

Abstract
Cephalostatin 1, OSW-1, ritterazine B and schweinfurthin A are natural products that potently, and in some cases selectively, inhibit the growth of cultured human cancer cell lines. The cellular targets of these small molecules have yet to be identified. We have discovered that these molecules target oxysterol binding protein (OSBP) and its closest paralog, OSBP-related protein 4L (ORP4L)—proteins not known to be involved in cancer cell survival. OSBP and the ORPs constitute an evolutionarily conserved protein superfamily, members of which have been implicated in signal transduction, lipid transport and lipid metabolism. The functions of OSBP and the ORPs, however, remain largely enigmatic. Based on our findings, we have named the aforementioned natural products ORPphilins. Here we used ORPphilins to reveal new cellular activities of OSBP. The ORPphilins are powerful probes of OSBP and ORP4L that will be useful in uncovering their cellular functions and their roles in human diseases.
Cephalostatin 1 (1)1, OSW-1 (2)2, ritterazine B (3)3, schweinfurthin A (4)4, schweinfurthin B (5)4 and stellettin E5 (Fig. 1) are structurally diverse, naturally occurring small molecules that inhibit the growth of human cancer cell lines with half-maximal inhibitory concentrations (GI50) in the nanomolar range5–8. All five compounds induce a similar pattern of sensitivity against the National Cancer Institute 60 cancer cell lines (NCI-60). The NCI-DTP COMPARE algorithm can be used to assess the similarity of compound sensitivity to the NCI-60, expressed as Pearson correlation coefficients (r). Compounds with high COMPARE correlations (r > 0.6) tend to have related mechanisms9. COMPARE analyses between cephalostatin 1 and OSW-1, ritterazine B and schweinfurthin A reveal r values of 0.60–0.83 (ref. 10), 0.93 (ref. 3) and 0.59 (ref. 4), respectively. The stellettin family of natural products is linked to cephalostatin 1, OSW-1, ritterazine B and schweinfurthin A through a pattern of cytotoxicity shared with schweinfurthin A (r = 0.75 for schweinfurthin A and stellettin A4). The highly correlative cancer cell line sensitivities of these four compounds and stellettin E suggest that all five molecules share a cellular target or affect the same cellular pathway(s)9. Furthermore, the shared cell line sensitivity pattern of these compounds against the NCI-60 is unique compared to the pattern of the other ~40,000 growth-inhibitory small molecules evaluated, indicating that the five compounds have new cellular target(s)11. However, the cellular target(s) of these compounds has not been identified.
Figure 1. Chemical structures.

Natural products cephalostatin 1 (1), OSW-1 (2), ritterazine B (3), schweinfurthin A (4), schweinfurthin B (5) and stellettin E. OSW-1 analogs 6–10 and the OSW-1 affinity reagent (OAR). PMbz, para-methoxy benzoyl.
The five compounds each have interesting cellular activities. Cephalostatin 1 induces apoptosis through an atypical mechanism12,13. OSW-1 is 30- to 150-fold more cytotoxic toward glioblastoma and leukemia cells compared to nontransformed astrocytes and lymphocytes, respectively8. Schweinfurthin A and stellettin E selectively inhibit growth of tumor cells deficient in NF-1 (ref. 14) or p21Cip/Waf1 (also known as CDKN1A, and hereafter referred to as p21)5, respectively. Repression of p21 expression15 and loss of NF-1 (ref. 14) is important for the development of some tumors. Therefore, small molecules that selectively inhibit the growth of tumor cells with these genetic alterations could be tumor-specific therapeutics.
Herein we reveal that OSBP and ORP4L are targets of cephalostatin 1, OSW-1, ritterazine B and schweinfurthin A. OSBP and ORP4L (also known as OSBP2 or HLM) belong to a 16-member protein superfamily16,17, OSBP and the ORPs, that has been implicated in lipid metabolism18,19, signaling20,21, vesicular traffic22 and nonvesicular sterol transport23,24. Although OSBP was the first protein discovered to bind endogenous oxysterols25,26, its function and that of its ORP paralogs remains largely unclear. Because these four natural products all target OSBP and ORP4L, we have named this family of compounds ORPphilins (ORP here stands for `OSBP and related proteins'). By uncovering the cellular targets of the ORPphilins, we have revealed that OSBP and ORP4L are involved in cancer cell survival. This discovery provides another link between cancer cell proliferation and lipid processing27.
RESULTS
OSBP and ORP4L are receptors of ORPphilins
We accessed cephalostatin 1 using our total synthesis procedure28, and we used OSW-1, ritterazine B and schweinfurthins A and B isolated from natural sources. We did not have access to stellettin E, and our small supply of ritterazine B limited its use in our experiments. As similar NCI-60 cytotoxicity profiles are only suggestive of a shared mechanism of action between compounds, we sought additional experimental support that the ORPphilins and stellettin E form a new class of mechanistically related anti-proliferative agents. Stellettin E is 117 times more growth-inhibitory to HCT-116 CDKN1A−/− (p21−/−) cells than to HCT-116 wild-type cells5. We determined that the growth-inhibitory activities of the ORPphilins were also significantly enhanced in HCT-116 p21−/− cells versus HCT-116 wild-type cells (Fig. 2a). Many of the other antiproliferative small molecules we tested did not show this p21−/− selectivity (Fig. 2a). The p21−/− selectivity was the first evidence beyond the NCI-60 COMPARE analysis that cephalostatin 1, OSW-1, ritterazine B, schweinfurthin A and stellettin E were mechanistically related. In addition, the p21−/− selectivity was a defining feature for this class of compounds, which we used to identify ORPphilin analogs that shared the same mechanism of action.
Figure 2. OSBP and ORP4L are high-affinity receptors of ORPphilins.

(a) Cephalostatin 1 (1), OSW-1 (2), ritterazine B (3), schweinfurthin A (4), stellettin E and OSW-1 analogs 6–10 are selectively growth-inhibitory toward HCT-116 cells lacking p21. GI50 values of the compounds were determined in HCT-116 p21+/+ and p21−/− cells after 48 h using a resazurin redox dye to measure cell viability. Values are mean ± s.d. of three individual experiments. Asterisk denotes value from ref. 5. (b) Affinity chromatography experiment with HeLa S-100 lysate using OSW-1 affinity reagent (OAR) in the absence (left lane) and presence (right lane) of excess OSW-1 (10 μM). The intense band indicated with a red arrow was excised from the gel and identified as oxysterol binding protein (OSBP). (c) Scatter plot depicting 121 proteins identified and quantified by itRAQ-based quantitative proteomics. Competition is plotted on the y axis as log10 fold change for 10 μM OSW-1 over DMSO control, and the P value representing statistical significance is plotted on the x axis. P values are arbitrarily set to 1 for nonsignificant single peptide quantitations. (d,e) Representative competition-binding experiments of [3H]25-OHC (20 nM) with ORPphilins in S100 lysate made from HEK-293t cells overexpressing either OSBP-Myc-His (d) or ORP4L-Myc-His (e). Ki values are mean ± s.d. from at least three independent experiments. An accurate Ki value for ritterazine B with ORP4L could not be determined because of insufficient quantities of compound. 25-OHC, 25-hydroxycholesterol; BFA, brefeldin A; 5-FU, 5-fluorouracil.
We used affinity purification to identify putative targets of the ORPphilins. Because OSW-1 is the most abundant of the natural products, we prepared several OSW-1 analogs (6–10, Fig. 1), and we subsequently determined their potencies and p21−/− selectivities. We discovered that an allyloxycarbonyl (alloc)-protected amine (6), prepared through carbamate formation at the C3-hydroxyl of the xylosyl saccharide, maintained the antiproliferative potency of OSW-1 (Supplementary Results, Supplementary Fig. 1a) and, critically, also showed p21−/− selectivity commensurate with that of OSW-1 (Fig. 2a).
Amine 6 was then deprotected and covalently linked to Sepharose resin to prepare OSW-1 affinity reagent (OAR) (Fig. 1). Affinity chromatography of HeLa-S3 cell lysate with OAR followed by protein gel electrophoresis afforded a highly enriched band at ~90 kDa (Fig. 2b). Critically, pretreatment of the HeLa-S3 lysate with 10 μM of OSW-1 competed away this band (Fig. 2b), indicating that this protein specifically bound OSW-1. Analysis of the full gel lane by iTRAQ-based quantitative mass spectrometry identified this band as OSBP. Among the 121 identified proteins enriched in the OAR pulldown, OSBP and its paralog ORP4L showed the largest extent of competition by soluble OSW-1 (Fig. 2c). Expression from two alternative transcription start sites produces two ORP4 isoforms: a long version (ORP4L) of 916 amino acids and a short version (ORP4S) of 461 amino acids. The peptide coverage in our iTRAQ experiment contains ORP4L-specific sequences, but we cannot exclude the possibility that ORP4S or another as-yet-unidentified ORP4 isoform was also pulled down. Additionally, vesicle-associated membrane protein–associated protein A (VAP-A), which is known to interact with both OSBP29 and ORP4L30, was significantly enriched and competed away by OSW-1 pretreatment. OSBP and ORP4L are believed to physically interact31. Therefore, any of these three proteins—OSBP, ORP4L or VAP-A—could have been the direct binding partner of the OAR, with the other two proteins being copurified. However, as OSBP and ORP4L are high-affinity receptors of oxysterols31, and OSW-1 comprises an oxysterol moiety, we chose to investigate OSBP and ORP4L as putative targets of the ORPphilins.
To determine whether the ORPphilins directly bind OSBP or ORP4L, we performed competition-binding assays with OSBP-Myc-His and ORP4L-Myc-His using [3H]25-hydroxycholesterol (25-OHC, Supplementary Fig. 2), a high-affinity ligand of both proteins (OSBP KD = 32 ± 14 nM; ORP4L KD = 54 ± 23 nM, Supplementary Fig. 1b). We found that cephalostatin 1, OSW-1, ritterazine B and schweinfurthin A each bound OSBP-Myc-His (Ki = 39 ± 10 nM, 26 ± 9 nM, 28 ± 4 nM and 68 ± 23 nM, respectively) and ORP4L-Myc-His (Ki = 78 ± 15 nM, 54 ± 11 nM, >360 nM and 2,600 ± 570 nM, respectively) (Fig. 2d,e). Schweinfurthin A was an outlier compared to the other ORPphilins in that its affinity for ORP4L was considerably lower (by a factor of ~40) than its affinity for OSBP.
OSBP and ORP4L levels affect ORPphilin activity
We performed shRNA knockdown of OSBP expression and then measured its effect on the growth-inhibitory activity of cephalostatin 1, OSW-1 and schweinfurthin A. Reduction of OSBP levels by 85% (Fig. 3a) did not affect HCT-116 p21−/− cell growth (Supplementary Fig. 3), but OSBP shRNA knockdown sensitized cells to cephalostatin 1, OSW-1 and schweinfurthin A by four-, six- and nine-fold, respectively (Fig. 3b). Sensitivity to taxol was not affected by shRNA knockdown of OSBP, revealing that OSBP protein levels do not nonspecifically enhance the activity of cytotoxic small molecules. Similar results were obtained in HeLa cells (Supplementary Fig. 5). Unfortunately, antibodies to ORP4L, either from commercial sources or developed in our lab, did not recognize endogenous levels of ORP4L in various cell lines, which prohibited conducting ORP4L knockdown experiments. However, we performed both OSBP and ORP4L overexpression and cytotoxicity experiments (Fig. 3c). OSBP and ORP4L overexpression each desensitized HeLa cells to the antiproliferative activity of cephalostatin 1, OSW-1 or schweinfurthin A by a factor of approximately 2–5 (Fig. 3d). Taxol-induced growth inhibition was not affected by overexpression of either OSBP-Myc-His or ORP4L-Myc-His (Fig. 3d).
Figure 3. shRNA knockdown, protein expression and SAR experiments support OSBP and ORP4L as targets of ORPphilins.

(a) Quantitation of OSBP concentrations in nontargeting shRNA (shNT) and OSBP-targeting shRna (shOSBP) cells. Mean ± s.d. of seven individual experiments. ****P ± 0.0001 (n = 7) compared to shNT cells (two-tailed Student's t-test). (b) Knockdown and dose-shift experiment in HCT-116 p21−/− cells stably transfected with either shOSBP or shNT. Cells were treated with compounds for 72 h, and GI50 values were determined as described in Figure 2. Values are mean ± s.d. of three or four individual experiments. *P = 0.0002 (n = 3), **P = 0.0006 (n = 4), ***P = 0.0003 (n = 3) compared to shNT cells (two-tailed Student's t-test). (c) Western blot of HeLa lysates expressing lacZ-, OSBP- or ORP4L-Myc-His. (d) Effect of overexpression of lacZ, OSBP or ORP4L on the growth-inhibitory activity (48 h) of cephalostatin 1, OSW-1, schweinfurthin A and taxol in HeLa cells. Relative GI30 values are averages from at least three individual experiments. (e,f) Correlation between binding affinity (Ki) to OSBP-Myc-His (e) or ORP4L-Myc-His (f) and growth inhibition (GI50) in HCT-116 cells. Data points represent mean ± s.d. from three individual experiments. Pearson correlation coefficients (r) were calculated using GraphPad Prism 5 (P < 0.001). In f, schweinfurthin A (red) was omitted from the calculation. Uncut, full-gel images of panels a and c are shown in Supplementary Figure 4.
Correlation of OSBP and ORP4L binding to ORPphilin activity
If the antiproliferative activity of the ORPphilins is mediated by OSBP, ORP4L or both, a positive correlation between their affinity for OSBP and ORP4L and their antiproliferative activity should exist. Indeed, plotting the OSBP Ki values versus the HCT-116 GI50 values of the ORPphilins and four OSW-1 analogs (6–9) revealed a strong positive correlation (r = 0.9644) (Fig. 3e). The same analysis with the ORP4L Ki values also revealed a strong positive correlation (r = 0.9069) but only when schweinfurthin A was excluded (Fig. 3f). We obtained similar SAR correlation results for both OSBP and ORP4L in the A549 and HCT-116 p21−/− cell lines (Supplementary Fig. 6). Further supporting this correlation, analog 10, which bears a tert-butyldimethylsilyl group on the C3 carbinol (Fig. 1), was approximately 1,400-fold less cytotoxic in HCT-116 cells (GI50 = 2,300 nM), 700-fold less cytotoxic in A549 cells and 2,100-fold less cytotoxic in HCT-116 p21−/− cells compared to OSW-1 (Supplementary Fig. 1a). Notably, analog 10 also did not compete with [3H]25-OHC for binding to OSBP or ORP4L (Supplementary Fig. 1c).
25-Hydroxycholesterol suppresses ORPphilin activity
25-OHC is not cytotoxic at concentrations up to 10 μM in most cell lines. ORPphilins competed with 25-OHC for binding to OSBP and ORP4L with Ki values similar to the 25-OHC KD (Fig. 2d,e). Therefore, in cells, administration of sublethal doses of 25-OHC with the ORPphilins should inhibit binding of the ORPphilins to OSBP and ORP4L, thereby desensitizing cells to the cytotoxic effects of the ORPphilins. We found that addition of 25-OHC caused a dose-dependent suppression of the antiproliferative activity of cephalostatin 1, OSW-1 and schweinfurthin A in the HCT-116 and HeLa cell lines (Fig. 4a and Supplementary Fig. 7a,b). Administration of 10 μM 25-OHC provided a nearly complete desensitization of HCT-116 cells treated with 250 nM of cephalostatin 1 or 10 nM OSW-1 (Fig. 4a)—doses that caused >75% growth inhibition when administered alone. 25-OHC also desensitized cells to schweinfurthin A, although to a lesser degree than for cephalostatin 1 and OSW-1 (Supplementary Fig. 7b). Notably, 25-OHC did not affect the antiproliferative activity of brefeldin A or taxol, demonstrating that 25-OHC does not nonspecifically desensitize cells to cytotoxic small molecules (Supplementary Fig. 7a,b).
Figure 4. Suppression of antiproliferative activity by 25-OHC administration and effects of lipoprotein-deficient media on proliferation.

(a) Effect of 25-OHC coadministration on growth-inhibitory activity (48 h) of cephalostatin 1 or OSW-1 in HCT-116 cells. (b) Removal of lipoproteins from the growth medium sensitizes CHO-7 cells to 25-OHC but not cephalostatin 1, OSW-1 or schweinfurthin A. Graphs represent mean ± s.d. of two individual experiments. CHO-7 is a CHO-K1 subclone that tolerates growth media depleted of lipoproteins.
In addition to OSBP and ORP4L, NPC1 (ref. 32) and Insig33 are also cellular receptors of 25-OHC. NPC1 is involved in the egress of lipoprotein-derived cholesterol from lysosomes34. Insig is a chief regulator of SREBP-dependent cholesterol homeostasis, and 25-OHC exerts much of its ability to inhibit cholesterol biosynthesis through its binding to Insig33. However, we observed no change in the antiproliferative activity of cephalostatin 1, OSW-1 or schweinfurthin A in NPC1−/− (M12) CHO cells compared to wild-type CHO cells, demonstrating that the activity of ORPphilins does not depend upon NPC1 (Supplementary Fig. 8). In addition, we evaluated the antiproliferative activity of the natural products in the CHO-7 cell line, which is adapted to grow in lipoprotein-deficient media and is dependent on Insig-mediated cholesterol biosynthesis for survival35. Unlike that of 25-OHC, which is much more cytotoxic in the CHO-7 cell line, the antiproliferative activity of cephalostatin 1, OSW-1 and schweinfurthin A in CHO-7 cells was not enhanced in lipoprotein-deficient media. This result indicates that the ORPphilins do not affect cholesterol biosynthesis and are therefore not ligands for Insig (Fig. 4b).
Mutant OSBP alleles desensitize cells to ORPphilins
To further support the idea that OSBP and ORP4L are targets of the ORPphilins, we mutated 15 OSBP residues both inside and outside the reported sterol binding region of the protein (Supplementary Fig. 9)36. ORPphilin growth inhibition in HeLa cells expressing nearly equivalent amounts of either wild-type or mutant OSBP-Myc-His protein (Supplementary Fig. 10a) was determined (Table 1). Two mutations in OSBP-Myc-His were revealed to confer ORPphilin resistance. Expression of OSBPM446W-Myc-His caused an 18-fold reduction in the antiproliferative activity of cephalostatin 1 at the GI30 level compared to expression of wild-type OSBP-Myc-His (Table 1, Supplementary Fig. 10c). The GI30 values are reported because they provide the clearest differential activities in the OSBP mutant overexpression experiments (Supplementary Fig. 10c,d). The same mutant caused a 3.9-fold reduction in the antiproliferative activity of OSW-1 and a 2.7-fold reduction in the activity of schweinfurthin A at the respective GI30 levels (Table 1). A second OSBP mutant, V582M, reduced the antiproliferative activity of cephalostatin 1 by 9.3-fold when compared to wild-type OSBP-Myc-His, yet this mutant did not affect the antiproliferative activity of OSW-1 or schweinfurthin A (Table 1, Supplementary Fig. 10c). The antiproliferative activity of taxol was unaffected by expression of these two OSBP mutant proteins.
Table 1.
| OSBP points mutants that desensitize to ORPphilins
| KD (nM) |
Ki (nM) |
Relative Gl30 |
||||||
|---|---|---|---|---|---|---|---|---|
| 25-OHC | Ceph 1 | OSW-1 | Schwf A | Ceph 1 | OSW-1 | Schwf A | Taxol | |
| Wild-type OSBP | 32 (±14) | 39 (±10) | 26 (±9) | 68 (±23) | 1.0 | 1.0 | 1.0 | 1.0 |
| OSBPM446W | 41 (±13) | 31 (±6) | 18 (±6) | 60 (±23) | 18 | 3.9 | 2.7 | 1.1 |
| OSBPV582M | 82 (±26) | 31 (±10) | 46 (±15) | 37 (±11) | 9.3 | 1.0 | 0.7 | 1.0 |
A table showing the following characteristics of wild-type OSBP, OSBPM446W and OSBPV582M: dissociation constants (Kd) for 25-OHc, inhibition constants (Ki) for cephalostatin 1, OSW-1 and schweinfurthin A in competition with 25-OHc, relative Gl30 values for cephalostatin 1, OSW-1, schweinfurthin a and taxol in HeLa cells overexpressing either wild-type OSBP or OSBP point mutants. Ki and Kd values are mean ± s.d. from three individual experiments. Gl30 values are the average of two individual experiments.
We then determined the binding affinity of cephalostatin 1, OSW-1 and schweinfurthin A to the OSBPM446W and OSBPV582M mutants using [3H]25-OHC competition binding (Table 1, Supplementary Fig. 11). The binding affinity of 25-OHC for OSBPM446W was identical to that of the wild type. The OSBPV582M mutant had a slightly higher 25-OHC KD than the wild type. Within error limits, the binding affinities (Ki) of cephalostatin 1, OSW-1 and schweinfurthin A for these two OSBP mutants were unchanged compared to that of wild-type OSBP-Myc-His (Table 1, Supplementary Fig. 11). This suggests that these mutations provide resistance to the ORPphilins not by affecting compound binding but by altering the cellular function of OSBP. Based on the sequence alignment of OSBP and ORP4L, we then made mutations in ORP4L at positions equivalent to OSBPM446W and OSBPV582M. Unlike expression of the two aforementioned OSBP mutants, expression of ORP4LM550W and ORP4LV686M did not reduce cell sensitivity to the ORPphilins when compared to wild-type ORP4L-Myc-His (Supplementary Fig. 10d).
ORPphilins perturb the cellular localization of OSBP
OSBP translocates from a cytoplasmic distribution (Fig. 5a) to the trans-Golgi network upon addition of 25-OHC to cells37. We treated HCT-116 cells with ORPphilins for short time periods (4 h) and monitored the effects on OSBP localization using immunofluorescence microscopy38. Much like 25-OHC37,38 (Fig. 5b), schweinfurthin A caused OSBP to accumulate at perinuclear positions with a large degree of co-localization with the trans-Golgi marker p230 (Fig. 5c). OSW-1 caused OSBP to localize to a perinuclear position that did not localize with p230 (Fig. 5d). Instead, OSW-1 treatment caused the p230 marker to appear as small, dispersed vesicles, which indicates trans-Golgi dismemberment. As with OSW-1, cells treated with cephalostatin 1 also resulted in OSBP occupying a perinuclear position with little colocalization of the dispersed p230 marker (Fig. 5e). However, unlike 25-OHC, OSW-1 or schweinfurthin A, cephalostatin 1 also caused OSBP to partially localize to the plasma membrane (Fig. 5e). This effect was also observed with ritterazine B, although there appeared to be greater localization of OSBP and p230 with ritterazine B than with cephalostatin 1 (Supplementary Fig. 12a). We also observed changes in OSBP localization upon treatment with ORPphilins in A549 cells (Supplementary Fig. 12b). Treatment with schweinfurthin B resulted in perinuclear OSBP staining similar to that seen with 25-OHC, whereas cephalostatin 1 and OSW-1 in this cell line induced dispersal or vesiculation of OSBP.
Figure 5. ORPphilins induce OSBP translocation in cells.

(a–e) OSBP (green) and trans-Golgi protein P230 (red) were visualized in HCT-116 cells using indirect immunofluorescence microscopy. Cells were treated with vehicle (a), 5 μM 25-OHC (b), 5 μM schweinfurthin A (c), 10 nM OSW-1 (d) or 20 nM cephalostatin 1 (e) for 4 h and then stained using specific primary antibodies against OSBP (green) and trans-Golgi protein P230 (red), followed by secondary staining with fluorescent antibodies. Nuclei (blue) were stained with Hoescht 33258. Scale bars are 5 μm. In a–c, arrows show colocalization of OSBP and p230. In e, the arrow shows OSBP localized at the plasma membrane.
Cephalostatin 1 and OSW-1 reduce OSBP levels
During our immunofluorescence studies, we noticed a lower fluorescent OSBP signal in cells treated for more than 8 h with either cephalostatin 1 or OSW-1. To determine if treatment with cephalostatin 1 or OSW-1 caused a reduction in the amount of OSBP in cells, we assessed OSBP concentrations in compound-treated HCT-116 cells by western blotting. Treatment with vehicle (DMSO) or 25-OHC (10 μM) did not affect OSBP levels (Fig. 6a). However, treatment with cephalostatin 1 or OSW-1 induced a dramatic, time-dependent reduction in OSBP concentrations (Fig. 6a). Treatment with 50 nM of cephalostatin 1 or 10 nM of OSW-1 caused an approximately 75% reduction in OSBP protein levels at 24 h in HCT-116 cells (Fig. 6a), with a similar effect recorded in A549 cells (Supplementary Fig. 13a). Notably, there was no effect on OSBP concentrations with schweinfurthin A (Fig. 6b). Coadministration of 10 μM or 20 μM of 25-OHC, which does not affect OSBP levels (Fig. 6a,b), significantly suppressed the reduction of OSBP levels induced by a 24-h treatment with 1 nM of OSW-1 (Fig. 6b). This result further demonstrated the interference of 25-OHC with ORPphilin activity in cells, presumably by blocking ORPphilin binding to OSBP. Notably, administration of 5 μM schweinfurthin A with 10 nM of OSW-1 for 24 h completely suppressed the reduction of OSBP concentrations (Fig. 6b). This result reveals that although OSW-1 and schweinfurthin A both bind and perturb OSBP in cells, they have considerably different effects on its activity.
Figure 6. Cephalostatin 1 and OSW-1 cause proteasome-dependent reduction of cellular OSBP concentrations.

(a) Cephalostatin 1 (50 nM) and OSW-1 (10 nM) induce a time-dependent reduction in OSBP protein levels in HCT-116 cells as judged by western blotting. (b) Schweinfurthin A and 25-OHC do not induce reduction in OSBP concentrations after 24 h of treatment but can block the reduction induced by OSW-1. (c) The reduction in OSBP protein concentrations (at 24-h time point) is blocked by addition of the proteasome inhibitors MG-132 or lactacystin. *P < 0.05 (n = 3), **P < 0.01 (n = 3), ***P< 0.001 (n = 3), ****P < 0.0001 (n = 3) relative to vehicle-treated cells; #P = 0.0006 (n = 3), ##P = 0.0196 (n = 3), ###P = 0.0025 (n = 3) (two-tailed Student's t-test). Uncut, full-gel image of a is shown in Supplementary Figure 4.
To determine if cephalostatin 1 and OSW-1 induced proteasome-dependent degradation39 of OSBP, we administered proteasome inhibitors MG-132 or lactacystin with cephalostatin 1 and OSW-1 (Fig. 6c). MG-132 or lactacystin alone did not affect OSBP concentrations, but these inhibitors completely blocked the OSBP degradation induced by cephalostatin 1 and OSW-1 in HCT-116 cells (Fig. 6c). Similar results were obtained in A549 cells (Supplementary Fig. 13a).
ORPphilins affect sphingomyelin biosynthesis
Sphingomyelin is biosynthesized from ceramide at the Golgi40 and is primarily transported from the ER to the Golgi by ceramide transport protein (CERT)41, and the transport activity of CERT is dependent on both OSBP and VAP-A40–42. In CHO-K1 cells, 25-OHC induces an increase in sphingomyelin biosynthesis in an OSBP-dependent fashion43. To determine whether the ORPphilins affect sphingomyelin synthesis, we performed pulse labeling with [3H] serine in CHO-K1 and HCT-116 cells, then measured the amount of tritium incorporated into sphingomyelin and other lipids. As expected based on previous reports43, treatment of CHO-K1 cells with 25-OHC (5 μM) induced an approximately four-fold increase in the biosynthesis of sphingomyelin (Fig. 7). Paralleling the effect of 25-OHC in CHO-K1 cells, low dose treatment (GI10) with the ORPphilins induced an increase in sphingomyelin biosynthesis, similar to but not to the extent of 25-OHC (Fig. 7). ORPphilin treatment did not significantly perturb the incorporation of radiolabel into phosphatidylethanolamine, a control lipid (Supplementary Fig. 14a). In HCT-116 cells, treatment with a range of OSW-1 concentrations had no effect on ceramide labeling (Supplementary Fig. 14b). This result is consistent with the ORPphilins altering the conversion of ceramide into sphingomyelin, a process that 25-OHC binding to OSBP is known to affect.
Figure 7. ORPphilins block sphingomyelin biosynthesis.
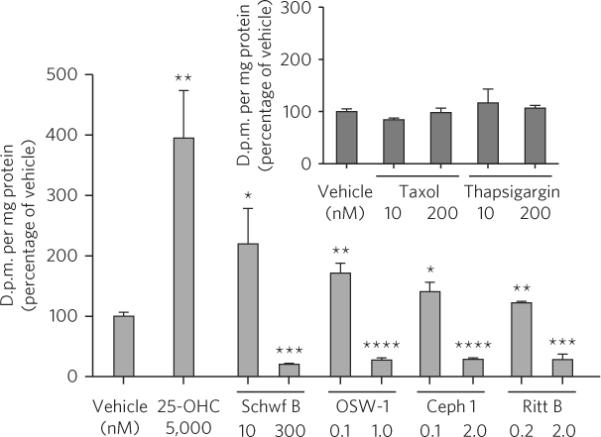
Pulse-labeling of sphingomyelin with [3H]serine in CHO-K1 cells. Cells were treated with vehicle or compound for 6 h, and in the last 2 h the cells were pulsed with 10 μCi [3H]serine. Label incorporation into sphingomyelin was measured by scintillation counting after lipid isolation by thin layer chromatography. All data are mean ± s.d. from three individual experiments. *P < 0.05 (n = 3), **P < 0.01 (n = 3), ***P < 0.001 (n = 3), ****P < 0.0001 (n = 3) relative to vehicle-treated cells (two-tailed Student's t-test).
However, unlike 25-OHC, exposure of CHO-K1 cells to high doses (GI90) of the ORPphilins caused a significant reduction in the biosynthesis of sphingomyelin (Fig. 7). Contrasting with this observed effect, exposure of a GI90 dose of 25-OHC (15 μM) caused a robust stimulation of sphingomyelin biosynthesis (Supplementary Fig. 14c). Inhibition of sphingomyelin biosynthesis did not occur upon exposure of cells to taxol or the ER stress-inducing small molecule thapsigargin, demonstrating that the ORPphilins do not inhibit sphingomyelin biosynthesis solely through the induction of cytotoxicity or ER stress. Suppression of CERT expression is reported to cause a significant reduction in basal sphingomyelin biosynthesis40. Therefore, we determined that the amount of CERT protein was not affected in HCT-116 or A549 cells treated with a GI90 concentration of cephalostatin 1 or OSW-1 (Supplementary Fig. 13a,b), unlike the observed effect on the amounts of OSBP.
DISCUSSION
We identified OSBP and its closest paralog ORP4L as high-affinity receptors of cephalostatin 1, OSW-1, ritterazine B and schweinfurthin A that mediate the phenotypic effects of these compounds. Although there are indications that stellettin E has biological activities similar to those of these compounds, there is insufficient evidence to conclude that stellettin E is a member of the ORPphilins. Convergence of the ORPphilins on the same protein targets validates the indication of a shared mechanism of action provided by the similar pattern of growth inhibition in the NCI-60 cell line panel.
Our results clearly showed that the cytotoxic ORPphilins interact with OSBP and ORP4L, but OSBP and ORP4L were not known to be involved in cancer cell survival. This led us to obtain multiple lines of evidence showing that OSBP and ORP4L mediate the antiproliferative activities of the ORPphilins in cancer cell lines. Our results are consistent with the ability of the ORPphilins to induce an OSBP and ORP4L loss-of-function effect in cells: OSBP shRNA knockdown sensitizes cells to the ORPphilins, while OSBP and ORP4L overexpression confers ORPphilin resistance. However, these results alone did not rule out the possibility that OSBP and ORP4L were `decoy proteins', that is, high-affinity receptors that prevent the ORPphilins from reaching the actual efficacy targets that cause their antiproliferative effects. Therefore, we obtained three additional lines of evidence in support of OSBP and ORP4L as efficacy targets of ORPphilins. First, we measured a high correlation between ORPphilin affinity for OSBP and ORP4L and ORPphilin cytotoxic potency. Second, we showed that ORPphilin activity was suppressed by administration of nontoxic doses of 25-OHC, a high-affinity ligand of OSBP and ORP4L. Finally, we discovered two dominant ORPphilin-resistant alleles of OSBP. Interestingly, these mutants did not show altered binding to any of the compounds. The effects of these mutations on OSBP function will not be understood until a more thorough characterization of OSBP biochemistry is achieved. However, a possible explanation for the desensitization of cells overexpressing these OSBP mutants is that these mutations affect protein-protein interactions, and the interaction of OSBP and other proteins is involved in the antiproliferative activity of the ORPphilins. The thyroid hormone β-receptor provides a precedent for this possibility. Known mutants of this protein suppress ligand-induced transactivation by disrupting protein-protein interactions rather than by affecting ligand binding44. OSBP has several known interacting partners16,17, and OSBP is reported to function as a sterol-sensing scaffolding protein that is capable of making multiprotein complexes in a ligand binding–dependent fashion20,21. Therefore, it is reasonable to speculate that the activity of the ORPphilins was reliant on the interaction of OSBP with other proteins and that mutations altering OSBP protein-protein interactions modulated the activity of these compounds.
Taken as a whole, the aforementioned results strongly supported OSBP and ORP4L as efficacy targets. However, our finding that the ORPphilins caused an apparent loss of OSBP function at first appeared to be inconsistent with the lack of growth inhibition caused by OSBP shRNA knockdown. A possible cause for this discrepancy is that a complete or nearly complete ablation of OSBP expression is required for cell growth inhibition; the OSBP shRNA knockdown experiments reduced OSBP levels only by ~85%. In contrast, the ORPphilins, highlighting the power of small-molecule effectors, could inhibit total OSBP function in the cell. This is especially true for cephalostatin 1 and OSW-1, which have a dual means of disrupting OSBP function. Specifically, both compounds are high-affinity ligands (low nM Ki) of OSBP and induced OSBP degradation. A second explanation for this discrepancy is that depletion of both OSBP and ORP4L is required to phenocopy the effects of ORPphilin treatment. Lacking an effective ORP4L antibody, however, we could not perform a simultaneous knockdown of both proteins to test this.
A third possible explanation for the discrepancy is that the ORPphilins perturb the cellular functions of multiple members of the OSBP and ORP superfamily other than OSBP and ORP4L, and that the inhibition of these other family members contributes to the antiproliferative activity of the ORPphilins. This possibility would be more likely if a critical cellular function is performed redundantly by OSBP and many ORPs, which would mirror the situation in yeast. The yeast homologs of OSBP and the ORPs are called Osh proteins16,17. Yeast have seven Osh proteins and can survive the loss of any six of these but not the loss of all seven45. Furthermore, the OSBP and ORP family has significant sequence homology in the lipid-binding domain16,17, and most of its members have been shown to bind 25-OHC46. Given that the ORPphilins competed with 25-OHC for binding to OSBP and ORP4L, other OSBP and the ORPs could also bind these compounds. The significant desensitization achieved by administration of 25-OHC could be due to displacement of the ORPphilins from other ORPs beyond OSBP and ORP4L. Superfamily wide inhibition of OSBP and the ORPs, or inhibition of the subset of these proteins required for cell survival, would also explain the failure of genetic perturbation methods to uncover a role of the individual ORPs in cell proliferation, with a possible exception being the reported cytotoxicity upon inducible ORP9S expression22.
Excepting schweinfurthin A, 25-OHC and the ORPphilins had comparable binding affinities for OSBP and ORP4L, but the GI50 values of the ORPphilins in human cancer cell lines are generally 1,000 times lower than for 25-OHC. This could be because ORPphilins bind at a different site from 25-OHC or because they have different effects on OSBP and ORP4L conformations. Additionally, 25-OHC is known to have many protein targets in cells, which could buffer the amount of 25-OHC reaching OSBP and ORP4L. Also, 25-OHC is rapidly esterified in cells, which could lower its affinity for OSBP and ORP4L and therefore reduce its cytotoxic effects47.
That the functions of OSBP and its paralogs, the ORPs, are not well understood is in part due to a lack of reagents capable of selectively altering the function of these proteins in cells. Although 25-OHC is often used to perturb OSBP and the ORPs in cells, its promiscuous interactions with NPC-1, Insig and other proteins complicates interpretation of these experiments. Our discovery that ORPphilins selectively target OSBP and ORP4L revealed new, high-affinity small-molecule probes of these proteins. Moreover, OSBP and ORP4L are implicated in atherosclerosis16,18,19, Alzheimer's disease48 and breast cancer metastasis49. Thus, ORPphilins could be powerful reagents to study the role of OSBP and ORP4L in these processes.
Our discovery that the antiproliferative ORPphilins target OSBP and ORP4L poses the question of whether endogenous, highly cytotoxic OSBP and ORP4L ligands exist. If so, these ligands could be involved in lipotoxicity and human disease. Furthermore, as ORPphilins are selectively growth-inhibitory to tumor cells lacking p21, and schweinfurthin A is selective toward tumor cells lacking NF-1 (ref. 14), to our knowledge our results provide the first indication that loss of these tumor suppressor proteins renders cells highly sensitive to perturbation of OSBP and ORP4L. Thus, our findings indicate that OSBP and ORP4L are new targets for achieving synthetic lethality with NF-1 and p21, which could result in tumor selective cancer therapeutics. We expect that the ORPphilins will be powerful reagents in helping uncover the function of OSBP and ORP4L, especially as the ORPphilins comprise three structurally diverse compounds, all targeting OSBP and ORP4L. The ORPphilins will also be useful in studying the newly identified role of OSBP and ORP4L in cancer cell survival.
METHODS
Natural product compounds
Cephalostatin 1 (~2 mg) was supplied via total synthesis (carried out in the Shair laboratory28). OSW-1 was obtained through isolation from nature and also through total synthesis in the Shair laboratory. OSW-1 analogs 6–10 were prepared as detailed in the Supplementary Methods section. Schweinfurthin A (~3 mg) and ritterazine B (0.2 mg) were obtained from the NCI. All compounds used were of analytical purity.
Antibodies
Polyclonal OSBP-specific (N terminus) rabbit antibodies (gift from H. Arai, University of Tokyo); polyclonal actin-specific rabbit antibodies (Sigma); monoclonal tubulin-specific (DSHB E7) mouse antibodies (Developmental Studies Hybridoma Bank); polyclonal c-Myc–specific (N-262) rabbit antibodies (Santa Cruz Biotechnologies); monoclonal c-Myc–specific (9E10) mouse antibodies (Santa Cruz Biotechnologies); polyclonal COL4A3BP/CERT–specific (ab72536) rabbit antibodies (Abcam); monoclonal p230-specific mouse antibodies (BD Biosciences).
Cell lines
HCT-116 wild type and HCT-116 p21−/−cell lines were supplied by the Vogelstein laboratory (Johns Hopkins University). CHO-7 cells were a gift from M. Brown and J. Goldstein (University of Texas Southwestern Medical Center). M12 cells were a gift from D. Ory (Washington University, St. Louis). All other cell lines were purchased from ATCC.
Plasmids, vectors and OSBP shRNAs
The pcDNA 3.1/Myc-His vector and the pcDNA3.1/Myc-His lacZ control vector were purchased from Invitrogen. OSBP cDNA was purchased from Open Biosystems. ORP4L cDNA was cloned from HeLa total RNA extract using standard molecular biology techniques. The OSBP and ORP4L cDNAs were cloned into the pcDNA3.1/Myc-His vector (Invitrogen). OSBPM446W, OSBPV582M, ORP4LM550W and ORP4LV686M genetic mutants were prepared with the QuikChange II XL Site-directed Mutagenesis Kit (Stratagene) as instructed by the supplier. OSBP shRNAs in the pSIREN-RetroQ vector (BD Biosciences) were generously provided by G. Romeo (Joslin Diabetes Center).
Affinity chromatography experiment—iTRAQ
25 mg HeLa-S3 S100 lysate was pre-incubated with DMSO or 10 μM OSW-1 and then incubated overnight at 4 °C with 30 μl of packed OSW-1 affinity reagent (OAR) per pulldown. The resin was transferred to a Mobicol 1 ml column (Boca Scientific). The resin was washed extensively with buffer, and the bound protein was eluted via denaturation in NuPage LDS sample loading buffer under reducing conditions, alkylated with iodoacetamide and separated on a NuPage 4–12% Bis-Tris gradient gel (Invitrogen). Gel lanes were excised using a LEAP 2DiD robot and digested in-gel digested with trypsin (Tecan Freedom EVO 200), and the resulting eight samples per lane were labeled with iTRAQ reagent (AB/Sciex). Samples were mixed and analyzed using an Eksigent NanoLC 1D+ HPLC coupled to a Thermo LTQ-Orbitrap XL mass spectrometer operated in Pulsed-Q Dissociation mode. Peptide and protein identification and validation and quantitative data analysis were done via an automated workflow incorporating a forward and reverse version of IPIv3.55, Mascot v2.2 (Matrix Science) and Transproteomic pipeline v3.3sqall (Institute for Systems Biology) Spotfire DXP. Protein fold changes were derived as median peptide fold change, P values were calculated using a one-way t-test, and data were visualized for further analysis using Spotfire DXP. See Supplementary Methods for more details.
Cell viability assay
Cells were plated into 96-well plates in 75 μl of medium. After 24 h, a time-zero plate was produced by adding 25 μl of medium and 20 μl of CellTiter-Blue (Promega) to the wells and then incubating the plates at 37 °C for 90 min. Fluorescence (544 nm excitation; 590 nm emission) was detected using a SPECTRAmax Gemini XS to establish cell viability at time of dosing. Then, compounds were serially diluted in medium and delivered to the cells as 4× solutions in 25 μl of medium. At either 48 h or 72 h, CellTiter-Blue was added, and the fluorescence was recorded as described above. Growth relative to untreated cells was calculated, and this data was fitted to a four-parameter dose-response curve using GraphPad Prism 5.
Overexpression-viability assays with wild-type and mutant OSBP and ORP4L constructs were carried out by transfecting HeLa cells with the appropriate plasmids using Lipofectamine LTX with PLUS reagent (Invitrogen) as instructed, and after 8 h of transfection, the cells were replated for the cell viability assay, performed as described above. The amount of expression was evaluated by immunoblotting at the time of dosing.
[3H]25-OHC charcoal-dextran binding assay
OSBP, OSBPM446W, OSBPV582M, ORP4L, ORP4LM550W, ORP4LV686M and lacZ were cloned into the pcDNA3.1 vector (Invitrogen), which encodes a C-terminal Myc-His tag. These vectors were transfected into HEK-293T cells using Lipofectamine2000 (Invitrogen), and 48 h after transfection, on-plate lysis of the transfected cells with M-Per with HALT and EDTA (ThermoScientific) reagent was performed, followed by ultracentrifugation at 100,000g. The S100 lysate was diluted to 0.2 mg ml−1, aliquoted and stored at −80 °C until use. The HEK-293T OSBP–, ORP4L- or lacZ-Myc-His lysate was incubated with [3H]25-OHC at 4 °C for 16 h in 96-well plates. After incubation, charcoal in dextran (C/D) was added to the wells for 30 min of shaking at room temperature (18–25 °C), followed by centrifugation for 15 min at 1,900g to pellet the C/D. The C/D-cleared lysate was added to MicroScint-20 (PerkinElmer) scintillation fluid, and the remaining protein-bound radiolabel was quantified using a TopCount scintillation counter. [3H]25-OHC binding curves and competition curves were repeated at least three times. Competition binding experiments to generate Ki values for compounds 1–4, 6–8 and 10 were performed with 20 nM of [3H]25-hydroxycholesterol. Additional details are described in Supplementary Methods.
Immunofluorescence microscopy
HCT-116 or A549 cells were plated on coverslips in six-well plates. After 72 h, the culture medium was removed and replaced with medium containing either compound or vehicle control. At the indicated time points, the cells were fixed for 10 min using 3.7% formaldehyde in PBS. The coverslips were washed with PBS and permeabilized for 5 min with 0.5% Triton X-100 in PBS. Following PBS washing, the coverslips were blocked for 30 min using Image-iT FX (Invitrogen), washed with PBS, then blocked for 30 min at room temperature with 1% BSA in PBS (BSA/PBS). The coverslips were then incubated overnight at 4 °C with a primary antibody solution in BSA/PBS. The coverslips were washed with BSA/PBS and incubated for 60 min at room temperature with Alexa Fluor–conjugated secondary antibodies (Invitrogen) in BSA/PBS, including incubation with Hoechst 33258 in the final 30 min. The coverslips were then washed sequentially with BSA/PBS, PBS and water and mounted on microscope slides using Aqua Poly/Mount (Polysciences). Imaging data was acquired using Zeiss LSM 510 META scanning confocal microscope.
Other methods
Any remaining experimental procedures are described in Supplementary Methods.
Supplementary Material
Acknowledgments
We thank the NCI for the generous gifts of schweinfurthin A. (J.A. Beutler) and ritterazine B. We gratefully acknowledge the aforementioned reagents provided by G. Romeo (Joslin Diabetes Center), M. Brown and J. Goldstein (University of Texas Southwestern Medical Center), H. Arai (University of Tokyo), B. Vogelstein (Johns Hopkins University) and D. Ory (Washington University, St. Louis). The beta-tubulin monoclonal antibody (E7) developed by M. Klymkowsky was obtained from the Developmental Studies Hybridoma Bank (National Institute of Child Health and Human Development, US National Institutes of Health (NIH) and University of Iowa, Department of Biology). R. King is acknowledged for helpful comments and discussions. T.B.P. would like to thank the Lundbeck foundation for a post-doctoral fellowship and the Danish Council for Independent Research–Natural Sciences for additional financial support. A.W.G.B gratefully thanks the Susan G. Komen for the Cure Foundation for providing a postdoctoral research fellowship. Financial support from the Novartis Institutes of Biomedical Research, the US NIH (grant no. R01GM090068), the Harvard Catalyst and The Harvard Clinical and Translational Science Center (NIH award no. UL1RR025758) is acknowledged. This work was conducted with support and financial contributions from Harvard University and its affiliated academic health care centers.
Footnotes
Author contributions M.D.S., A.W.G.B., T.B.P., K.W. and D.R.A. conceived and designed the study. A.W.G.B., T.B.P., K.W. and D.R.A. implemented experiments. K.C.F. prepared cephalostatin 1. The OSBP and ORP4L molecular biology was done by K.W. and C.K. K.S. prepared OSW-1. S.O. performed early studies on p21 selectivity. Y.M. and M.K. supplied OSW-1 isolated from nature. A.W.G.B., D.R.A., M.S., J.P.M., E.C.P., D.J.S., I.C.-T. and J.A.T. performed the OSW-1 affinity chromatography experiments and subsequent quantitative mass spectrometry analysis. M.D.S., A.W.G.B., T.B.P. and K.W. wrote the paper.
Competing financial interests The authors declare no competing financial interests.
Additional information Supplementary information and chemical compound information is available online at http://www.nature.com/naturechemicalbiology/. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Pettit GR, et al. Antineoplastic agents. 147. Isolation and structure of the powerful cell growth inhibitor cephalostatin 1. J. Am. Chem. Soc. 1988;110:2006–2007. [Google Scholar]
- 2.Kubo S, et al. Acylated cholestane glycosides from the bulbs of Ornithogalum saundersiae. Phytochemistry. 1992;31:3969–3973. [Google Scholar]
- 3.Komiya T, et al. Ritterazine B, a new cytotoxic natural compound, induces apoptosis in cancer cells. Cancer Chemother. Pharmacol. 2003;51:202–208. doi: 10.1007/s00280-002-0558-8. [DOI] [PubMed] [Google Scholar]
- 4.Beutler JA, Shoemaker RH, Johnson T, Boyd MR. Cytotoxic geranyl stilbenes from Macaranga schweinfurthii. J. Nat. Prod. 1998;61:1509–1512. doi: 10.1021/np980208m. [DOI] [PubMed] [Google Scholar]
- 5.Tasdemir D, et al. Bioactive isomalabaricane triterpenes from the marine sponge Rhabdastrella globostellata. J. Nat. Prod. 2002;65:210–214. doi: 10.1021/np0104020. [DOI] [PubMed] [Google Scholar]
- 6.Moser BR. Review of cytotoxic cephalostatins and ritterazines: isolation and synthesis. J. Nat. Prod. 2008;71:487–491. doi: 10.1021/np070536z. [DOI] [PubMed] [Google Scholar]
- 7.Beutler JA, et al. The schweinfurthins. In: Bogers RJ, et al., editors. Medicinal and Aromatic Plants. Agricultural, Commercial, Ecological, Legal, Pharmacological and Social Aspects. Springer; 2006. Ch. 22. [Google Scholar]
- 8.Zhou Y, et al. OSW-1: A natural compound with potent anticancer activity and a novel mechanism of action. J. Natl. Cancer Inst. 2005;97:1781–1785. doi: 10.1093/jnci/dji404. [DOI] [PubMed] [Google Scholar]
- 9.Rabow AA, Shoemaker RH, Sausville EA, Covell DG. Mining the national cancer institute's tumor-screening database: identification of compounds with similar cellular activities. J. Med. Chem. 2002;45:818–840. doi: 10.1021/jm010385b. [DOI] [PubMed] [Google Scholar]
- 10.Mimaki Y, et al. Cholestane glycosides with potent cytostatic activities on various tumor cells from Ornithogalum saundersiae bulbs. Bioorg. Med. Chem. Lett. 1997;7:633–636. [Google Scholar]
- 11.Boyd MR. The NCI Human Tumor Cell Line (60-Cell) Screen: Concept, Implementation, and Applications. In: Teicher BA, et al., editors. Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval. Humana Press; 2004. Ch. 3. [Google Scholar]
- 12.Dirsch VM, et al. Cephalostatin 1 selectively triggers the release of Smac/DIABLO and subsequent apoptosis that is characterized by an increased density of the mitochondrial matrix. Cancer Res. 2003;63:8869–8876. [PubMed] [Google Scholar]
- 13.Rudy A, López-Antón N, Dirsch VM, Vollmar AM. The cephalostatin way of apoptosis. J. Nat. Prod. 2008;71:482–486. doi: 10.1021/np070534e. [DOI] [PubMed] [Google Scholar]
- 14.Turbyville TJ, et al. Schweinfurthin A selectively inhibits proliferation and Rho signaling in glioma and neurofibromatosis type 1 tumor cells in a NF1-GRD–dependent manner. Mol. Cancer Ther. 2010;9:1234–1243. doi: 10.1158/1535-7163.MCT-09-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan D, Olkkonen VM. Characteristics of oxysterol binding proteins. Int. Rev. Cytol. 2008;265:253–285. doi: 10.1016/S0074-7696(07)65007-4. [DOI] [PubMed] [Google Scholar]
- 17.Fairn GD, McMaster CR. Emerging roles of the oxysterol-binding protein family in metabolism, transport, and signaling. Cell. Mol. Life Sci. 2008;65:228–236. doi: 10.1007/s00018-007-7325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan D, et al. Oxysterol binding protein induces upregulation of SREBP-1c and enhances hepatic lipogenesis. Arterioscler. Thromb. Vasc. Biol. 2007;27:1108–1114. doi: 10.1161/ATVBAHA.106.138545. [DOI] [PubMed] [Google Scholar]
- 19.Yan D, et al. Expression of human OSBP-related protein 1L in macrophages enhances atherosclerotic lesion development in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007;27:1618–1624. doi: 10.1161/ATVBAHA.107.144121. [DOI] [PubMed] [Google Scholar]
- 20.Romeo GR, Kazlauskas A. Oxysterol and diabetes activate STAT3 and control endothelial expression of profilin-1 via OSBP1. J. Biol. Chem. 2008;283:9595–9605. doi: 10.1074/jbc.M710092200. [DOI] [PubMed] [Google Scholar]
- 21.Wang P-Y, Weng J, Anderson RGW. OSBP is a cholesterol-regulated scaffolding protein in control of ERK 1/2 activation. Science. 2005;307:1472–1476. doi: 10.1126/science.1107710. [DOI] [PubMed] [Google Scholar]
- 22.Ngo M, Ridgway ND. Oxysterol binding protein-related protein 9 (ORP9) is a cholesterol transfer protein that regulates Golgi structure and function. Mol. Biol. Cell. 2009;20:1388–1399. doi: 10.1091/mbc.E08-09-0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngo MH, Colbourne TR, Ridgway ND. Functional implications of sterol transport by the oxysterol-binding protein gene family. Biochem. J. 2010;429:13–24. doi: 10.1042/BJ20100263. [DOI] [PubMed] [Google Scholar]
- 24.Schulz TA, et al. Lipid-regulated sterol transfer between closely apposed membranes by oxysterol-binding protein homologues. J. Cell Biol. 2009;187:889–903. doi: 10.1083/jcb.200905007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor FR, Kandutsch AA. Oxysterol binding protein. Chem. Phys. Lipids. 1985;38:187–194. doi: 10.1016/0009-3084(85)90066-0. [DOI] [PubMed] [Google Scholar]
- 26.Dawson PA, Ridgway ND, Slaughter CA, Brown MS, Goldstein JL. cDNA cloning and expression of oxysterol-binding protein, an oligomer with a potential leucine zipper. J. Biol. Chem. 1989;264:16798–16803. [PubMed] [Google Scholar]
- 27.DeBerardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fortner KC, Kato D, Tanaka Y, Shair MD. Enantioselective synthesis of (+)-cephalostatin 1. J. Am. Chem. Soc. 2010;132:275–280. doi: 10.1021/ja906996c. [DOI] [PubMed] [Google Scholar]
- 29.Wyles JP, McMaster CR, Ridgway ND. Vesicle-associated membrane protein-associated protein-A (VAP-A) interacts with the oxysterol-binding protein to modify export from the endoplasmic reticulum. J. Biol. Chem. 2002;277:29908–29918. doi: 10.1074/jbc.M201191200. [DOI] [PubMed] [Google Scholar]
- 30.Wyles JP, Ridgway ND. VAMP-associated protein-A regulates partitioning of oxysterol-binding protein-related protein-9 between the endoplasmic reticulum and Golgi apparatus. Exp. Cell Res. 2004;297:533–547. doi: 10.1016/j.yexcr.2004.03.052. [DOI] [PubMed] [Google Scholar]
- 31.Wyles JP, Perry RJ, Ridgway ND. Characterization of the sterol-binding domain of oxysterol-binding protein (OSBP)-related protein 4 reveals a novel role in vimentin organization. Exp. Cell Res. 2007;313:1426–1437. doi: 10.1016/j.yexcr.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 32.Infante RE, et al. Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J. Biol. Chem. 2008;283:1052–1063. doi: 10.1074/jbc.M707943200. [DOI] [PubMed] [Google Scholar]
- 33.Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. USA. 2007;104:6511–6518. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Infante RE, et al. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. USA. 2008;105:15287–15292. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metherall JE, Goldstein JL, Luskey KL, Brown MS. Loss of transcriptional repression of three sterol-regulated genes in mutant hamster cells. J. Biol. Chem. 1989;264:15634–15641. [PubMed] [Google Scholar]
- 36.Wang P-Y, Weng J, Lee S, Anderson RGW. The N terminus controls sterol binding while the C terminus regulates the scaffolding function of OSBP. J. Biol. Chem. 2008;283:8034–8045. doi: 10.1074/jbc.M707631200. [DOI] [PubMed] [Google Scholar]
- 37.Ridgway ND, Dawson PA, Ho YK, Brown MS, Goldstein JL. Translocation of oxysterol binding protein to Golgi apparatus triggered by ligand binding. J. Cell Biol. 1992;116:307–319. doi: 10.1083/jcb.116.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishimura T, et al. Inhibition of cholesterol biosynthesis by 25-hydroxycholesterol is independent of OSBP. Genes Cells. 2005;10:793–801. doi: 10.1111/j.1365-2443.2005.00879.x. [DOI] [PubMed] [Google Scholar]
- 39.El Khissiin A, Leclercq G. Implication of proteasome in estrogen receptor degradation. FEBS Lett. 1999;448:160–166. doi: 10.1016/s0014-5793(99)00343-9. [DOI] [PubMed] [Google Scholar]
- 40.Perry RJ, Ridgway ND. Oxysterol-binding protein and vesicle-associated membrane protein–associated protein are required for sterol-dependent activation of the ceramide transport protein. Mol. Biol. Cell. 2006;17:2604–2616. doi: 10.1091/mbc.E06-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawano M, Kumagai K, Nishijima M, Hanada K. Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J. Biol. Chem. 2006;281:30279–30288. doi: 10.1074/jbc.M605032200. [DOI] [PubMed] [Google Scholar]
- 42.Peretti D, Dahan N, Shimoni E, Hirschberg K, Lev S. Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol. Biol. Cell. 2008;19:3871–3884. doi: 10.1091/mbc.E08-05-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ridgway ND. 25-Hydroxycholesterol stimulates sphingomyelin synthesis in Chinese hamster ovary cells. J. Lipid Res. 1995;36:1345–1358. [PubMed] [Google Scholar]
- 44.Collingwood TN, et al. A natural transactivation mutation in the thyroid hormone β receptor: Impaired interaction with putative transcriptional mediators. Proc. Natl. Acad. Sci. USA. 1997;94:248–253. doi: 10.1073/pnas.94.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beh CT, Cool L, Phillips J, Rine J. Overlapping functions of the yeast oxysterol-binding protein homologues. Genetics. 2001;157:1117–1140. doi: 10.1093/genetics/157.3.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suchanek M, et al. The mammalian oxysterol-binding protein-related proteins (ORPs) bind 25-hydroxycholesterol in an evolutionarily conserved pocket. Biochem. J. 2007;405:473–480. doi: 10.1042/BJ20070176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olkkonen VM, Hynynen R. Interactions of oxysterols with membranes and proteins. Mol. Aspects Med. 2009;30:123–133. doi: 10.1016/j.mam.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 48.Zerbinatti CV, et al. Oxysterol-binding protein-1 (OSBP1) modulates processing and trafficking of the amyloid precursor protein. Mol. Neurodegener. 2008;3:5. doi: 10.1186/1750-1326-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fournier MV, et al. Identification of a gene encoding a human oxysterol-binding protein-homologue: a potential general molecular marker for blood dissemination of solid tumors. Cancer Res. 1999;59:3748–3753. erratum 61, 792 (2001) [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.