5697–5700. doi: 10.1021/jf071064x. [DOI] [PubMed] [Google Scholar]
- (21).Orlov VD, Desenko SM, Potekhin KA, Struchkov YT. Cyclocondensation of α,β-unsaturated ketones with 3-amino-1,2,4-triazole. Chem. Heterocycl. Compd. 1988;24:192–196. [Google Scholar]
- (22).Desenko SM, Orlov VD, Lipson VV, Shishkin OV, Lindeman SV, Struchkov YT. Imine-enamine tautomerism of dihydroazolopyrimidines. 3. 5-aryl-substituted 4,7(6,7)-dihydro-1,2,4-triazolo[1,5-α]pyrimidines. Chem. Heterocycl. Compd. 1991;27:1242–1246. [Google Scholar]
- (23).Desenko SM, Orlov VD, Lipson VV. Chemical conversions of 5,7-disubstituted dihydro-1,2,4-triazolo[1,5-a]pyrimidines. Chem. Heterocycl. Compd. 1990;26:1362–1366. [Google Scholar]
- (24).Desenko SM, Shishkin OV, Orlov VD, Lipson VV, Lindeman SV, Struchkov YT. Synthesis and structure of 4,5,6,7-tetrahydro-1,2,4-triazolo[1,5-α]pyrimidines. Chem. Heterocycl. Compd. 1994;30:851–855. [Google Scholar]
- (25).The absolute stereochemistry of enantiomer 1 and 2 of compound 1a has not been established.
- (26).Tipples GA, Ma MM, Fischer KP, Bain VG, Kneteman NM, Tyrrell DL. Mutation in HBV RNA-dependent DNA polymerase confers resistance to lamivudine in vivo. Hepatology. 1996;24:714–717. doi: 10.1002/hep.510240340. [DOI] [PubMed] [Google Scholar]
- (27).Colonno RJ, Rose R, Baldick CJ, Levine S, Pokornowski K, Yu CF, Walsh A, Fang J, Hsu M, Mazzucco C, Eggers B, Zhang S, Plym M, Klesczewski K, Tenney DJ. Entecavir resistance is rare in nucleoside naïve patients with hepatitis B. Hepatology. 2006;44:1656–1665. doi: 10.1002/hep.21422. [DOI] [PubMed] [Google Scholar]
- (28).Tenney DJ, Levine SM, Rose RE, Walsh AW, Weinheimer SP, Discotto L, Plym M, Pokornowski K, Yu CF, Angus P, Ayres A, Bartholomeusz A, Sievert W, Thompson G, Warner N, Locarnini S, Colonno RJ. Clinical emergence of entecavir-resistant hepatitis B virus requires additional substitutions in virus already resistant to Lamivudine. Antimicrob. Agents Chemother. 2004;48:3498–3507. doi: 10.1128/AAC.48.9.3498-3507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zoulim F. Entecavir: a new treatment option for chronic hepatitis B. J. Clin. Virol. 2006;36:8–12. doi: 10.1016/j.jcv.2006.01.010. [DOI] [PubMed] [Google Scholar]
- (30).Papatheodoridis GV, Manolakopoulos S, Dusheiko G, Archimandritis AJ. Therapeutic strategies in the management of patients with chronic hepatitis B virus infection. Lancet Infect. Dis. 2008;8:167–178. doi: 10.1016/S1473-3099(07)70264-5. [DOI] [PubMed] [Google Scholar]
- (31).Seeger C, Mason WS. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yotsuyanagi H, Koike K. Drug resistance in antiviral treatment for infections with hepatitis B and C viruses. J. Gastroenterol. 2007;42:329–335. doi: 10.1007/s00535-007-2034-z. [DOI] [PubMed] [Google Scholar]
- (33).Guidotti LG, Matzke B, Schaller H, Chisari FV. High-level hepatitis B virus replication in transgenic mice. J. Virol. 1995;69:6158–6169. doi: 10.1128/jvi.69.10.6158-6169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Utoh R, Tateno C, Yamasaki C, Hiraga N, Kataoka M, Shimada TC,K, Yoshizato K. Susceptibility of chimeric mice with livers repopulated by serially subcultured human hepatocytes to hepatitis B virus. Hepatology. 2008;47:435–446. doi: 10.1002/hep.22057. [DOI] [PubMed] [Google Scholar]
- (35).Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J. Virol. 2007;81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.