

Abstract
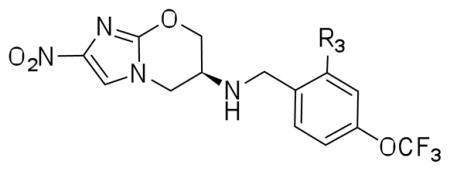
The (S)-2-nitro-6-(4-(trifluoromethoxy)benzyloxy)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine named PA-824 (1) has demonstrated antitubercular activity in vitro and in animal models and is currently in clinical trials. We synthesized derivatives at three positions of the 4-(trifluoromethoxy)benzylamino tail and these were tested for whole-cell activity against both replicating and non-replicating Mycobacterium tuberculosis (Mtb). In addition, we determined their kinetic parameters as substrates of the deazaflavin-dependent nitroreductase (Ddn) from Mtb that reductively activates these pro-drugs. These studies yielded multiple compounds with 40nM aerobic whole cell activity and 1.6μM anaerobic whole cell activity - ten fold improvements over both characteristics from the parent molecule. Some of these compounds exhibited enhanced solubility with acceptable stability to microsomal and in vivo metabolism. Analysis of the conformational preferences of these analogs using quantum chemistry suggests a preference for a pseudoequatorial orientation of the linker and lipophilic tail.
Introduction
The nitroimidazo-oxazines, including PA-824 (1)1, and nitroimidazo-oxazoles, including OPC-676832, 3 (Figure 1) are candidates for the treatment of tuberculosis (TB) that are currently in Phase 2 clinical trials4. These compounds have shown good activity in animal models of disease both as single agents and in combination studies5–8. This class has attracted attention because of its ability to not only kill rapidly dividing bacteria but also to kill quiescent bacteria maintained under hypoxic conditions1. It is hoped that this activity against non-replicating organisms may allow the shortening of the 6–8 month duration of TB chemotherapy required to achieve a durable cure9.
Figure 1.

These nitroimidazoles are pro-drugs and require bioreductive activation to exert their bactericidal effect10. In Mycobacterium tuberculosis (Mtb) this reduction is mediated by a deazaflavin-dependent nitroreductase, Ddn11 and reduction occurs on the imidazole ring resulting in the elimination of bactericidal reduced nitrogen species such as nitric oxide12. The anaerobic activity of compounds in this series correlates with the extent of production of reduced nitrogen species and both aerobic and anaerobic activities correlate roughly with the efficiency of enzymatic reduction by Ddn13.
Recently a significant amount of information on structure-activity relations (SAR) within this series of molecules has begun to emerge both in terms of whole cell activity and Ddn substrate preferences. The key determinants of these molecules as substrates include: an S configuration at C-6 of the oxazine ring; the nitro group; an 8-oxy substituted bicyclic nitroimidazole; and a lipophilic trifluoromethoxybenzyl tail14. The lipophilic tail has been the subject of considerable optimization chemistry since the series was first reported in the patent literature15, 16. A wide variety of substituted aromatics have been employed, in general with the most significant improvements in activity occurring with para substituents. The coincidental finding that p-trifluoromethoxy substitution (which appears in both candidate drugs) with linkers that vary in length by 4–5Å consistently provided the most active derivatives, prompted us to explore that more systematically with a homologous series of linkers increasing in carbon length. This study showed an optimal spacing of 4 carbons between the ether oxygen and the aromatic ring14. These findings and the extended lipophilic tail present in some analogs inspired further exploration of various substituted biphenyl analogs of this tail17. This study reported a correlation between lipophilicity of the side chain and identified analogs with significantly improved in vivo activity in a mouse model.
We have previously derived a 3D-Quantitative Structure-Activity Relationship (QSAR) pharmacophore model that was reasonably predictive for aerobic MIC amongst a series of 21 training and 22 test nitroimidazoles14. Although the best model suggested only one hydrophobic feature, other models suggested the possibility of two distinct hydrophobes in the tail region of 1 and raised the prospect that additional analogs could be synthesized, which would simultaneously engage both features. In this manuscript we describe our attempts to explore this hypothesis with three series of analogs synthesized primarily from the more soluble 6-S-amino series of compounds. The first (R1) employs the amine as an attachment site, the second (R2) focuses on the benzylic carbon and the third (R3) explores additional diversity on the aromatic nucleus of the trifluoromethoxybenzyl moiety of the parent compound.
Chemistry
R1 modifications
2 was synthesized as previously reported14 and utilized as a starting point for R1 (Scheme 1). Amide derivatives, 4a–f, were synthesized by reacting 2 with the corresponding acyl chlorides in presence of NaH in DMF. Formylation of 2 with formic acid in the presence of acetic anhydride in THF yielded the N-formamide derivative (3) in 55% yield. The tertiary amine derivatives (5a–c) were synthesized by reductive amination of formaldehyde, propionaldehyde or acetone with amine 2 in moderate yield. Reaction of 2 with triphosgene followed by ethylamine hydrochloride in the presence of triethylamine afforded the urea derivative 6 in 66% yield.
Scheme 1.

Reaction conditions: i) HCO2H, Ac2O, THF, 0 °C, 1h, 55%; ii) R1COCl, NaH, DMF rt − 70 °C; iii) R1CHO, NaBH(OAc)3, MeOH/AcOH; iv) triphosgene, EtNH2·HCl, Et3N, THF, 0 °C – rt, 66%.
R2 modifications
Oxazol-2-yl(4-(trifluoromethoxy)phenyl)methanone (9a) was prepared by copper mediated acylation18 of oxazol-2-yl zinc chloride using 4-trifluoromethoxybenzoyl chloride and then subjected to reductive amination with amine (7) in the presence of Ti(iPrO)4/AcOH/THF/NaCNBH3 to provide 17a as diastereomeric mixture (Scheme 2). Synthesis of the R2 methyl analog 17b was accomplished by reductive amination of the commercially available 4-trifluoromethoxyacetophenone 9b. Imidizolium ylide generated from N-benzylimidazole using diisopropylcarbamyl chloride and diisopropylethylamine was treated with trifluoromethoxybenzaldehyde in refluxing CH3CN to produce the carbamate, which upon hydrolysis afforded the benzylic alcohol (11). N-debenzylation using Pd/C followed by mesylation of the benzylic alcohol and subsequent reaction with amine 7 in THF in the presence of NaH at room temperature produced the final product (17c) as a mixture of diastereomers. The three alkyl derivatives (17d–f) were all prepared in moderate yield by alkylation of amine 7, using the corresponding bromides 15a–c, as illustrated in Scheme 2. Addition of ethyl and n-propyl Grignard reagents to 4-trifluoromethoxybenzaldehyde (13) afforded the corresponding benzylic alcohols (14a–b) while 1-(4-(trifluoromethoxy)phenyl)butan-1-ol (14c) was prepared by the addition of n-BuLi to trifluoromethoxybenzaldehyde. Alcohols 14a–c were converted to their corresponding bromides 15a–c) using PBr3 in ether and treated with amine (7) in DMF in the presence of K2CO3/KI at 90 °C to provide the required compounds 17d–f as a mixture of diastereomers. Synthesis of carboxamide derivative 17g was achieved by nucleophilic addition of TMSCN to the imine formed between the amine 7 and 4-trifluoromethoxybenzaldehyde (13) at 100 °C and subsequent hydrolysis in ethanolic HCl. Reaction of 4-((trifluoromethoxy)phenyl)oxirane 18 with 4-trifluoromethoxybenzyl alcohol in presence of KOtBu at 60 °C afforded alcohol 19. Further oxidation of the ring opened product, followed by removal of the p-methoxybenzyl group using TBDMSOTf afforded 2-hydroxy-1-(4-(trifluoromethoxy)phenyl)ethanone (20). 3-Hydroxy-1-(4-(trifluoromethoxy)-phenyl)propan-1-one (22) was synthesized from 13 in three steps involving a Reformatsky reaction with ethylbromoacetate in presence of Zn, LiAlH4 mediated reduction of the ester group to yield 21, and finally oxidation of the benzylic to alcohol using MnO2 to provide the hydroxy ketone 22. Reductive amination of 20 and 22 with amine 7 afforded nitroimidazooxazines 17h–i respectively.
Scheme 2.

Reaction conditions: n-BuLi, ZnCl2, CuI, THF, −78 °C – rt, 1.5 h, then 4-trifluoromethoxybenzoyl- chloride, rt, 1 h, 40%; ii) diisopropylcarbamyl chloride, DIPEA, 4-trifluoromethoxybenzaldehyde, CH3CN, reflux, 19 h, 73%; iii) 50 % TFA in water, THF, reflux, 15 h, 82%; iv) H2, Pd/C, MeOH, 1 atm, 81%; v) MsCl, Et3N, CH2Cl2, rt, 1 h; vi) 7, NaH, THF, rt, 40 h, 15%.; vii) RMgBr, THF, 0 °C – rt when R = Et and nPr; nBuLi, THF, −78 °C – rt when R = nBu; viii) PBr3, ether, 0 °C – rt; ix) 7, K2CO3, DMF, KI, 90 °C.; x) 4-trifluoromethoxybenzaldehyde, neat, 100 °C, 5 min. then TMSCN, 100 °C, 30 min, 50%; xi) EtOH/HCl, −10 °C, 38%; xii) 7, NaCNBH3, AcOH, EtOH, 5%; xiii) 4-methoxybenzylalcohol, KOtBu, 60 °C, 2 h, 34%; xiv) PDC, CH2Cl2, rt, 24 h, 62%; xv) TBDMSOTf, CH2Cl2, rt, 5 min, 86%; xvi) ethylbromoacetate, Zn, CH2Cl2, rt, 3 h; xvii) LiAlH4, THF, 0 °C - rt, 2 h, 25% over two steps; xviii) MnO2, CH2Cl2, rt, 6 h, 50%; xix) 7, Ti(iOPr)4, AcOH, NaBH3CN, 9%.
R3 modifications
R3 modified nitroimidazooxazines were synthesized by the reductive amination of substituted trifluoromethoxybenzaldehydes with amine (7). Most of these aldehydes were not available commercially and were synthesized as described in Schemes 3, 4 and 5. Thus 2-hydroxy-4-trifluoromethoxybenzoic acid (23)19 was used as a key intermediate in the synthesis of 2-hydroxy bearing analogs and the corresponding ether derivatives (32a–c, Scheme 3). Esterification of (23) with ethyl alcohol followed by alkylation with BnBr and cyclopropylmethylbromide gave the respective ether derivatives (27b–c) in excellent yields. Protection of phenolic-OH group of 24 as its O-methoxymethyl ether was carried out using MOMCl/DIPEA to afford 27a. LiAlH4 mediated reduction of the ester group of 27a–c and subsequent oxidation using PCC in CH2Cl2 afforded the required aldehydes 28a–c in good yield.
Scheme 3.
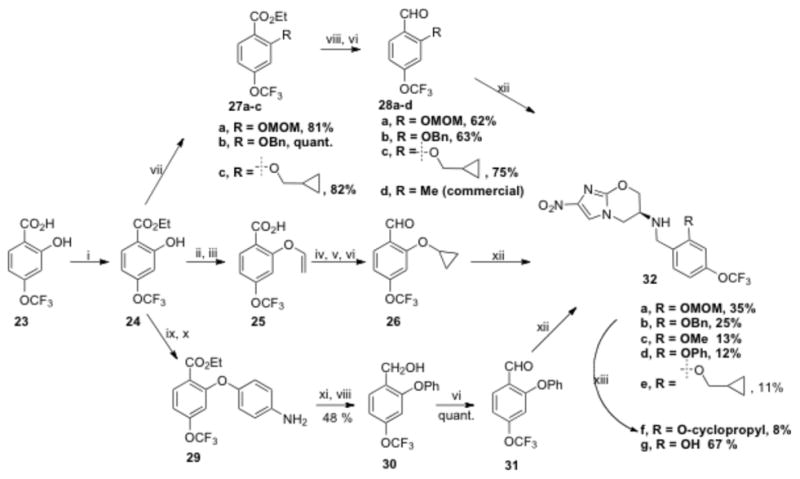
Reaction conditions: i) EtOH/H+, 80 °C, 80 %; ii) 1-bromo-2-chloroethane, K2CO3, DMF, rt, 15 h, 80%; iii) KOtBu, THF, 1 h, 20 °C, 78 % over two steps; iv) Et2Zn, CH2I2, ClCH2CH2Cl, toluene, rt, 17 h, 51%; v) BH3-DMS, THF, reflux, 1.5 h; vi) PCC, CH2Cl2, rt, 0.5 h, 80 % over two steps; vii) RX, K2CO3, DMF, 70 °C, 30 min - 2 h, when R = Bn, cyclopropylmethyl; MOMCl, DIPEA, CH2Cl2, rt, 3 h; viii) LiAlH4, THF, 0 °C – rt, 1 h; ix) 4-fluoronitrobenzene, NaH, DMF, 100 °C, 2 h, 67%; x) H2, Pd/C, EtOAc, rt, 1.5 h, 87%; xi) NaNO2, H3PO2, 6N HCl, 50 °C, 1 h; xii) NaBH(OAc)3, AcOH, DMF, 20 h; xiii) 6N HCl, THF, 1h, rt.%.
Scheme 4.

Reaction conditions: i) styrene, Pd(OAc)2, Et3N, 95 °C, 16 h, 64%; ii) OsO4, NaIO4, acetone-water, rt, 16 h, 16%; iii) 7, NaCNBH3, AcOH, DMF, 20 h; iv) ethylene glycol, p-TSA, benzene, 80 °C, 8 h, 86%; v) Pd(OAc)2, Cs2CO3, Xantphos, dioxane, amine, 90 °C, 8 h; vi) THF, 6 N HCl, 30 min., room temperature; vii) n-BuLi, THF, −78 °C, DMF, 15 min.
Scheme 5.

Reaction conditions: i) TBSCl, imidazole, CH2Cl2, rt, 60%; ii) s-BuLi, TMEDA, THF, −78 °C, 1 h, then FB(OMe)2, −78 °C, 30 min followed by alk, H2O2, 30 min, 28%; iii) K2CO3, MeI, DMF, 70 °C; iv) MOMCl, DIPEA, DMF, rt, 16 h, 82 %; v) TBAF, THF, rt, 1.5 h; vi) PCC, CH2Cl2, rt, 1 h; vii) 7, NaCNBH3, AcOH, DMF; viii) 4-fluoronitrobenzene, NaH, DMF, 100 °C, 2 h, 30 %; ix) Fe/NH4Cl, EtOAc-water, reflux, 1.5 h; x) NaNO2, H3PO2, 6N HCl, 50 °C, 1 h, 52 % over two steps; xi) ethylene glycol, p-TSA, benzene, 80 °C, 8 h, 72 %; xii) Pd(OAc)2, Cs2CO3, Xantphos, dioxane, amine, 90 °C, 8 h; xiii) THF, 6 N HCl, 30 min., rt; xiv) sec-BuLi, MeOCOCl, THF, −78 °C – rt, 3 h.
2-Cyclopropyloxy-4-trifluoromethoxy benzaldehyde (26) was synthesized in five steps20 from ethyl-2-hydroxy-4-trifluoromethoxybenzoate (24). Alkylation of 24 with 1-bromo-2-chloroethane using K2CO3 in DMF followed by saponification with KOtBu in THF at 20 °C resulted in the 4-(trifluoromethoxy)-2-(vinyloxy)benzoic acid (25) in 63% yield. Cyclopropanation of 25 under Simmons-Smith conditions followed by reduction of the carboxylic acid group using BH3·DMS and subsequent oxidation of the benzylic alcohol using PCC provided 2-O-cyclopropyl-4-trifluoromethoxybenzaldehyde (26) in 40% yield.
Reaction of ethyl-2-hydroxy-4-trifluoromethoxybenzoate (24) with 4-fluoronitrobenzene in the presence of NaH in DMF followed by Pd/C mediated reduction of the nitro group afforded the amine (29). Removal of the amino group by diazotization and subsequent reduction of the ester group using LiAlH4 provided the benzylic alcohol derivative (30) with 48% yield. The required aldehyde 31 was then obtained by PCC mediated oxidation of (30). Reductive amination of these aldehydes 26, 28a–d and 31 with amine 7 in presence of NaBH(OAc)3 in DMF-AcOH provided the nitroimidazooxazines 32a–f. Deprotection of the O-methoxymethyl ether using 6N HCl in THF provided 32g in 67% yield.
2-fluoro and 2-chloro-4-trifluoromethoxybenzaldehyde (34a–b) were readily synthesized from commercially available 2-chloro and 2-Fluoro substituted 4-trifluoromethoxyiodobenzene (33a–b) by lithiation using n-BuLi at −78 °C followed by quenching with DMF in 72% and 57% yield respectively (Scheme 4). 2-Bromo-4-trifluoromethoxybenzaldehyde 37 was synthesized by oxidative cleavage of the product 36 of Pd(0) mediated Heck coupling between 2-bromo-4-trifluoromethoxyiodobenzene (35) and styrene. Buchwald coupling of 36 with morpholine and piperidine followed by oxidative cleavage of the olefin afforded aldehydes 40a–b in moderate yields. 2-(4-Methylpiperazin-1-yl)-4-(trifluoromethoxy)benzaldehyde 39 was synthesized by Buchwald coupling of N-methylpiperazine with 2-[2-bromo-4-(trifluoromethoxy)phenyl]-1,3-dioxolane (38) followed by deprotection of the acetal. The reductive amination of aldehydes 34a–b, 37, 39 and 40a–b with amine 7 afforded nitroimidazooxazines 41a–f.
4-Trifluoromethoxybenzyl alcohol (42) was protected as a TBS ether (43) in 80% yield (Scheme 5) which was then reacted with s-BuLi at −78 °C in the presence of TMEDA and subsequently treated with FB(OMe)2 followed by alkaline H2O2 hydrolysis affording the required phenol (44) in 30% yield. Conversion of 44 to the corresponding O-methyl derivative (45) and O-methoxymethyl ether (46) was achieved by using MeI/K2CO3 and MOMCl/Et3N respectively. Cleavage of the TBS ether using TBAF followed by PCC-mediated oxidation produced the corresponding aldehydes 47a–b. Synthesis of 3-phenoxy-4-trifluoromethoxybenzaldehyde 52 followed a similar protocol used for the synthesis of 2-phenoxy derivative 31. Thus, 5-((tert-butyl(dimethyl)silyl)oxymethyl)-2-(trifluoromethoxy)phenol (44) was converted to the 3-(4-nitrophenoxy) derivative 50. Reduction to the corresponding aniline derivative followed by diazotization gave 3-phenoxy-4-trifluoromethoxybenzylic alcohol 51 in 52% yield. Oxidation of 51 with PCC in CH2Cl2 produced the required aldehyde 52.
3-Morpholino- and 3-(1-pipiridyl)-substituted 4-trifluoromethoxybenzaldehydes 55a–b were synthesized following a similar sequence of reaction used in the synthesis of 40a–b. Thus, Buchwald coupling of the morpholine and piperidine with 2-(3-chloro-4-(trifluoromethoxy)phenyl)-[1,3]-dioxolane 54 and subsequent deprotection of the acetal afforded the required aldehydes 55a–b in moderate yields. Methyl-5-formyl-2-(trifluoromethoxy)benzoate 48 was synthesized from 42 in three steps. Lithiation of 42 with sec-BuLi at −78 °C followed by the addition of methylchloroformate, subsequent deprotection of the TBS ether with TBAF and PCC mediated oxidation of the benzylic alcohol afforded 48.
Reductive amination of these aldehydes (47a–b, 48, 49a–b, 52, 55a–b) with amine 7 was carried out in presence of NaCNBH3 in DMF containing AcOH to provide the nitroimidazole derivatives 56a–h in 15–55 % yields. Deprotection of the O-methoxymethyl ether 56d was carried out as mentioned previously to provide 56i. Syntheses of 1 derivatives 59a–c with R3 modification having 3-F, 3-OMe and 3-OMOM substitutions was achieved by alkylation of alcohol 57 with the corresponding benzylic bromides 58a–b (Scheme 6). Deprotection of the O-methoxymethyl ether in 59a was carried out using 6N HCl in THF to afford 59d.
Scheme 6.

Reaction conditions: i) NaH, DMF, −78 °C – rt; ii) 6N HCl, THF, rt, 4 h.
Results and Discussion
R1 modifications: SAR of amides, ureas and tertiary amines
Both benzyl ether and amine analogues of nitroimidazooxazines have been shown to be equipotent against M. tuberculosis. The amenability of the benzylic amine in 2 allowed us to explore further modifications in the series shown in Table 1. N-formylation (3) reduced both cellular activity and efficiency as a substrate for Ddn by two-fold compared to the parent compound 2 whereas N-acetylation of the amino group was detrimental to both MIC and MAC and reduced the catalytic efficiency of this as a substrate for Ddn by nearly ten-fold. The N-propionyl derivative 4b was insoluble and could not be evaluated. N-aroyl amide derivatives behaved in a similar fashion with both benzoyl (4c) and -chlorobenzoyl (ortho, meta and para) derivatives (4d–f) resulting in compounds that were significantly less and potent against replicating and non-replicating Mtb. We also explored one urea derivative (6) but did not elaborate on the series after the observation that reducing the basicity of nitrogen resulted in less potent compounds.
Table 1.
Minimum Inhibitory Concentration (MIC) and Minimum Anaerobicidal Concentration (MAC) of R1 modifications:
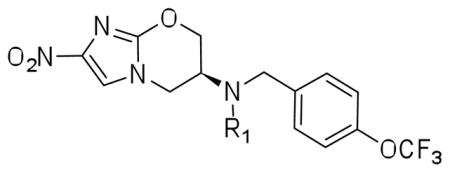 | ||||
|---|---|---|---|---|
| No | R1 | Mtb MIC99 μM (±SD) | Mtb MAC μM (±SD) | Ddn kcat/KM |
| 1 | -- | 0.63±0.22 | 8.8±3.4 | 0.15 |
| 2 | −H | 0.36±0.05 | 14±2 | 0.17 |
| 3 | N-Formyl | 0.79±0.01 | 12.5±8.8 | 0.07 |
| 4a | N-Acetyl | 50±0.0 | >50 | 0.02 |
| 4b | N-propionyl | Insoluble | ND | ND |
| 4c | Benzoyl | 8.8±3.4 | >50 | 0.16 |
| 4d | o-Chlorobenzoyl | 5.6±1.4 | >50 | 0.13 |
| 4e | m-Chlorobenzoyl | 7.5±2.8 | 21±7 | 0.25 |
| 4f | p-Chlorobenzoyl | 1.3±0.5 | 7.8±3.1 | 0.37 |
| 5a | Me | 0.15±0.05 | 2.5±0.9 | 0.25 |
| 5b | nPr | 0.11±0.06 | 3.8±1.4 | 0.32 |
| 5c | iPr | 0.34±0.05 | 11±3 | 0.04 |
| 6 |
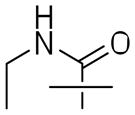
|
17±7 | 69±38 | 0 |
The catalytic activity of the simple amide substituted molecules as substrates for the Ddn enzyme, measured as kcat/KM for reoxidation of reduced F420, was generally lower than that of the 1, however, within this group there was in general only a weak correlation between enzymatic activity and MIC. The same was not true of the N-aroyl derivatives, some of which seem to be significantly better substrates for the enzyme. However this improvement in substrate efficiency did not translate into improvements in MIC. One potential explanation for this phenomenon could be that the aroyl substituent, with its similarity to the p-trifluoromethoxybenzyl amine substituent, competes with this group and alters the binding mode of the substrate, altering product formation and hence efficacy. Alternatively these could be transported into the cell less efficiently reducing the effective intracellular concentration.
The tertiary amine analogues (5a–c) showed significantly improved potency in comparison with the amide derivatives. Higher alkyl derivatives such as N-n-Pr derivative 5b showed a six-fold increase in the potency compared to 1 and was similar in potency to the N-Me derivative (5a). Both 5a and 5b were 2-fold more potent against anaerobic Mtb. However, the N-i-Pr derivative 5c was less active against both replicating and non-replicating Mtb when compared to N-nr-Pr 5b, which indicates that branched chains may not be suitable at this position. For 5a and 5b improvements in MIC were paralleled by increases in kcat/KM suggesting that the binding mode of the substrate was maintained to optimize aerobic activity. While these tertiary amines showed improvements in the overall activity profile, the intrinsic clearance of these compounds was unfortunately very high in mouse liver microsomes and we discontinued exploration of this series (Table 4).
Table 4.
Stability in mouse liver microsomes and solubility
| No | Structure | ClogP | Solubility pH 6.8 μg/mL | Mouse liver microsomes
|
||
|---|---|---|---|---|---|---|
| CL μl.min −1mg−1 | t1/2 min. | Predicted hepatic extraction ratio21 (%) | ||||
| 5a | N-Me | 3.2 | 10 | 124.9 | 11 | 85 |
| 5b | N-nPr | 4.2 | <2 | 407 | 3.4 | 95 |
| 5c | N-iPr | 4.1 | <2 | 433 | 3.2 | 95 |
| 32d | 2-OPh | 4.7 | 3 | 216.6 | 6.4 | 90 |
| 32e | 2-O-cyclopropylmethyl | 3.7 | 6 | 866.3 | 1.6 | 97 |
| 32f | 2-O-cyclopropyl | 3.3 | 11 | 150.7 | 9.2 | 87 |
| 32g | 2-OH | 2.1 | 25 | 96.9 | 14.3 | 81 |
| 41a | 2-F | 2.9 | 72 | 95.6 | 14.5 | 81 |
| 41b | 2-Cl | 3.4 | 30 | 69.7 | 19.9 | 75 |
| 56a | 3-F | 2.7 | 43 | 58.7 | 23.6 | 72 |
| 56b | 3-Cl | 3.2 | 77 | 103.4 | 13.4 | 82 |
| 56i | 3-OH | 1.9 | <2 | 61.9 | 22.4 | 73 |
| 56e | 3-OPh | 4.3 | 56 | 149 | 9.3 | 87 |
R2 modifications: flat SAR and a potential site for metabolic tailoring
Our efforts to systematically optimize the benzylic position by incorporating lower alkyl groups (17b, 17d–f) resulted in compounds with either comparable or very slightly enhanced activity compared to 1 against both replicating and non-replicating Mtb (Table 2). Elongation of the alkyl chain from methyl to n-butyl resulted in very slight enhancements in potency (two-fold) suggesting the presence of a hydrophobic pocket but the SAR here was notably flat, and the introduction of a hydroxymethyl group at this position resulted in compound (17h) which had activity profile similar to 2. The derivatives with hydroxyethyl (17i) and carboxamide (17g) were 5- and 1.5-fold more potent respectively against replicating Mtb although their potency against anaerobic bacilli was compromised. Introduction of heterocycles such as oxazole (17a) or imidazole (17c) led to compounds with similar potency against both replicating and non-replicating Mtb. In general, the presence of more polar groups resulted in the reduction of activity against non-replicating Mtb whereas the potency against replicating Mtb remained in the range of 0.15–0.4 μM. Although the stereochemistry of the C-6 substituent is critical for 1 activity (the S-isomer being 100 fold more potent than the R isomer), the introduction of another chiral centre at the benzylic position does not seem to affect the activity as testing the individual diastereoisomers showed equivalent activity in all assays. This flatness of the SAR is mirrored in the kinetics of F420 reoxidation when they were tested as substrates for Ddn. The kcat/KM was comparable to the parent compound except for the compounds (17e, 17f) whose kcat/KM was 0.31 and 0.4 respectively (Table 2). This two-fold increase in substrate efficiency translated into a slight improvement in MIC only for compound 17f.
Table 2.
Minimum Inhibitory Concentration (MIC) and Minimum Anaerobicidal Concentration (MAC) of R2 modifications:
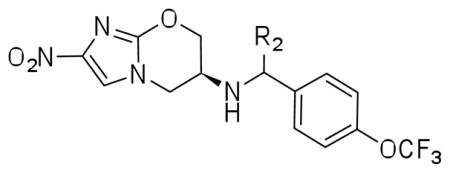 | ||||
|---|---|---|---|---|
| No | R2 | Mtb MIC99 μM (±SD) | Mtb MAC μM (±SD) | Ddn kcat/KM |
| 1 | −H | 0.63±0.22 | 8.8±3.4 | 0.15 |
| 2 | −H | 0.36±0.05 | 14±2 | 0.17 |
| 17a | 2-Oxazole | 0.40±0.18 | 7.8±3.1 | 0.20 |
| 17b | Methyl | 0.44±0.16 | 5.6±1.4 | 0.09 |
| 17c | 2-Imidazole | 0.52±0.23 | 17±7 | 0.17 |
| 17d | Et | 0.36±0.18 | 3.1±2.2 | 0.14 |
| 17e | nPr | 0.27±0.07 | 3.8±1.4 | 0.31 |
| 17f | nBu | 0.15±0.00 | 4.4±1.7 | 0.40 |
| 17g | CONH2 | 0.41±0.35 | 30±11 | 0.12 |
| 17h | −CH2OH | 0.30±0.22 | 11±3 | 0.15 |
| 17i | −CH2CH2OH | 0.13±0.04 | 23±6 | 0.21 |
Benzylic positions in general are susceptible to metabolism (Metasite predictions, for example, highlight the benzylic carbon of 1 as the primary site of metabolic liability). Blocking of benzylic position would make this position metabolically more stable. The flat SAR observed at this position suggests that this does not form an important contact site for Ddn and therefore raises the possibility of further manipulation of this center to alter metabolic stability or other metabolic properties without compromising activity.
R3 modifications: Hydrophobic groups preferred
Significant improvements in potency were realized upon substituting at the 2-position of the aromatic ring of the benzyl ether (Table 3A). Substitution using halogens (41a–c) as well as hydroxyl (32g) groups improved MIC to lower than 100nM. Phenoxy (32d) and cyclopropyloxy (32f) produced the most potent compounds in this series, showing MIC values down to 60nM. The corresponding benzyloxy derivative (32b) was less potent against both aerobic and anaerobic Mtb, but still three times as potent as 1. Similarly, cyclopropylmethoxy (32e) group was tolerated less well in both cases suggesting an optimal spacing between a cyclic hydrophobe and the ether oxygen that may play a critical role in binding to Ddn. Consistent with this hypothesis, both cyclopropylmethoxy and benzyloxy substituents are poorer substrates for Ddn by comparison with their correspondingly shorter analogs. In fact, phenoxy substituted 32d was the best substrate for Ddn seen in the present study with a kcat/KM of 0.42. Compounds 41d–e bearing a piperidine and morpholine group also showed improved MIC, while N-methylpiperazine analogue 41f resulted in the least aerobically potent compound among these amine-bearing substitutions. All three compounds in this series resulted in disproportionate loss of anaerobic activity and an inconsistent correlation with their efficiency as substrates for Ddn.
Table 3A.
Minimum Inhibitory Concentration (MIC) and Minimum Anaerobicidal Concentration (MAC) of R3 modifications:
 | ||||
|---|---|---|---|---|
| No | R3 | Mtb MIC99 μM (±SD) | Mtb MAC μM (±SD) | Ddn kcat/KM |
| 1 | −H | 0.63±0.22 | 8.8±3.4 | 0.15 |
| 2 | −H | 0.36±0.05 | 14±2 | 0.17 |
| 32b | −OBn | 0.24±0.19 | 7.5±2.8 | 0.31 |
| 32c | −OMe | 0.23±0.07 | 5.2±1.8 | 0.15 |
| 32d | −OPh | 0.06±0.02 | 4.4±1.7 | 0.42 |
| 32e |
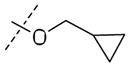
|
0.10±0.04 | 4.2±1.8 | 0.37 |
| 32f |
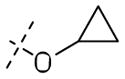
|
0.06±0.02 | 2.0±0.8 | 0.31 |
| 32g | −OH | 0.09±0.01 | 4.4±1.7 | 0.18 |
| 41a | −F | 0.13±0.06 | 2.5±0.9 | 0.20 |
| 41b | −Cl | 0.12±0.05 | 2.1±0.9 | 0.27 |
| 41c | −Br | 0.09±0.01 | 2.0±0.8 | 0.25 |
| 41d |
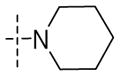
|
0.21±0.13 | 11±3 | 0.42 |
| 41e |
|
0.15±0.05 | 10±4 | 0.19 |
| 41f |
|
2.2±0.5 | 33±14 | 0.17 |
The 3-position of the trifluoromethoxyphenyl ring likewise tolerated most of the substituents tested, resulting in compounds with superior potency to 1. Halogen substitutions (56a–b, 59b) at the 3-position of the phenyl ring exhibited improved potency similar to that seen in the case of the corresponding 2-halo substituted analogs (Table 3B). Compounds 56i and 59d both bear 3-OH moieties and differ in being the analogous benzyl ether and benzyl amines and behave identically in all assays, showing comparable aerobic and anaerobic potency to 1. Compounds 56c and 59c are the corresponding methyl ethers and unremarkable except in the distinction that the amino analog was slightly superior to the ether analog. This compound, 56c however does not show a significant increase in kcat/KM suggesting that the effect may be complex. Surprisingly, the 3-phenoxy substituted compound 56e showed relatively similar activities to the hydroxy and methoxy substituents. The methyl carboxylate (56f), morpholine (56g) and piperazine (56h) all showed comparable cellular activity as well as enzymatic activity resulting in a very flat picture of the SAR at the 3-position.
Table 3B.
Minimum Inhibitory Concentration (MIC) and Minimum Anaerobicidal Concentration (MAC) of R3 modifications:
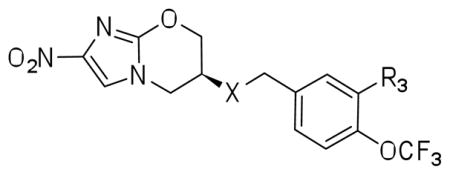 | |||||
|---|---|---|---|---|---|
| No | R3 | X | Mtb MIC99 μM (±SD) | Mtb MAC μM (±SD) | Ddn kcat/KM |
| 1 | −H | O | 0.63±0.22 | 8.8±3.4 | 0.15 |
| 2 | −H | NH | 0.36±0.05 | 14±2 | 0.17 |
| 56a | −F | NH | 0.12±0.07 | 4.2±1.8 | 0.21 |
| 59b | −F | O | 0.17±0.03 | 7.5±2.8 | 0.33 |
| 56b | −Cl | NH | 0.12±0.07 | 4.2±1.8 | 0.31 |
| 56c | −OMe | NH | 0.07±0.02 | 2.1±0.9 | 0.19 |
| 59c | −OMe | O | 0.13±0.04 | 4.2±1.8 | 0.24 |
| 56i | −OH | NH | 0.16±0.06 | 7.5±2.8 | 0.13 |
| 59d | −OH | O | 0.14±0.03 | 8.3±3.6 | 0.14 |
| 56e | −OPh | NH | 0.14±0.06 | 3.9±1.6 | 0.13 |
| 56f | −COOMe | NH | 0.14±0.03 | 11±3 | 0.17 |
| 56g |
|
NH | 0.15±0.05 | 7.0±3.9 | 0.10 |
| 56h |
|
NH | 0.17±0.02 | 7.8±3.1 | 0.16 |
In general, ortho and meta positions of the trifluoromethoxyphenyl ring exhibited similar activity profile for the groups tested, except for –OMe and N-methylpiperazine groups, for which a preference for meta position was observed. The loss of potency against non-replicating Mtb was more significant when amine substituents were used in the R3 modification. The activity of these analogs was not dependent on the nature of the substituents tested. Although benzylic ether analogs and their amine derivatives exhibited similar profile in their activities against both replicating and non-replicating Mtb, the amino series exhibited a better solubility profile and a 2-fold improvement in potency.
Solubility, microsomal stability and in vivo clearance rates in mice
Solubility and stability in mouse liver microsomes were determined for selected potent compounds (Table 4). Hydroxyl group and halogen substitution on the aryl ring resulted in compounds with reasonable solubility while N-alkyl derivatives such as 5a–c and ether derivatives such as 32d–f and 56e had significantly reduced solubility. This is probably due to the higher lipophilicity of these derivatives as reflected in their ClogP values. Metabolic stability studies in mouse liver microsomes revealed high clearance rates for the more lipophilic compounds. Although the 3-Cl derivative 56b and 2-F derivative 41a were the most soluble compounds, their intrinsic clearance was comparatively higher than 56a and 56i (3-F and 3-OH derivatives respectively) that exhibited the most attractive metabolic stability. We used these intrinsic clearance rates to calculate a predicted hepatic extraction ratio21. Based on these values we attempted to verify whether low in vitro clearance would be translated in vivo. We therefore performed in vivo pharmacokinetic studies with compounds 56a, 56b and 56i. Following intravenous injection at 5mg/kg, compounds 56a and 56b showed low to moderate clearance with good systemic exposure and elimination half-lives of 1.3 and 4.3h, respectively. Compound 56i on the other hand was cleared more rapidly with a half-life of 0.4h resulting in low systemic exposure. This relative disconnect between in vitro and in vivo clearance for 56i is suspected to be due to glucuronidation which is not captured in microsomal stability studies that only measure Phase I metabolism.
Ddn may prefer substrates in a pseudo-equatorial conformation
We have previously shown that in this bicyclic system the lipophilic tail can adopt a pseudo-axial or pseudo-equatorial conformation at C-6 and that the preferred form in crystalline 1 was pseudo-axial. In addition, we found that 7R-methylated 1 crystallized in a pseudo-equatorial conformation22. In order to investigate the energetics of 1 in solution, we have calculated the Gibbs free energy of both conformers of 1 using density functional theory at the level of B3LYP/6-31G* 23 in a solvent reaction field of cyclohexane. These calculations reveal that the pseudo-axial form is 0.9 kcal/mol more stable than the pseudo-equatorial form indicating that only about 18% of 1 would be in the pseudo-equatorial form in solution. Furthermore, the calculated energy barrier between the two conformers is less than 5 kcal/mol suggesting that both conformers exist in solution at room temperature with rapid inter-conversion on the nanosecond time scale. Interestingly, the ortho substituted R3 derivatives yielded the largest improvements in potency with two candidates in the 60 nM range with substantial improvements in their activity as substrates for Ddn. Fig. 2A (showing the pseudo-equatorial conformer) and Fig. 2B (showing the pseudo-axial conformer) show an overlay of the geometry optimized ortho substituted analogs including two promising molecules (2-phenoxy-(32d) and cyclopropyloxy-(32f)) as well as the methoxy-(32c), chloro-(41b), and N-methylpiperazino-(41f) analogs. Fig. 2A and Fig. 2B depict an overlay of the head portion of each ortho analog with 1 to illustrate the conformational deviation of the tail portion of each derivative. In general the tail group of the pseudo-equatorial conformers better overlap with 1 than the tail groups of the pseudo-axial conformers. In terms of energetics, relative to 1, each ortho substitution stabilizes the equatorial form from 0.5 kcal/mol (32c) to 1.6 kcal/mol (41f) as seen in the values of ΔH. Additional stabilization of the equatorial conformation from 0.1 kcal/mol (32d) to 0.9 kcal/mol (32C) arises from the larger vibrational entropy as seen in the calculated ΔG values; the equatorial conformation, which is more extended in molecular shape than the axial, is more flexible and thus tends to have larger vibrational entropy. This stabilization energy directly translates into an increase in the concentration of the pseudo-equatorial conformer at equilibrium; for example, about 70 % of compound 41b would be in the pseudo-equatorial form in cyclohexane. Accordingly, it can provide a rationale at a qualitative level for the 10 to 20 fold enhanced potency of the ortho-substituted compounds listed in Table 3a provided that Ddn preferentially recognizes the pseudo-equatorial conformation. With larger and more polar substituents such as 41f there might be other factors (e.g., steric repulsion) at the binding site of Ddn contributing to its relative inactivity. Besides the increase in the concentration of the pseudo-equatorial conformer, the improved catalytic efficiency of several of these compounds, including 2-phenoxy (32d), cyclopropoxy (32f), Cl (41b), and Br (41c) suggests that these groups might well increase either the polarizability of the tail group by donating π-electron density (e.g., cyclopropoxy) and/or the separation of the electrostatic charge on the tail group by an electron withdrawing group (e.g., Cl). This increased polarizability and/or charge separation might be responsible for enhancing potential-π stacking interactions24,25 with an aromatic residue at the binding site, resulting in improved catalytic efficiency of 32d, 32f, 41b, and 41c.
Figure 2.
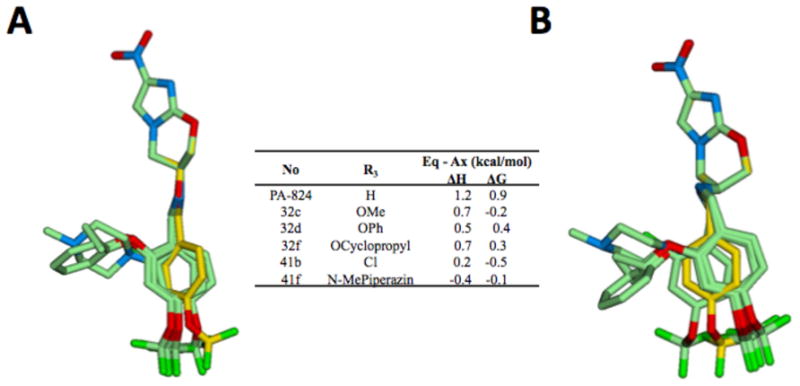
Superpositions of PA-824 (1) and some R3 derivatives that were geometry optimized at the level of B3LYP/61-31G*. In each the head, tail and linker region of 1 is shown with yellow carbons, A the tail is shown in the pseudo-equatorial conformation while in B the tail is shown in the pseudo-axial conformation. A and B show R3 analogs 32c, 32d, 32f, 41b, and 41f. The inset table shows the difference in enthalpy (ΔH) and Gibbs free energy (ΔG) at 298.15 K calculated for the pseudo-equatorial conformer compared with the pseudo-axial conformer for each compound with a solvent reaction field of cyclohexane. Atoms represented by colors are as follows: green, carbon; dark green, fluorine or chlorine; blue, nitrogen; red, oxygen. Hydrogen atoms are not shown.
Conclusions
We have explored three positions of diversity in the 4-(trifluoromethoxy)benzylamino tail region of 1 using our previously described 3D-QSAR pharmacophore model for predicting MICs of compounds in this series. All of these analogs were predicted by our previously described QSAR model to be highly active (MIC <1μM) and in general this was the case. In the case of the N-acylated and N-alkylated derivative in Table 1, however, there were some notable exceptions. Two of the three positions (R1 and R3) provided substantial SAR and improved lead compounds in terms of both potency and solubility, while one site provided little SAR (R2). This is nonetheless a useful observation since this position could be potentially manipulated to modulate the pharmacological properties of additional derivatives. A recent report on a series of biphenyl analogs of these compounds highlighted the importance of microsomal stability in determining in vivo activity of compounds in this series17. In this study, we have confirmed good in vitro metabolic stability with in vivo pharmacokinetic studies. Based on our results, a subset of these compounds are both stable and soluble enough to merit further testing in animals. In general the QSAR model was successful in predicting active compounds with the exception of compounds substituted at the benzylic nitrogen that are likely to affect the conformation of the oxazine ring. The present study provides an additional insight into the preferential binding mode of 1 to Ddn based on the conformational energetics, and this should facilitate further optimization of this exciting class of molecules for the treatment of TB.
Experimental Section
General methods
Reagents and solvents were purchased from Aldrich, Acros, or other commercial sources and used without further purification. Thin layer chromatography (TLC) was carried out on precoated silica gel 60 F254 plates from Merck. Compounds were visualized under UV light, or phosphomolybdic acid (PMA) stain. NMR spectra were obtained on a Varian 400 MHz Oxford NMR. Preparative HPLC separation was performed using Agilent reverse phase HPLC with Atlantis dC18 column, 19×250mm, 10μm. Unless otherwise noted, purity of compounds was established to be >95% by LC/MS (Aquity UPLC with PDA detector and ELSD using Waters Quattro Micro-API Micromass with multimode ionization as detector and Acquity BEH C18, 50 × 2.1 mm, 1.7 μm column) and Waters Aquity UPLC with PDA detector, using Acquity BEH C18, 100 × 2.1 mm, 1.7 μm column. Column chromatography was carried out on silica gel (100–200 mesh). MIC, MAC and enzyme kinetic assays were carried out as previously described14. MIC, MAC and enzyme assays were carried out multiple times in two independent laboratories and discrepancies were repeated until concordant results were obtained. Values reported are averages of multiple measurements and reported errors are standard deviations.
(S)-N-(2-Nitro-6,7-dihydro-5H-imidazo[2,1−b][1,3]oxazin-6-yl)-N-(4-trifluoromethoxybenzyl)-formamide (3)
Acetic anhydride (26 μL, 0.28 mmol) was added to formic acid (0.11 mL, 2.78 mmol) at 0 °C. After 30 min at 0 °C, a solution of ((S)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-yl)-(4-trifluoromethoxybenzyl)amine 2 (50 mg, 0.14 mmol) in THF (1 mL) was added to the mixture and stirred for 1h. The solvent was removed in vacuo and the residue partitioned between EtOAc and water. The organic layer was washed with water, brine and dried (anh. Na2SO4) and concentrated under reduced pressure. Chromatographic purification of the residue on silica gel eluting with 1% MeOH in CH2Cl2 gave 3 (30 mg, 55%). 1H NMR (CDCl3): δ 4.03 (dd, 1H, J = 5.6, 12.0 Hz), 4.30–4.40 (m, 2H), 4.52–4.54 (m, 1H), 4.55 (s, 2H), 4.79 (dd, 1H, J = 6.8, 12.0 Hz), 7.17 (s, 1H), 7.23–7.24 (s, 4H), 8.37 (s, 1H); 13C NMR (CDCl3): δ 44.8, 46.2, 50.9, 67.3, 114.8, 121.7, 121.9, 128.6, 129.1, 134.4, 147.0, 149.7, 164.0. HRMS calcd for C15H13F3N4O5 [M+H+] 387.0916, found 387.0926.
(S)-N-(2-Nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-yl)-N-(4-trifluoromethoxybenzyl)-acetamide (4a)
To a solution of 2 (50 mg, 0.14 mmol) in dry DMF (0.5 mL) was added NaH (60% dispersion in mineral oil, 18 mg, 0.42 mmol) at 0 °C. After 0.5 h, acetyl chloride (29 μL, 0.42 mmol) was added and stirring continued at room temperature for 2 h. The reaction mixture was diluted with water (5 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layer was washed with 1N aqueous HCl (2 × 5 mL), water (10 mL), brine (10 mL), dried (anh. Na2SO4) and concentrated under reduced pressure. Chromatographic purification of the residue on silica gel eluting with 1% MeOH in CH2Cl2 gave 4a (28 mg, 50%). 1H NMR (CDCl3): δ 2.24 (s, 3H), 4.08 (dd, 1H, J = 5.6, 12.0 Hz), 4.30–4.40 (m, 2H), 4.52–4.54 (m, 1H), 4.55 (s, 2H), 4.79 (dd, 1H, J = 6.8, 12.0 Hz), 7.19 (d, 2H, J = 8.0 Hz), 7.23 (d, 2H, J = 8.0 Hz), 7.24 (s, 1H).
(S)-N-(2-Nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-yl)-N-(4-trifluoromethoxybenzyl)-propionamide (4b)
To a solution of 2 (50 mg, 0.14 mmol) in dry THF (1 mL) was added NaH (18 mg, 0.42 mmol) at 0 °C. After 10 min, propionyl chloride (36 μl, 0.42 mmol) was added and the reaction mixture heated at 70 °C for 3h. The reaction mixture was diluted with water (5 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layer was washed with water (10 mL), brine (10 mL), dried (anh. Na2SO4) and concentrated under reduced pressure. Chromatographic purification of the residue on silica gel eluting with 1% MeOH in CH2Cl2 gave 4b (30 mg, 52%). 1H NMR (CDCl3): δ 1.19 (t, 3H, J = 7.2 Hz), 2.45 (q, 2H, J = 7.2, 14.4 Hz), 4.08 (dd, 1H, J = 6.0, 12.5 Hz), 4.40 (dd, 2H, J = 7.2, 12.0 Hz), 4.61 (m, 3H), 4.77 (m, 1H), 7.17 (d, 2H, J = 8.0 Hz), 7.24–7.27 (m, 3H).
Using the same procedure, the following compounds were synthesized:
(S)-N-(2-Nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-yl)-N-(4-trifluoromethoxybenzyl)benzamide (4c)
48% yield. 1H NMR (CDCl3): δ 4.08 (dd, 1H, J = 4.8, 12.0 Hz), 4.43–4.46 (m, 2H), 4.60–4.75 (m, 3H), 4.95 (m, 1H), 7.21 (m, 5H), 7.45 (m, 5H). HRMS calcd for C21H17F3N4O5 [M+H+] 463.1229, found 463.1232.
(S)-2-Chloro-N-(2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-yl)-N-(4-trifluoromethoxybenzyl)-benzamide (4d)
51 % yield. 1H NMR (DMSO-d6): δ 4.10–4.20 (m, 1H), 4.20–4.70 (m, 4H), 4.70–5.05 (m, 2H), 7.00–7.70 (m, 8H), 7.91 (s, 1H). HRMS calcd for C21H16ClF3N4O5 [M+H+] 497.0840, found 497.0847. HPLC purity: 88%.
(S)-3-Chloro-N-(2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-yl)-N-(4-trifluoromethoxybenzyl)-benzamide (4e)
55 % yield. 1HNMR (CDCl3): δ 4.09 (dd, 1H, J = 4.8, 12.0 Hz), 4.43–4.61 (m, 2H), 4.65–4.73 (m, 3H), 4.93 (m, 1H), 7.18–7.26 (m, 6H), 7.32 (d, 1H, J = 7.2 Hz), 7.83 (t, 1H, J = 7.6 Hz), 7.45 (br s, 1H). HRMS calcd for C21H16ClF3N4O5 [M+H+] 497.0840, found 497.0829.
(S)-4-Chloro-N-(2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-yl)-N-(4-trifluoromethoxybenzyl)-benzamide (4f)
53% yield. 1H NMR (CDCl3): δ 4.11 (m, 1H), 4.42–4.73 (m, 5H), 4.94 (m, 1H), 7.21–7.41 (m, 9H); 13C NMR (CDCl3): δ 44.8, 49.5, 67.6, 115.1, 122.0, 124.9, 127.3, 128.3, 130.7, 131.2, 134.9, 136.7. HRMS calcd for C21H16ClF3N4O5 [M+H+] 497.0840, found 497.0827. HPLC purity: 91.3%.
(S)-N-Methyl-2-nitro-N-4-trifluoromethoxybenzyl-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (5a)
To solution of 2 (70 mg. 0.196 mmol) in a mixture of methanol (1 mL) and AcOH (0.2 mL) was added aqueous formaldehyde (5.9 μL, 0.196 mmol) followed by NaBH(OAc)3 (24,6 mg, 0.392 mmol). After stirring at room temperature for 3h, the solvent was removed under reduced pressure and the residue partitioned between water and ethyl acetate. The organic layer was washed with water, brine, dried (anh. Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using 10–20% gradient mixture of ethyl acetate/chloroform as eluent to give 5a (20 mg, 27%). 1H NMR (CDCl3): δ 2.35 (s, 3H), 3.34 (m, 1H), 3.74 (ABq, 2H, J = 14.0, 19.0 Hz), 4.15 (m, 2H), 4.53 (m, 2H), 7.18 (d, 2H, J = 8.0 Hz), 7.29 (d, 2H, J = 8.0 Hz), 7.40 (s, 1H); 13C NMR (CDCl3): δ 38.6, 45.3, 53.5, 58.4, 68.0, 115.0, 121.4, 129.7, 136.8, 147.8, 148.8. HRMS calcd for C15H15F3N4O4 [M+H+] 373.1124, found 373.1110. HPLC purity: 93.5%.
(S)-2-Nitro-N-propyl-N-(4-trifluoromethoxybenzyl)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (5b)
To a stirred solution of 2 (75 mg, 0.3 mmol) in THF (2 mL) containing AcOH (0.5 mL) was added propionaldehyde (53 μL, 0.73 mmol)) and Ti(i-OPr)4 (0.27 mL, 0.9 mmol) at room temperature. After 30 min, NaBH(OAc)3 (190 mg, 0.9 mmol) was added and the reaction was stirred for 15h. The reaction mixture was neutralized with sat. aq. NaHCO3 and extracted with EtOAc (3 × 20 mL). The combined organic layer was washed with water (10 mL), brine (10 mL), dried (anh. Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using gradient mixture of 0.5–1% MeOH in CHCl3 to afford 75 mg of 5b (64%). 1H NMR (DMSO-d6): 0.77 (t, 3H, J = 7.2 Hz), 1.38 (m, 2H), 2.50 (m, 2H), 3.44 (m, 1H), 3.77 (ABq, 2H, J = 14.8, 20.4 Hz), 4.13 (m, 2H), 4.52 (d, 2H, J = 6.0 Hz), 7.29 (d, 2H, J = 8.0 Hz), 7.39 (d, 2H, J = 8.0 Hz), 7.98 (s, 1H). HRMS calcd for C17H19F3N4O4 [M+H+] 401.1437, found 401.1434.
(S)-N-Isopropyl-2-nitro-N-(4-trifluoromethoxybenzyl)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (5c)
To a stirred solution of 2 (100 mg, 0.279 mmol) and acetone (0.2 ml, 1.39 mmol) in methanol (1.5 mL) containing AcOH (1.5 mL) was added titanium isopropoxide (0.5 mL, 1.67 mmol) at room temperature. After stirring for 2h, NaBH(OAc)3 (235 mg, 1.1 mmol) was added and the reaction was stirred for 3 days. Saturated aq. NaHCO3 was added and the reaction mixture extracted with ethyl acetate (2 × 75 mL). The organic layer was washed with water, brine, dried (anh. Na2SO4) and then concentrated under reduced pressure. The residue was purified over silica gel column chromatography using 1% MeOH in CHCl3 as eluent to give 5c (30 mg, 29%). 1H NMR (CDCl3): δ 1.13, 1.14 (2 d, 6H, J = 6.8 Hz), 3.15 (m, 1H), 3.55 (m, 1H), 3.73 (d, 1H, J = 15.2, Hz), 3.86 (d, 1H, J = 15.2 Hz), 3.99 (m, 2H), 4.31 (dd, 1H, J = 8.8, 11.2 Hz), 4.47 (dd, 1H, J = 2.4, 11.2 Hz), 7.16 (d, 2H, J = 8.0 Hz), 7.25 (s, 1H), 7.29 (d, 2H, J = 8.0 Hz); 13C NMR (CDCl3): δ 19.3, 20.8, 46.2, 49.1, 49.2, 68.7, 115.0, 119.0, 121.0, 128.6, 138.7, 143.6, 147.5, 148.3. HRMS calcd for C17H19F3N4O4 [M+H+] 401.1437, found 401.1451.
(S)-3-Ethyl-1-(2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-yl)-1-4-trifluoromethoxybenzyl)-urea (6)
To a stirred solution of ethylamine hydrochloride (42 mg, 0.52 mmol) and triphosgene (312 mg, 1.05 mmol) in dry THF (4 mL) at 0°C was added Et3N (0.5 mL, 2.1 mmol). After 15 min. a solution of 2 (75 mg, 0.2 mmol) in THF (2 mL) was added and the resulting mixture was stirred at room temperature for 2h. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layer was washed with water (10 mL), brine (10 mL), dried (anh. Na2SO4) and then concentrated under reduced pressure. The residue thus obtained (50 mg) was re-dissolved in DMF (3 mL) and treated with Et3N (83 μL, 0.6 mmol) followed by ethylamine hydrochloride (49 mg, 0.6 mmol). After stirring for 3h at room temperature, the reaction mixture was diluted with water (10 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layer was washed with brine (10 mL), dried (anh. Na2SO4) and then concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel using 1–2% MeOH in CHCl3 as eluent to afford 35 mg (66%) of 6. 1H NMR (DMSO-d6): 0.99 (t, 3H, J = 7.2 Hz), 3.08 (m, 2H), 4.15 (d, 2H, J = 7.2 Hz), 4.36 (dd, 1H, J = 3.2, 10.4 Hz), 4.46–4.68 (m, 4H), 6.78 (t, 1H, J = 4.8 Hz), 7.26 (d, 2H, J = 8.8 Hz), 7.30 (d, 2H, J = 8.8 Hz), 7.98 (s, 1H); 13C NMR (CDCl3): δ 15.4, 36.1, 45.8, 48.4, 49.1, 68.5, 115.2, 122.1, 127.3, 135.2, 143.9, 157.6.
Oxazol-2-yl-(4-trifluoromethoxyphenyl)methanone (9a)
To a solution of oxazole (600 mg, 8.69 mmol) in dry THF (10 mL) at −78 °C was added n-BuLi (2.9 M in hexane, 3.3 mL, 9.57 mmol). After 30 min at −78° C, ZnCl2 (0.5M in THF, 13.7 mL, 17.39 mmol) was added and the mixture warmed to room temperature. After 45 min, CuI (1.65 g, 8.69 mmol) was added and stirring was continued for 10 min. A solution of 4-trifluoromethoxybenzoylchloride [obtained by refluxing 4-trifluoromethoxybenzoic acid (2.14g, 9.57 mmol) in dry THF (15 mL) containing oxalyl chloride (4.16 mL) and cat. pyridine for 3h followed by evaporation] in THF (15 mL) was added at room temperature and the mixture was stirred for 1h. The reaction mixture was diluted with water (5 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layer was washed with water (10 mL), brine (10 mL), dried (anh. Na2SO4) and concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel using gradient of 1–2% MeOH in CHCl3 to afford 9a (900 mg, 40%). 1H NMR (CDCl3): δ 7.35 (d, 2H, J = 8.0 Hz), 7.44 (s, 1H), 7.93 (s, 1H), 8.60 (d, 2H, J = 8.0 Hz). ESI MS: m/z 258.0 (M+H).
(6S)-2-Nitro-N-(oxazol-2-yl(4-trifluoromethoxyphenyl)methyl)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (17a)
To a stirred solution of oxazol-2-yl-(4-(trifluoromethoxy)phenyl)methanone (280 mg, 1.09 mmol) and amine 7 (200 mg, 1.09 mmol) in dry THF (4 mL) containing AcOH (2 mL) was added Ti(i-OPr)4 (0.98 mL, 3.27 mmol) and stirred at room temperature. After 15 h, NaBH3CN (203 mg, 3.27 mmol) was added and stirring continued for 2 h. The reaction mixture was quenched with water, neutralized to pH 7 (sat. aq. NaHCO3) and extracted with EtOAc (3 × 20 mL). The combined organic layer was washed with water (10 mL), brine (10 mL) dried (anh. Na2SO4) and concentrated under reduced pressure. The residue was purified using preparative HPLC to afford 17a (40 mg, 9%) as a mixture of diastereomers. 1H NMR (DMSO-d6): δ 3.20 (m, 1H), 3.51–3.57 (m, 1H), 4.06–4.14 (m, 2H), 4.37–4.46 (m, 2H), 5.39 (d, 1H, J = 9.0 Hz), 7.19 (s, 1H), 7.34–7.37 (m, 2H), 7.55–7.587 (m, 2H), 7.97 and 8.00 (2s, 1H), 8.07 (s, 1H). HRMS calcd for C17H14F3N5O5 [M+H+] 426.1025, found 426.1025.
(6S)-2-Nitro-N-(1-(4-trifluoromethoxyphenyl)ethyl)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (17b) was prepared in a similar fashion to 17a as a mixture of diastereomers from 9b and amine 7 in 28% yield. 1H NMR (DMSO-d6): δ 1.26 (d, 3H, J = 6.4 Hz), 2.77 (m, 1H), 3.01 (br s, 1H), 3.78 (m, 1H), 3.92–4.07 (m, 2H), 4.19–4.35 (m, 2H), 7.31 (t, 2H, J = 8.0 Hz), 7.49 (t, 2H, J = 8.0 Hz), 7.94 and 8.01 (2s, 1H). FT-ICR HRMS calcd for C15H15F3N4O4 [M+H+] 373.1118, found 373.1120.
(1-Benzyl-1H-imidazol-2-yl)(4-trifluoromethoxyphenyl)methanol (11)
To a stirred solution of N-benzylimidazole (1.0 g, 6.32 mmol) in acetonitrile (15 mL) was added diisopropylcarbamoyl chloride (1.24g, 7.59 mmol), 4-trifluoromethoxybenzaldehyde (1.4 mL, 9.49 mmol) and DIPEA (3.4 mL, 19.61 mmol) at 0 °C under nitrogen atmosphere. The reaction mixture was heated at reflux for 19h, quenched with water (30 mL) and extracted with EtOAc (20 mL × 2). The combined organic layer was washed with water (20 mL), brine (10 mL), dried (anh. Na2SO4) and concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel using a gradient mixture of 0–15% of EtOAc in hexane as eluent to afford 2.2g (73%) of (1-benzyl-1H-imidazol-2-yl)(4-(trifluoromethoxy)phenyl)-methyldiisopropylcarbamate. 1H NMR (DMSO-d6): δ 1.00–1.15 (m, 12H), 3.81 (br s, 2H), 5.24–5.40 (m, 2H), 6.92 (d, 2H, J = 8.0 Hz), 7.06 (d, 2H, J = 8.0 Hz), 7.21 (s, 1H), 7.22–7.35 (m, 5H), 7.41 (d, 2H, J = 8.0 Hz). ESI MS: m/z 476.1 (M+H). To a stirred solution of the above product (2.2 g, 4.6 mmol) in THF (20 mL) was added TFA (2 mL) and water (2 mL). After heating at reflux for 15h, the mixture was quenched with aqueous NaHCO3 (30 mL) and extracted with EtOAc (2 × 20 mL). The combined organic layer was washed with water (20 mL), brine solution (10 mL), dried (anh. Na2SO4) and concentrated under reduced pressure to afford 1.32 g (82%) of 11. 1H NMR (DMSO-d6): δ 5.21 (s, 2H), 5.92 (d, 1H, J = 4.0 Hz), 6.38 (d, 1H, J = 4.0 Hz), 6.83 (br s, 1H), 6.95–7.10 (m, 3H), 7.23–7.25 (m, 5H), 7.39 (d, 2H, J = 8.0 Hz). ESI MS: m/z 349.1 (M+H).
(1H-Imidazol-2-yl)(4-trifluoromethoxyphenyl)methanol (12)
To a stirred solution of 11 (500 mg, 1.43 mmol) in MeOH (10 mL) was added 5% Pd/C (100 mg) and two drops of AcOH and stirred under hydrogen (balloon) atmosphere at room temperature for 2.5h. The reaction mixture was filtered through celite pad and washed with EtOAc (50 mL). The filtrate was washed with aqueous NaHCO3 solution (20 mL), water (10 mL) and brine solution (10 mL). The organic phase was dried (anh. Na2SO4) and concentrated under reduced pressure to afford 12 (300 mg, 81%). 1H NMR (DMSO-d6): δ 5.76 (s, 1H), 6.26 (s, 1H), 6.89 (s, 2H), 7.30 (d, 2H, J = 8.4 Hz), 7.51 (d, 2H, J = 8.4 Hz), 11.94 (br s, 1H). ESI MS: m/z 259.0 (M+H).
(6S)-N-((1H-Imidazol-2-yl)(4-trifluoromethoxyphenyl)methyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (17c)
To a stirred solution of 12 (300 mg, 1.20 mmol) in CH2Cl2 (10 mL) was added methanesulfonyl chloride (0.11 mL, 1.45 mmol) and triethylamine (0.25 mL, 1.81 mmol) at 0 °C under nitrogen atmosphere and stirred at room temperature for 1h. The reaction mixture was diluted with CH2Cl2 (20 mL) and washed with water (10 mL) and brine solution (10 mL). The organic phase was dried (anh. Na2SO4) and concentrated under reduced pressure to afford 400 mg of mesylate which was re-dissolved in dry THF (3 mL) and added to a previously stirred solution of (2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-yl)-(4-trifluoromethoxybenzyl)amine (0.19 mg, 1.03 mmol) in THF (10 mL) containing NaH (85 mg, 60% dispersion in mineral oil, 2.06 mmol) at room temperature under the nitrogen atmosphere. After 40h, the reaction was quenched with aqueous solution of NaHCO3 (20 mL) and extracted with EtOAc (20 mL × 2). The combined organic layer was washed with water (20 mL), brine (10 mL) and dried (anh. Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using a gradient mixture of 0–10% of MeOH in EtOAc as eluent to afford 17c (65 mg, 15%) as a mixture of diastereomers. 1H NMR (DMSO-d6): δ 3.10–3.30 (m, 2H), 3.90–4.10 (m, 1H), 4.13 (dd, 1H, J = 4.0, 12.0 Hz), 4.30–4.45 (m, 2H), 5.17 (d, 1H, J = 7.6 Hz), 6.60–7.10 (br s, 2H), 7.25–7.35 (m, 2H), 7.51 (dd, 2H, J = 2.4, 8.8 Hz), 7.98 (d, 1H, J = 2.4 Hz), 11.8 (br s, 1H). HRMS calcd for C17H15F3N6O4 [M+H+] 425.1185, found 425.1187.
1-(4-Trifluoromethoxyphenyl)propan-1-ol (14a)
To stirred solution of 4-trifluoromethoxybenzaldehyde (5 g, 26.31 mmol) in THF was added ethylmagnesium bromide (2 M solution, 32.8 mL, 65.7 mmol) at −15 °C and warmed to room temperature over 30 min. After 3h, the reaction mixture was quenched with aqueous NH4Cl and extracted with ethyl acetate. The organic layer was washed with water (2 × 200 mL), brine (200 mL) dried over (anh. Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using a solvent gradient mixture of 0–5% EtOAc in hexane as eluent to afford 14a (3.5 g, 62%). 1H NMR (DMSO-d6): δ 0.81 (t, 3H, J = 7.6 Hz), 1.50–1.70 (m, 2H), 4.48 (m, 1H), 5.24 (d, 1H, J = 4.4 Hz), 7.29 (d, 2H, J = 8.4 Hz), 7.42 (d, 2H, J = 8.4 Hz).
In a similar fashion, the following compounds were synthesized:
1-(4-Trifluoromethoxyphenyl)butan-1-ol (14b)
65% yield. 1H NMR (DMSO-d6): δ 0.88 (t, 3H, J = 7.2 Hz), 1.22–1.36 (m, 2H), 1.34–1.61 (m, 2H), 4.53–4.58 (m, 1H), 5.20 (d, 1H, J = 4.0 Hz), 7.29 (d, 2H, J = 8.0 Hz), 7.43 (d, 2H, J = 8.0 Hz).
1-(4-Trifluoromethoxyphenyl)pentan-1-ol (14c)
To solution of 4-trifluoromethoxy benzaldehyde (3 g, 15.78 mmol) in THF at −78 °C was added n-BuLi (1.6M in hexane, 11 mL, 17.36 mmol) and warmed to room temperature over 30 min. After 2h, the reaction mixture was quenched with ice and extracted with diethyl ether (2 × 60 mL). The combined organic layer was washed with water, brine solution, dried over (anh. Na2SO4) and concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel using a solvent gradient mixture of 0–2% EtOAc in hexane as eluent to afford 14c (3.1 g, 79%). 1H NMR (DMSO-d6): δ 0.82–0.89 (m, 3H), 1.15–1.41 (m, 4H), 1.50–1.63 (m, 2H), 4.51–4.57 (m, 1H), 5.21–5.24 (m, 1H), 7.26–7.32 (m, 2H), 7.40–7.45 (m, 2H).
1-(1-Bromopropyl)-4-trifluoromethoxybenzene (15a)
To a stirred solution of 14a (3.5 g, 15.98 mmol, 1eq) in ether was added PBr3 (0.5 g, 0.75 mL, 7.99 mmol) at 0 °C. After stirring at 0 °C for 1h, the reaction mixture was diluted with cold water and extracted into ether (800 mL). The organic layer was washed with water (150 mL), brine (150 mL), dried over (anh.Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel (100–200mesh) using a 5–10% gradient mixture of EtOAc in hexane as eluent to afford 15a (2.5 g, 51%). 1H NMR (CDCl3): δ 1.00 (t, 3H, J = 6.8 Hz), 2.10–2.30 (m, 2H), 4.80–4.96 (m, 1H), 7.18 (d, 2H, J = 8.4 Hz), 7.41 (d, 2H, J = 8.4 Hz).
In a similar fashion, the following compounds were synthesized:
1-(1-Bromobutyl)-4-trifluoromethoxybenzene (15b)
50% yield. 1H NMR (CDCl3): δ 0.86 (t, 3H, J = 7.6 Hz), 1.26–1.57 (m, 2H), 2.03–2.12 (m, 1H), 2.18–2.29 (m, 1H), 4.94 (t, 1H, J = 7.6 Hz), 7.18 (d, 2H, J = 8.0 Hz), 7.42 (d, 2H, J = 8.0 Hz).
1-(1-Bromopentyl)-4-trifluoromethoxybenzene (15c)
52% yield. 1H NMR (CDCl3): δ 0.87–0.98 (m, 3H), 1.25–1.49 (m, 4H), 2.00–2.40 (m, 2H), 4.90–5.00 (m, 1H), 7.16–7.20 (m, 2H), 7.39–7.44 (m, 2H).
(6S)-2-Nitro-N-(1-(4-trifluoromethoxyphenyl)propyl)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (17d)
To a stirred solution of amine 7 (200 mg, 1.086 mmol) and 1-(1-bromopropyl)-4-(trifluoromethoxy)benzene (500 mg, 1.63 mmol) in acetonitrile was added K2CO3 (449 mg, 3.26 mmol) and KI (16 mg, 0.108 mmol). After heating at reflux for 48h, the solvent was evaporated and the residue partitioned between ethyl acetate and water. The organic layer was washed with brine (150 mL), dried (anh.Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel (100–200mesh) using a solvent gradient mixture of 0–10% MeOH in CHCl3 to afford 17d (100 mg, 25%) as a mixture of diastereomers. 1H NMR (DMSO-d6): δ 0.67, 0.72 (2 t, 3H, J = 7.6 Hz), 1.40–1.70 (m, 2H), 2.70–2.80 (m, 1H), 2.90–3.00 (m, 1H), 3.60–4.10 (m, 3H), 4.10–4.46 (m, 2H), 7.33 (d, 2H, J = 8.0 Hz), 7.47 (d, 2H, J = 8.0 Hz), 7.94 and 8.01 (2s, 1H). HRMS calcd for C16H17F3N4O4 [M+H+] 387.1280, found 387.1267.
In a similar manner, the following compounds were synthesized:
(6S)-2-Nitro-N-(1-(4-trifluoromethoxyphenyl)butyl)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (17e)
10% yield. 1H NMR (DMSO-d6): δ 0.70–0.85 (m, 3H), 1.00–1.25 (m, 2H), 1.35–1.70 (m, 2H), 2.72–2.79 (m, 1H), 2.95 (s, 1H), 3.74–3.83 (m, 1H), 4.01–4.05 (m, 2H), 4.15–4.36 (m, 2H), 7.32 (m, 2H), 7.46–7.49 (m, 2H), 7.96 and 8.02 (2 s, 1H). HRMS calcd for C17H19F3N4O4 [M+H+] 401.1437, found 401.1431.
(6S)-2-Nitro-N-(1-(4-trifluoromethoxyphenyl)pentyl)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (17f)
10% yield. 1H NMR (DMSO-d6): δ 0.75–0.79 (2 t, 3H, J = 5.2 Hz), 1.00–1.23 (m, 4H), 1.42–1.63 (m, 2H), 2.71–2.76 (m, 1H), 2.95 (brs, 1H), 3.67–3.80 (m, 2H), 4.01–4.05 (m, 1H), 4.16–4.34 (m, 2H), 7.30–7.43 (m, 2H), 7.46, 7.48 (2 dd, 2H, J = 8.0 Hz), 7.95 and 8.01 (2s, 1H). HRMS calcd for C18H21F3N4O4 [M+H+] 415.1593, found 415.1600.
2-((S)-2-Nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-ylamino)-2-(4-trifluoromethoxyphenyl)-acetamide (17g)
A mixture of amine 7 (100 mg, 0.543 mmol) and trifluoromethoxybenzaldehyde (206 mg, 1.086 mmol) were heated at 100°C for 5 min. The reaction mixture was brought to room temperature and added TMSCN (216 mg, 2.172 mmol) to the reaction mixture. After heating for 30 min., the reaction mixture was cooled to room temperature and diluted with ethyl acetate (50 mL). Organic layer was washed with water (2 × 10 mL), brine, dried (anh. Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel (100 – 200 mesh) using a solvent gradient of 10–60% EtOAc – hexane as eluent to afford 105 mg (50%) of 2-((S)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-ylamino)-2-(4-trifluoromethoxyphenyl)acetonitrile (16). 1H NMR (DMSO-d6): δ 3.46–3.52 (m, 1H), 3.80–3.95 (m, 1H), 4.00–4.10 (m, 1H), 4.17–4.20 (m, 1H), 4.42–4.59 (m, 2H), 5.33–5.42 (m, 1H), 7.42–7.47 (m, 2H), 7.55–7.61 (m, 2H), 8.00 and 8.08 (2s, 1H). ESI MS: m/z 384.0 (M+H). A solution of 16 (100 mg, 0.257 mmol) in ethanol (5 mL) was purged with HCl gas for 1h at −10 °C. After 15min, reaction mixture was neutralized with saturated NaHCO3 solution and extracted with ethyl acetate (2 × 30mL). The organic layer was washed with water, brine, dried over (anh.Na2SO4) and concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel using a solvent gradient of 15% MeOH in CHCl3 as eluent to afford 40 mg (38%) of 17g as a mixture of diastereomers. 1H NMR (DMSO-d6): δ 3.11–3.16 (m, 2H), 3.95–4.20 (m, 2H), 4.38–4.46 (m, 3H), 7.19 (s, 1H), 7.31–7.35 (m, 2H), 7.52–7.56 (m, 3H), 7.97 and 8.05 (2s, 1H). FT-ICR MS calcd for C15H14F3N5O5 [M+H+] 402.1019, found 402.1023. HPLC purity 95.6%.
2-(4-Methoxybenzyloxy)-1-(4-trifluoromethoxyphenyl)ethanol (19)
To a mixture of 2-(4-trifluoromethoxyphenyl)oxirane (2 g, 9.80 mmol) and p-methoxybenzyl alcohol (2.19 g, 19.60 mmol) was added KOtBu (2.19g, 19.6 mmol) at and heated at 60 °C for 2 h. The reaction mixture was diluted with water (100 mL) and extracted with ethyl acetate (150 mL). The organic layer was washed with water (2 × 50 mL), brine (1 × 50 mL), dried over (anh.Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel (100 – 200 mesh) using a solvent gradient of 0–5% EtOAc in hexane as eluent to afford 19 (1.3 g, 34%). 1H NMR (DMSO-d6): δ 3.35–3.55 (m, 2H), 3.73 (s, 3H), 4.42 (s, 2H), 4.74–4.77 (m, 1H), 5.52 (d, 1H, J = 4.8 Hz), 6.87 (d, 2H, J = 8.8 Hz), 7.19 (d, 2H, J = 8.0 Hz), 7.30 (d, 2H, J = 8.0 Hz), 7.47 (d, 2H, J = 8.8 Hz).
2-Hydroxy-1-(4-trifluoromethoxyphenyl)ethanone (20)
To a solution of 19 (1.3 g, 3.82 mmol) in CH2Cl2 (20 mL) was added pyridinium dichromate (2.6 g, 68.8 mmol) and 4 Å molecular sieves powder and stirred at room temperature for 24h. The reaction mixture was filtered through celite and filtrate concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel using a solvent gradient of 0–30% EtOAc in hexane as eluent to afford 800 mg (62%) of 2-(4-methoxybenzyloxy)-1-(4-(trifluoromethoxy)phenyl)ethanone. This compound (800 mg, 2.36 mmol) was dissolved in CH2Cl2 (10 mL) and added TBDMSOTf (1.08 mL, 4.73 mmol) at 0 °C. After stirring for 15 min. at room temperature, the reaction mixture was quenched with sat NaHCO3 solution and extracted with ethyl acetate (75 mL). The organic layer was washed with water (2 × 50 mL), brine (50 mL) and dried (over anh.Na2SO4) and concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel using a solvent gradient of 0–30% EtOAc in hexane as eluent to afford 20 (450 mg, 86%). 1H NMR (DMSO-d6): δ 4.80 (d, 2H, J = 5.6 Hz), 5.19 (t, 1H, J = 6.0 Hz), 7.51 (d, 2H, J = 8.0 Hz), 8.06 (d, 2H, J = 8.0 Hz).
2-((S)-2-Nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-ylamino)-2-(4-trifluoromethoxyphenyl)-ethanol (17h)
A mixture of amine 7 (300 mg, 1.63 mmol) and 2-hydroxy-1-(4-trifluoromethoxyphenyl)ethanone (430 mg, 1.95 mmol) in ethanol (2 mL) was heated at 70 °C for 1 h. After cooling the reaction mixture to room temperature, AcOH (1 drop) and NaCNBH3 (101.08 mg, 1.63 mmol) were added and stirring continued for 16h. The solvent was removed and the residue partitioned between ethyl acetate and water. The organic layer was washed with sat. aq. NaHCO3 solution (2 × 50 mL), water (50 mL), brine (1 × 50 mL), dried over (anh.Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatogrpahy eluting with 2% MeOH in CHCl3 to afford 30 mg (5%) of 17h as a mixture of diasteromers. 1H NMR (DMSO-d6): δ 2.69 (br s, 1H), 3.09 (s, 1H), 3.30–3.43 (m, 2H), 3.70–4.60 (m, 5H), 4.80–5.00 (m, 1H), 7.20–7.60 (m, 4H), 7.91 and 8.04 (2s, 1H). HRMS calcd for C15H15F3N4O5 [M+H+] 389.1073, found 389.1079.
1−(4−Trifluoromethoxyphenyl)propane−1,3−diol (21)
To a stirred solution of 4-trifluoromethoxybenzaldehyde (6.0 g, 31.57 mmol) in THF (40 mL) and methyl bromoacetate (11.5 mL, 126.31 mmol), Zn powder (16.5 g, 252.6 mmol) was added portion-wise at room temperature and sonicated for 3 h. The reaction mixture was filtered through celite and washed with CH2Cl2 (500 mL). The filtrate was concentrated under reduced pressure and the crude residue purified by column chromatography over silica gel using a solvent gradient of 0–5% EtOAc in hexane as eluent to afford 3 g of ethyl 3-hydroxy-3-(4-trifluoromethoxyphenyl)propanoate. This compound (3 g, 10.8 mmol) was dissolved in anhydrous THF (30 mL) and treated with LiAlH4 (514 mg, 15.15 mmol) at 0 °C. After 2 h, the reaction mixture was quenched at 0 °C with sat. aq. Na2SO4 solution and filtered. The filtrate was washed with water (2 × 75 mL), brine (75 mL), dried over (anh.Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using a solvent gradient of 0–40% EtOAc in hexane as eluent to afford 21 (1.8 g, 25%). 1H NMR (CDCl3): δ 1.80–2.10 (m, 2H), 2.31 (s, 1H), 3.31 (s, 1H), 3.90 (s, 2H), 4.80–5.05 (m, 1H), 7.10–7.30 (m, 2H), 7.35–7.45 (m, 2H). ESI MS: m/z 237.0 (M+H).
3−Hydroxy−1−(4−trifluoromethoxyphenyl)propan−1−one (22)
A solution of compound 21 (1.2 g, 5.08 mmol) in THF (20 mL) was treated with MnO2 (4.2 g, 50.84 mmol) at room temperature. After 6 h, the reaction mixture was filtered through celite, washed with excess CH2Cl2 (100 mL) and concentrated. The residue was purified by column chromatography over silica gel using a solvent gradient of 0–30% EtOAc in hexane as eluent to afford 22 (450 mg, 50%). 1H NMR (DMSO-d6): δ 3.14–3.18 (t, 2H, J = 6.0 Hz), 3.78 (m, 2H), 4.650 (t, 1H, J = 4.8 Hz), 7.51(d, 2H, J = 8.2 Hz), 8.10 (d, 2H, J = 8.2 Hz).
2−((S)−2−Nitro−6,7−dihydro−5H−imidazo[2,1−b][1,3]oxazin−6−ylamino)−2−(4−trifluoromethoxyphenyl)−ethanol (17i)
A mixture of amine 7 (300 mg, 1.630 mmol) and 22 (457 mg, 1.456 mmol) in ethanol (5 mL) was heated at 70 °C for 36 h. After cooling the reaction mixture to room temperature, AcOH (1 drop) and NaCNBH3 (202 mg, 3.26mmol) were added and stirring continued for 4 h. The solvent was removed and the residue partitioned between ethyl acetate and water. The organic layer was washed with sat. aq. NaHCO3 solution (2 × 50 mL), water (50 mL), brine (1 × 50 mL), dried over (anh.Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography eluting with 2% MeOH in CHCl3 to afford 30 mg (5%) of 17i as a mixture of diastereomers. 1H NMR (DMSO-d6): δ 1.69–1.70 (br s, 2H), 2.23 (s, 1H), 2.64 (brs, 2H), 3.24 (s, 1H), 3.92 (d, 1H, J = 12.0 Hz), 4.15 (d, 1H, J = 12.0 Hz), 4.29–4.33 (m, 1H), 4.39–4.45 (m, 1H), 4.66 (s, 1H), 5.39 (m, 1H), 7.28–7.31 (m, 2H), 7.40–7.44 (m, 2H), 8.06 (s, 1H). HRMS calcd for C16H17F3N4O5 [M+H+] 403.1229, found 403.1218.
Ethyl−2−benzyloxy−4−trifluoromethoxybenzoate (27b)
To a stirred solution of ethyl-2-hydroxy-4-trifluoromethoxybenzoate 24 (300 mg, 1.20 mmol) in DMF (4 mL) was added benzyl bromide (0.17 mL, 1.44 mmol) and K2CO3 (331 mg, 2.4 mmol) and heated at 70 °C for 30min. It was diluted with EtOAc (50 mL), washed with water (30 mL × 2) and brine solution (20 mL). The organic phase was separated, dried (anh. Na2SO4) and concentrated under reduced pressure to afford 400 mg (quant.) of 27b. 1H NMR (CDCl3): δ 1.37 (t, 3H, J = 7.2 Hz), 4.36 (q, 2H, J = 3.2, 14.4 Hz), 5.15 (s, 2H), 6.84 (m, 2H), 6.29–7.49 (m, 5H), 7.87 (d, 1H, J = 8.4 Hz). ESI MS: m/z 341.1 (M+H).
Ethyl 2−methoxymethoxy−4−trifluoromethoxybenzoate (27a)
To a solution of ethyl-2-hydroxy-4-trifluoromethoxybenzoate 24 (500 mg, 2.00 mmol) in CH2Cl2 (5 mL) was added diisopropylethylamine (1.09 mL, 6.72 mmol) followed by methoxymethylchloride (0.53 mL, 6.72 mmol) at room temperature. After 3h, the reaction mixture was poured into water and extracted with CH2Cl2 (10 mL). The organic layer was washed with water, brine, dried (Na2SO4) and concentrated under reduced pressure to give crude product which, upon purification by silica gel column chromatography using 2% ethyl acetate in hexane as eluent gave 480 mg (81%) 27a. 1H NMR (CDCl3): δ 1.38 (t, 3H, J = 7.2 Hz), 3.52 (s, 3H), 4.36 (q, 2H, J = 3.2, 14.4 Hz), 5.25 (s, 2H), 6.89 (d, 1H, J = 8.4 Hz), 7.05 (s, 1H), 7.82 (d, 1H, J = 8.4Hz). ESI MS: m/z 295.0 (M+H).
Ethyl (2−cyclopropylmethoxy)−(4−trifluoromethoxy)benzoate (27c)
To a solution of 24 (300 mg, 1.20 mmol) in DMF (4 mL) was added bromomethylcyclopropane (0.17 mL, 1.44 mmol) and K2CO3 (331 mg, 2.4 mmol). After heating at 100 °C for 3h, the reaction mixture was poured into water and extracted with EtOAc (50 mL). The organic layer was washed with water (30 mL × 2), brine solution (20 mL), dried (anh. Na2SO4) and concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel using a gradient mixture of 0–3% of EtOAc-hexane as eluent to afford 27c (300 mg, 82%). 1H NMR (CDCl3): δ 0.39–0.43 (m, 2H), 0.63–0.67 (m, 2H), 1.22–1.29 (m, 1H), 1.37 (t, 3H, J = 7.2 Hz), 3.89 (d, 2H, J = 6.8 Hz), 4.37 (q, 2H, J = 3.2, 14.4 Hz), 6.75 (s, 1H), 6.82 (d, 1H, J = 8.4 Hz), 7.82 (d, 1H, J = 8.4 Hz). ESI MS: m/z 305.0 (M+H).
2−Benzyloxy−4−trifluoromethoxybenzaldehyde (28b)
To a stirred solution of 27b (400 mg, 1.17 mmol) in THF (10 mL) was added LiAlH4 (66 mg, 1.76 mmol) at 0 °C under nitrogen atmosphere. After stirring 0 °C for 1 h, the reaction mixture was quenched with water (30 mL) and then extracted with EtOAc (20 mL × 2). The organic layer was washed with water (20 mL), brine solution (10 mL), dried (anh. Na2SO4) and concentrated under reduced pressure to afford 320 mg of (2-benzyloxy-4-trifluoromethoxyphenyl)methanol which was dissolved in CH2Cl2 (10 mL) and treated with PCC (300 mg) at room temperature under nitrogen atmosphere. After 1h, the reaction mixture was filtered through celite pad and the filtrate concentrated under reduced pressure. The residue was passed through a short pad of silica gel eluting with EtOAc:hexane (1:5) to afford 220 mg (63%) of 28b. 1H NMR (DMSO-d6): δ 5.34 (s, 2H), 7.08 (d, 1H, J = 8.4 Hz), 7.34 (s, 1H), 7.35–7.44 (m, 4H), 7.51 (d, 1H, J = 7.2 Hz), 7.84 (d, 1H, J = 8.4 Hz), 10.35 (s, 1H). ESI MS: m/z 297.0 (M+H).
2−(Methoxymethoxy)−4−(trifluoromethoxy)benzaldehyde (28a)
To a stirred solution of 27a (480 mg, 1.63 mmol) in THF (5 mL) at (0 °C) was added LiAlH4 (74.2 mg, 1.96 mmol) and stirred at room temperature for 30 min. The reaction mixture was quenched with moist ethyl acetate. The organic layer was washed with water, brine, dried (anhyd Na2SO4) and concentrated under reduced pressure. The crude product was filtered through short silica plug using 10% ethyl acetate in hexane as eluent to get 380 mg of (2-methoxymethoxy-4-trifluoromethoxyphenyl)-methanol which was dissolved (380 mg, 1.29 mmol) in CH2Cl2 (5 mL) and treated with pyridinium chlorochromate (760 mg) at room temperature for 1h. The reaction mixture was filtered through celite and the filtrate concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using gradient mixture of 0–2% ethyl acetate in hexanes as eluent to give 28a (350 mg, 62%). 1H NMR (CDCl3): δ 3.54 (s, 3H), 5.31 (s, 2H), 6.94 (d, 1H, J = 8.4 Hz), 7.08 (s, 1H), 7.89 (d, 1H, J = 8.4Hz), 10.43 (s, 1H).
2−Cyclopropylmethoxy−4−trifluoromethoxybenzaldehyde (28c)
To a stirred solution of 27c (280 mg, 0.92 mmol) in THF (10 mL) was added LiAlH4 (52 mg, 1.38 mmol) at 0 °C under nitrogen atmosphere. After stirring 0 °C for 1 h, the reaction mixture was quenched with water (30 mL) and extracted with EtOAc (20 mL × 2). The organic layer was washed with water (20 mL), brine solution (10 mL), dried (anh. Na2SO4) and concentrated under reduced pressure to afford 190 mg of (2-cyclopropylmethoxy-4-trifluoromethoxyphenyl)methanol. This compound was dissolved in CH2Cl2 (10 mL) and added PCC (400 mg) at room temperature under nitrogen atmosphere. After 2h, the reaction mixture was filtered through Celite pad and the filtrate concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using a gradient mixture of 0–2% EtOAc in hexane as eluent to afford 28c (180 mg, 75%). 1H NMR (CDCl3): δ 0.38–0.42 (m, 2H), 0.67–0.72 (m, 2H), 1.25–1.33 (m, 1H), 3.93 (d, 2H, J = 6.8 Hz), 6.75 (s, 1H), 6.86 (d, 1H, J = 8.4 Hz), 7.88 (d, 1H, J = 8.4 Hz), 10.48 (s, 1H). ESI MS: m/z 261.0 (M + H).
4−(Trifluoromethoxy)−2−(vinyloxy)benzoic acid (25)
To a stirred solution of 24 (800 mg, 3.20 mmol) in DMF (5 mL) was added K2CO3 (1.32 g, 9.60 mmol) and 1-bromo-2-chloroethane (1.32 mL, 16.00 mmol) under nitrogen atmosphere and stirred at room temperature for 15h. It was diluted with cold water (20 mL) and extracted with EtOAc (15 mL × 2). The combined organic layer was washed with water (10 mL) and brine. The organic phase was dried (anh. Na2SO4) then concentrated under reduced pressure to give crude compound. The residue was purified by column chromatography over silica gel using a gradient mixture of 0–5% EtOAc-Hexane as eluent to afford 800 mg (80%) of ethyl-[2-(2-chloro-ethoxy)-4-trifluoromethoxy]benzoate. 1H NMR (CDCl3): δ 1.39 (t, 3H, J = 6.8 Hz), 3.87 (t, 2H, J = 6.0 Hz), 4.28 (t, 2H, J = 6.0 Hz), 4.36 (q, 2H, J = 6.8 Hz), 6.77 (s, 1H), 6.89 (d, 1H, J = 8.4 Hz), 7.86 (d, 1H, J = 8.4 Hz); ESI MS: m/z 313.0 (M+H). To a stirred solution of ethyl-[2-(2-chloro-ethoxy)-4-trifluoromethoxy]benzoate (800 mg, 2.56 mmol) in THF (10 mL) was added KOtBu (575 mg, 5.12 mmol) at 0° C under nitrogen atmosphere. After stirring at 20 °C for 1h, saturated NH4Cl solution (20 mL) was added and the reaction mixture extracted with EtOAc (15 mL × 2). The combined organic layer was washed with water (10 mL) and brine. The organic phase was dried (anh. Na2SO4) and concentrated under reduced pressure to afford 25 (500 mg, 78%). 1H NMR (CDCl3): δ 4.86 (m, 1H), 5.09 (dd, 1H, J = 2.0, 13.2 Hz), 6.60 (dd, 1H, J = 5.6, 13.2 Hz), 6.94 (s, 1H), 7.07 (d, 1H, J = 8.0 Hz), 8.19 (d, 1H, J = 8.0 Hz). ESI MS: m/z 249.0 (M+ H).
2−Cyclopropoxy−4−trifluoromethoxybenzaldehyde (26)
To a solution of Et2Zn (4 mL, 4.01 mmol) in dichloroethane (5 mL) at −20 °C was added CH2I2 (0.32 mL, 4.01 mmol) under Nitrogen atmosphere and stirred for 10 min at −20 °C. A cold (0 °C) solution of 4-trifluoromethoxy-2-vinyloxybenzoic acid (500 mg, 2.01 mmol, solution in 30% toluene-dichloroethane) was added at −20 °C and the resulting mixture stirred at room temperature for 17h. It was quenched with 2N aqueous HCl (20 mL) then extracted with EtOAc (10 mL × 2). The combined organic layer was washed with water (10 mL), brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using a gradient mixture of 0–20% EtOAc in hexane as eluent to afford 270 mg (51%) of 2-cyclopropoxy-4-trifluoromethoxybenzoic acid. 1H NMR (DMSO-d6): δ 0.69 (m, 2H), 0.83 (m, 2H), 3.98 (m, 1H), 7.00 (d, 1H, J = 8.0 Hz), 7.35 (s, 1H), 7.76 (d, 1H, J = 8.0 Hz), 12.84 (s, 1H); ESI MS: m/z 261.0 (M–H). To a cold stirred solution of 2-cyclopropoxy-4-trifluoromethoxybenzoic acid (270 mg, 1.03 mmol) in THF (10 mL) at 0 °C was added BH3-DMS (0.19 mL, 2.06 mmol) under Nitrogen atmosphere. After refluxing for 1.5h at room temperature, the reaction mixture was cooled to 0 °C and quenched with saturated aqueous NH4Cl solution (20 mL). The crude product was extracted with EtOAc (10 mL × 2), organic layer washed with water (10 mL), brine, dried (anh. Na2SO4) and evaporated to afford 230 mg of (2-cyclopropoxy-4-trifluoromethoxyphenyl)methanol as colorless oil. This compound (230 mg, 0.927 mmol) was dissolved in CH2Cl2 (10 ml) and treated with PCC (500 mg) under Nitrogen atmosphere at room temperature. After 0.5h, the mixture was filtered through celite pad and the filtrate concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using a gradient mixture of 0–5% EtOAc in hexane as eluent to afford 26 (200 mg, 80%). 1H NMR (CDCl3): δ 0.89 (m, 4H), 3.84 (m, 1H), 6.89 (d, 1H, J = 8.0 Hz), 7.18 (s, 1H), 7.86 (d, 1H, J = 8.0 Hz), 10.32 (s, 1H). ESI MS: m/z 247.0 (M + H).
Ethyl 2-(4-aminophenoxy)-4-(trifluoromethoxy)benzoate (29)
To a stirred solution of 24 (250 mg, 1.0 mmol) in DMF (5 mL) was added NaH (45 mg, 1.20 mmol) at 0 °C under nitrogen atmosphere and stirred at 0 °C for 10 min. 1-fluoro-4-nitrobenzene (0.3 mL, 3.0 mmol) was added and the mixture heated at 100 °C. After 2h, the reaction mixture was cooled to 0 °C, diluted with water (50 mL) and then extracted with EtOAc (20 mL × 2). The combined organic layer was washed with water (50 mL), brine, dried (anh. Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using a gradient mixture of 0–4% EtOAc in hexane as eluent to afford 250 mg (67%) of ethyl-2-(4-nitrophenoxy)-4-trifluoromethoxybenzoate. 1H NMR (CDCl3): δ 1.81 (t, 3H, J = 7.2 Hz), 4.21 (q, 2H, J = 7.2, 14.0 Hz), 6.94–6.99 (m, 3H), 7.22 (d, 1H, J = 8.0 Hz), 8.10 (d, 1H, J = 8.0 Hz), 8.21–8.24 (m, 2H); ESI MS: m/z 370.0 (M-H). To a stirred solution of ethyl-2-(4-nitrophenoxy-4-trifluoromethoxybenzoate (250 mg, 0.65 mmol) in EtOAc (15 mL) was added 5% Pd/C (20 mg) and stirred under the hydrogen atmosphere. After 1.5h, the reaction mixture was filtered through celite pad and the filtrate concentrated under reduced pressure to afford 29 (200 mg, 87 %). 1H NMR (DMSO-d6): δ 1.23 (t, 3H, J = 7.2 Hz), 4.25 (q, 2H, J = 7.2, 14.0 Hz), 5.10 (brs, 2H), 6.59–6.63 (m, 3H), 6.78 (d, 2H, J = 8.2 Hz), 7.10 (d, 1H, J = 8.2 Hz), 7.86 (d, 1H, J = 8.2 Hz). ESI MS: m/z 342.0 (M+H).
(2-Phenoxy-4-(trifluoromethoxy)phenyl)methanol (30)
To a solution of 29 (190 mg, 0.55 mmol) in aqueous 6N HCl (5 mL) was added aqueous NaNO2 solution (50 mg, 0.72 mmol in 1 mL of water) at 0 °C. After stirring at 0 °C for 30 min, a cold solution of 50% H3PO2 (0.3 mL, 5.57mol) was added and stirred at 50 °C for 1h. The reaction mixture was diluted with water (15 mL) and extracted with EtOAc (10 mL × 2). The combined organic layer was washed with water (10 mL), brine (10 mL), dried (anh. Na2SO4) and concentrated to afford 130 mg of ethyl-2-phenoxy-4-trifluoromethoxybenzoate (0.39 mmol), which was dissolved in THF (10 mL) and treated with LiAlH4 (22 mg, 0.59 mmol) at 0 °C under nitrogen atmosphere. After stirring at room temperature for 30 min., the reaction mixture was quenched with ice and filtered. The filtrate was diluted with EtOAc (20 mL), washed with water (20 mL), brine solution (10 mL) and dried (anh. Na2SO4). Evaporation under reduced pressure and purification of the residue by column chromatography over silica gel using a gradient mixture of 0–10% EtOAc in hexane as eluent afforded 30 (55 mg, 48%). 1H NMR (CDCl3): δ 4.77 (d, 2H, J = 5.6 Hz), 6.67 (s, 1H), 6.97–6.99 (m, 2H), 7.03 (d, 1H, J = 8.0 Hz), 7.18 (t, 1H, J = 7.2Hz), 7.38 (t, 2H, J = 7.2Hz), 7.48 (d, 1H, J = 8.0 Hz). ESI MS: m/z 283.1 (M−H).
2-Phenoxy-4-trifluoromethoxybenzaldehyde (31)
To a stirred solution of 30 (55 mg, 0.19 mmol) in CH2Cl2 (10 mL) was added PCC (120 mg) at room temperature under nitrogen atmosphere. After 30 min, the reaction mixture was filtered through celite and the filtrate concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluting with 10% EtOAc in hexane to afford 31 (50 mg, quant). 1H NMR (CDCl3): δ 6.65 (s, 1H), 7.00 (d, 1H, J = 8.0 Hz), 7.11 (d, 2H, J = 8.2 Hz), 7.24–7.28 (m, 1H), 7.45 (t, 2H, J = 8.0 Hz), 7.98 (d, 1H, J = 8.2 Hz), 10.45 (s, 1H). ESI MS: m/z 283 (M+H).
(S)-N-(2-Benzyloxy-4-trifluoromethoxybenzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (32b)
To a stirred solution of 28b (100 mg, 0.33 mmol) in DMF-AcOH (5 mL, 1:1) was amine 7 (68 mg, 0.37 mmol) at 0 °C under the nitrogen atmosphere. After stirring at room temperature for 1 h, NaBH(OAc)3 (214 mg, 1.01 mmol) was added and stirring continued for 24 h. The reaction mixture was diluted with EtOAc (50 mL), washed with saturated aqueous NaHCO3 solution (20 mL), water (20 mL) and brine solution. The organic layer was dried (anh. Na2SO4), evaporated under reduced pressure and the residue purified by column chromatography over silica gel using a gradient mixture of 0–1% MeOH in CHCl3 as eluent to afford 32b (35 mg, 24%). 1H NMR (DMSO-d6): 3.78 (br s, 2H), 3.94–3.97 (m, 1H), 4.11–4.13 (m, 1H), 4.34–4.40 (m, 3H), 5.15 (s, 2H), 6.88 (d, 1H, J = 8.0 Hz), 7.04 (s, 1H), 7.30–7.40 (m, 5H), 7.44 (d, 1H, J = 7.8 Hz), 7.94 (s, 1H); 13C NMR (acetone-d6): δ 45.7, 48.3, 49.0, 70.3, 71.2, 106.6, 113.4, 117.4, 128.6, 129.1, 129.6, 131.2, 137.7, 149.8, 158.5. HRMS calcd for C21H19F3N4O5 [M+H+] 465.1386, found 465.1387. The following compounds were synthesized in a similar manner:
(S)-N-(2-Methoxy-4-trifluoromethoxybenzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (32c)
23% yield. 1H NMR (DMSO-d6): δ 3.20–3.30 (m, 1H), 3.75 (d, 3H, J = 9.2 Hz), 3.81 (s, 3H), 3.90–4.10 (dd, 1H, J = 5.6, 16.0 Hz), 4.16 (dd, 1H, J = 5.6, 16.0 Hz), 4.30–4.50 (m, 2H), 6.89 (d, 1H, J = 10.2 Hz), 6.95 (s, 1H), 7.39 (d, 1H, J = 10.2 Hz), 8.01 (s, 1H). HRMS calcd for C15H15F3N4O5 [M+H+] 389.1059, found 389.1061. HPLC purity: 94%.
(S)-N-(2-Methoxymethoxy-4-trifluoromethoxybenzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (32a)
35% yield. 1H NMR (CDCl3): δ 3.37–3.39 (m, 1H), 3.46 (s, 3H), 3.90 (s, 2H), 3.94 (dd, 1H, J = 4, 12.4 Hz), 4.15 (dd, 1H, J = 4, 12.4 Hz), 4.32–4.44 (m, 2H), 5.20 (s, 2H), 6.86 (d, 1H, J = 8.4 Hz), 7.00 (s, 1H), 7.27 (m, 1H), 7.38 (s, 1H). ESI MS: m/z 419.2 (M+H).
(S)-N-(2-Cyclopropylmethoxy-4-trifluoromethoxybenzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (32e)
11% yield. 1H NMR (DMSO-d6): δ 0.32–0.35 (m, 2H), 0.55–0.59 (m, 2H), 1.19–1.25 (m, 1H), 3.27 (m, 1H), 3.78 (d, 2H, J = 6.8 Hz), 3.87 (d, 2H, J = 6.8 Hz), 4.02 (dd, 1H, J = 3.2, 13.0 Hz), 4.17 (dd, 1H, J = 4.0, 13.0 Hz), 4.41 (m, 2H), 6.87 (d, 1H, J = 8.4 Hz), 6.91 (s, 1H), 7.39 (d, 1H, J = 8.4 Hz), 8.02 (s, 1H). HRMS calcd for C18H19F3N4O5 [M+H+] 429.1386, found 429.1391.
(S)-N-(2-Cyclopropoxy-4-trifluoromethoxybenzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (32f)
8% yield. 1H NMR (DMSO-d6): δ 0.69 (m, 2H), 0.81 (m, 2H), 3.21 (br s, 1H), 3.68 (d, 2H, J = 6.8 Hz), 3.92–4.00 (m, 2H), 4.14 (dd, 1H, J = 4.0, 12.0 Hz), 4.37–4.43 (m, 2H), 6.92 (d, 1H, J = 8.0 Hz), 7.19 (s, 1H), 7.39 (d, 1H, J = 8.0 Hz), 8.02 (s, 1H). 13C NMR (CDCl3): δ 6.3, 45.5, 47.5, 47.7, 51.4, 69.4, 106.2, 112.8, 115.2, 125.4, 130.1, 143.5, 147.3, 149.3, 157.6. HRMS calcd for C17H17F3N4O5 [M+H+] 415.1229, found 415.1231. HPLC purity: 93.88%.
(S)-2-Nitro-N-(2-phenoxy-4-trifluoromethoxybenzyl)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (32d)
12% yield. 1H NMR (DMSO-d6): δ 3.28 (br s, 1H), 3.83 (d, 2H, J = 6.0 Hz), 4.00 (br d, 1H, J = 12.0 Hz), 4.14 (dd, 1H, J = 3.2, 12.0 Hz), 4.35–4.43 (m, 2H), 6.73 (s, 1H), 7.02 (d, 2H, J = 8.0 Hz), 7.14–7.20 (m, 2H), 7.41 (t, 2H, J = 7.2 Hz), 7.60 (d, 2H, J = 8.0 Hz), 8.00 (s, 1H). HRMS calcd for C20H17F3N4O5 [M+H+] 451.1270, found 451.1251.
(S)-2-((2-Nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-ylamino)methyl)-5-trifluoromethoxyphenol (32g)
To a solution of 32a (200 mg, 0.47 mmol) in THF (2 mL) was added 6N aqueous HCl (5 mL) and stirred at room temperature for 4h. The reaction mixture was neutralized using aq. NaHCO3 solution and extracted with ethyl acetate (2 × 50 mL). The organic layer was washed with water, brine, dried (anh.Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using 0–5% MeOH in CHCl3 as eluent to give 32g (120 mg, 67%). 1H NMR (DMSO-d6): δ 3.25 (m, 1H), 3.76 (s, 2H), 4.01 (d, 1H, J = 12.0 Hz), 4.15 (d, 1H, J = 12.0 Hz), 4.41 (br s, 2H), 6.69–6.71 (m, 2H), 7.26 (d, 1H, J = 7.6 Hz), 8.01 (s, 1H), 10.21 (br s, 1H); 13C NMR (CD3OD): δ 47.3, 48.0, 48.7, 70.0, 109.1, 112.5, 117.8,123.1, 125.0, 131.6, 143.9, 149.2, 150.6, 158.8. HRMS calcd for C14H13F3N4O5 [M+H+] 375.0916, found 375.0916.
2-Chloro-4-trifluoromethoxybenzaldehyde (34b)
To a solution of 2-chloro-4-triflouromethoxyiodobenzene (100 mg, 0.310 mmol) in THF (4 mL) at 78 °C was added n-butyl lithium (1M in hexane, 0.62 mL, 0.021 mmol) and stirred at −78 °C for 30min. The pale yellow suspension was treated with a solution of DMF (0.048 mL, 0.621 mmol) in diethyl ether (1 mL) at −78 °C. The reaction mixture was stirred for 15 min and quenched with dilute aqueous H2SO4. The organic layer was separated. The aqueous layer was extracted with diethyl ether and the combined organic layer washed with water, brine, dried (anh. Na2SO4) and evaporated under reduced pressure. The residue was purified by column chromatography over silica gel by using mixture of 0–3% ethyl acetate in hexane to get 34b (50 mg, 72%). 1H NMR (CDCl3): δ 7.28–7.33(m, 1H), 7.45 (d, 1H, J = 8.0 Hz), 7.53 (t, 1H, J = 8.0 Hz), 10.45 (s, 1H).
2-Fluoro-4-trifluoromethoxybenzaldehyde (34a)
To a solution of 2-fluoro-4-triflouromethoxyiodobenzene (1.0 g, 3.86 mmol) in THF (20 mL) at −78 °C was added n-butyl lithium (1 solution, 7.7 mL, 7.72 mmol) and stirred at −78 °C for 30 min. The pale yellow suspension was treated with a solution of DMF (0.59 mL, 7.72 mmol) in diethyl ether (5 mL) at −78 °C. The reaction mixture was stirred for 15 min and quenched with dilute aqueous H2SO4. The organic layer was separated. The aqueous layer was extracted with diethyl ether and the combined organic layer washed with water, brine, dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by column chromatography over silica gel by using mixture of 0–3% ethyl acetate in hexane to get 460 mg (57%) of 34a. 1H NMR (CDCl3): δ 7.06 (d, 1H, J = 10.0 Hz), 7.13 (d, 1H, J = 8.0 Hz), 7.95 (t, 1H, J = 8.0 Hz), 10.32 (s, 1H).
(E)-2-Bromo-1-styryl-4-(trifluoromethoxy)benzene (36)
To a solution of 2-bromo-1-iodo-4-trifluoromethoxybenzene (200 mg, 0.544 mmol) in acetonitrile (5 mL) was added styrene (0.08 mL, 0.708 mmol) and triethylamine (0.1 mL, 0.71 mmol). After purging with argon for 30 min, Pd(OAc)2 (10 mg, 0.04 mmol) was added and the reaction mixture was stirred at 95 °C. After 16h, the reaction mixture was cooled to room temperature and poured into 10% aq. HCl (10 mL). The product was extracted using diethyl ether. The combined organic layer was washed with water, brine, dried (anh.Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel by using a solvent gradient mixture of 0–1% EtOAc in hexane to afford 36 (120 mg, 64%). 1H NMR (CDCl3): δ 7.02 (d, 1H, J = 16.0 Hz), 7.20 (m, 1H), 7.29–7.32 (m, 1H), 7.37–7.42 (m, 3H), 7.47 (m, 1H), 7.54 (d, 2H, J = 8.0 Hz), 7.68 (d, 1H, J = 8.0 Hz).
2-Bromo-4-(trifluoromethoxy)benzaldehyde (37)
To a solution of 36 (1.0 g, 2.91 mmol) in acetone (20 mL) and water (5 mL) at 0 °C was added OsO4 (0.4 M in t-BuOH, 0.072 mL, 0.029 mmol), NaIO4 (931 mg, 4.37 mmol). After 16h at room temperature, the reaction mixture was diluted with aq. sodium metabisulphite solution and extracted with EtOAc (2 × 50 mL). The combined organic layer was washed with water, brine solution, dried (Na2SO4) and concentrated under reduced pressure. The residue upon purification by column chromatography over silica gel using a gradient mixture of 0–2% EtOAc in hexane afforded 37 (130 mg, 16%). 1H NMR (CDCl3): δ 7.27–7.30 (m, 1H), 7.52 (dd, 1H, J = 1.2, 2.4 Hz), 7.98 (d, 1H, J = 8.5 Hz), 10.32 (s, 1H).
2-(Piperidin-1-yl)-4-(trifluoromethoxy)benzaldehyde (40a)
To a stirred solution of 36 (1.0 g, 2.91 mmol), piperidine (0.34 mL, 3.49 mmol) and Cs2CO3 (2.83 g, 8.73 mmol) in dioxane (15 mL) was added Xantphos (804 mg, 0.873 mmol) under argon. After degassing the reaction mixture with argon for 30 min, Pd(OAc)2 (65 mg, 0.291 mmol) was added and continued the argon purging for another 10 min. The resulting reaction mixture was stirred at 80 °C for 16h. After cooling to room temperature, the reaction mixture was diluted with water (20 mL) and extracted with diethyl ether. The organic layer was washed with water, brine, dried over (anh.Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel by using a solvent gradient of 0–10% EtOAc in hexane as eluent to afford 800 mg of (E)-1-(2-styryl-5-(trifluoromethoxyphenyl)piperidine. This compound (0.8 g, 2.30 mmol) was dissolved in acetone (30 mL), water (6 mL) was added OsO4 (1M solution in t-BuOH, 72 μL, 0.028 mmol), NaIO4 (920 mg, 4.32 mmol) was added at 0 °C and stirred at room temperature for 16h. The reaction mixture was quenched with aq. metabisulphite solution and extracted with ethyl acetate. The combined ethyl acetate layer was washed with aq. Na2S2O3, water, brine and dried over (anh.Na2SO4) and concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel (100 – 200 mesh) by using a solvent gradient mixture of 0–15% EtOAc in hexane as eluent to afford 40a (250 mg, 31%). 1H NMR (CDCl3): δ 1.58–1.77 (m, 6H), 3.10 (t, 4H, J = 5.2 Hz), 6.86–6.89 (m, 2H), 7.82 (d, 1H, J = 8.4 Hz), 10.18 (s, 1H).
2-Morpholino-4-(trifluoromethoxy)benzaldehyde (40b)
To a stirred solution of 36 (1.5 g, 4.37 mmol) in 1,4-dioxane (20 mL) was added morpholine (0.42 mL, 5.24 mmol) and Cs2CO3 (4.2 g, 13.11 mmol). After degassing the reaction mixture with argon, Pd(OAc)2 (98 mg, 0.43 mmol) was added and stirred at 100 °C. After 18h, the reaction mixture was filtered through celite. The filtrate was diluted with water (40 mL) and extracted with EtOAc (2 × 30 mL). The organic layer was washed with water (20 mL), brine, dried over (anh. Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using a gradient of 0–7% EtOAc in hexane as eluent to afford 900 mg (59%) of (E)-4-(2-styryl-5-(trifluoromethoxy)phenyl)morpholine. 1H NMR (CDCl3): δ 2.99 (t, 4H, J = 4.8 Hz)), 3.89 (t, 4H, J = 4.8 Hz), 6.85 (br s, 1H), 6.94 (d, 1H, J = 8.8 Hz), 7.03 (d, 1H, J = 16.0 Hz), 7.29 (d, 1H, J = 7.2 Hz), 7.35 (d, 1H, J = 8.4 Hz), 7.39 (t, 2H, J = 7.2 Hz), 7.52 (d, 2H, J = 7.2 Hz), 7.58 (d, 1H, J = 8.8 Hz). ESI MS: m/z 350.1 (M+H). To a solution of (E)-4-(2-styryl-5-(trifluoromethoxy)phenyl)morpholine (900 mg, 2.57 mmol) in acetone (15 mL), water (30 mL) at 0 °C was added NaIO4 (823 mg, 3.86 mmol) followed by OsO4 (1M solution in t-BuOH, 0.05 mL, 0.02 mmol). After stirring at room temperature for 19 h, the reaction mixture was quenched with aq. sodium metabisulfite solution (20 mL) and extracted with EtOAc (2 × 15 mL). The combined organic layer was washed with water (10 mL), brine, dried (anh.Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using a gradient of 0–5% EtOAc in hexane as eluent to afford 40b (350 mg, 49%). 1H NMR (CDCl3): δ 3.10 (t, 4H, J = 4.8 Hz), 3.91 (t, 4H, J = 4.8 Hz), 6.88 (s, 1H), 6.97 (d, 1H, J = 8.4 Hz), 7.85 (d, 1H, J = 8.4 Hz), 10.23 (s, 1H).
2-(2-Bromo-4-(trifluoromethoxy)phenyl)-1,3-dioxolane (38)
To a stirred solution 2-bromo-4-trifluoromethoxybenzaldehyde (1.5 g, 5.57 mmol) in benzene (30 mL) was added p-TSA (15 mg) and ethylene glycol (0.94 mL, 16.72 mmol). After heating at reflux using Dean-Stark apparatus for 8 h, the reaction mixture was cooled to room temperature. The organic layer was diluted with ethyl acetate, washed with sat. aq. NaHCO3 (50 mL), water and brine. The organic layer was dried (anh. Na2SO4), evaporated under reduced pressure and the residue purified by column chromatography over neutral alumina using a solvent gradient of 0–1% EtOAc in hexane as eluent to afford 38 (1.5 g, 86%). 1H NMR (CDCl3): δ 4.06–4.17 (m, 4H), 6.06 (s, 1H), 7.21 (d, 1H, J = 8.0 Hz), 7.45 (br s, 1H), 7.64 (d, 1H, J = 8.0 Hz).
In a similar manner, 2-(3-chloro-4-(trifluoromethoxy)phenyl)-1,3-dioxolane (54) was prepared in 72% yield from 3-chloro-4-trifluoromethoxybenzaldehyde. 1H NMR (CDCl3): δ 4.00–4.15 (m, 4H), 5.80 (s, 1H), 7.32 (d, 1H, J = 8.4 Hz), 7.39–7.42 (m, 1H), 7.61 (d, 1H, J = 1.6 Hz).
2-(4-Methylpiperazin-1-yl)-4-(trifluoromethoxy)benzaldehyde (39)
To a stirred solution of 38 (200 mg, 0.63 mmol), N-methylpiperzine (0.085 mL, 0.766 mmol), Cs2CO3 (622 mg, 1.914 mmol) in dioxane (3 mL) was added Xantphos (110 mg, 0.191 mmol) and degassed with argon for 20 min. Pd(OAc)2 (14 mg, 0.063 mmol) was added and continued the argon purging for another 10 min. After stirring at 90 °C for 8 h, the reaction mixture was cooled to room temperature, diluted with ethyl acetate (10 mL) and washed with water, brine and dried (anh.Na2SO4). Evaporation of the organic solvent under reduced using a solvent gradient of 0–1% MeOH in CHCl3 as eluent to afford 1-(2-(1,3-dioxolan-2-yl)-5-(trifluoromethoxyphenyl)-4-methylpiperazine (120 mg). 1H NMR (DMSO-d6): δ 2.22 (s, 3H), 2.48 (m, 4H), 2.96 (m, 4H), 3.95–4.11 (m, 4H), 5.95 (s, 1H), 7.01 (br s, 1H), 7.06 (d, 1H, J = 8.0 Hz), 7.56 (d, 1H, J = 6.4 Hz); ESI MS: 333.0 (M+H). This compound (120 mg, 0.361 mmol) was dissolved in THF (5 mL) and treated with 6 N HCl (10 mL) at room temperature for 30 min. The reaction mixture was neutralized with solid NaHCO3 and extracted with ethyl acetate. The organic layer was washed with water, brine, dried (anh.Na2SO4) and concentrated under reduced pressure to afford 39 (100 mg, 55%). 1H NMR (DMSO-d6): δ 2.24 (s, 3H), 2.52 (m, 4H), 3.10 (m, 4H), 7.08 (m, 2H), 7.81 (d, 1H, J = 9.6 Hz), 10.09 (s, 1H). ESI MS: 289.0 (M+H).
Adopting a similar procedure, the following compounds were synthesized:
3-Morpholino-4-(trifluoromethoxy)benzaldehyde (55a)
21 % yield. 1H NMR (CDCl3): δ 3.12 (t, 4H, J = 5.2 Hz), 3.87 (t, 4H, J = 5.2 Hz), 7.38 (dd, 1H, J = 2.0, 8.0 Hz), 7.50–7.60 (m, 2H), 9.96 (s, 1H).
3-(4-Methylpiperazin-1-yl)-4-(trifluoromethoxy)benzaldehyde (55b)
15% yield. 1H NMR (CDCl3): δ 2.36 (s, 3H), 2.59 (t, 4H, J = 4.4 Hz), 3.16 (t, 4H, J = 4.4 Hz), 7.35 (dd, 1H, J = 1.2, 8.0 Hz), 7.50 (dd, 1H, J = 2.0, 8.0 Hz), 7.53 (d, 1H, J = 1.6 Hz), 9.95 (s, 1H).
Tert-butyldimethyl-(4-(trifluoromethoxy)benzyloxy)silane (43)
To a solution of (4-(trifluoromethoxy)phenyl)methanol (1.49 g, 7.76 mmol) in CH2Cl2 (20 mL) at 0 °C was added imidazole (686 mg, 10.08 mmol) followed by TBDMSCl (1.40 g, 9.31 mmol). After stirring for 16 h at room temperature, the reaction mixture was diluted with CH2Cl2, washed with water, brine and dried (anh. Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using 10% ethyl acetate in hexane as eluent to give 43 (1.70 g, 60%). 1H NMR (CDCl3): δ 0.10 (s, 6H), 0.94 (s, 9H), 4.73 (s, 2H), 7.17 (d, 2H, J = 8.0 Hz), 7.34 (d, 2H, J = 8.0 Hz).
5-((Tert-butyldimethylsilyloxy)methyl)-2-(trifluoromethoxy)phenol (44)
To a solution of 43 (500 mg, 1.63 mmol) in THF (10 ml) at −78 °C was added TMEDA (0.245 mL, 1.63 mmol) followed by sec-BuLi (1.4 M in cyclohexane, 1.16 mL, 1.63 mmol). After stirring at −78 °C for 1 h, fluorodimethoxyborane-dimethyl ether complex (prepared by adding BF3.OEt2 (0.17 mL, 1.40 mmol) and trimethylborate (0.311 mL, 2.81 mmol) in a separate flask followed by diethyl ether (0.27 mL, 2.81 mmol) was added at −78 °C. After warming the reaction mixture to room temperature over 30 min, alkaline H2O2 was added and stirred for 20 min. The reaction mixture was poured into 1N HCl and extracted with ether. The organic layer was washed with water, brine, dried (anh. Na2SO4) and concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel using 10% ethyl acetate in hexane as eluent to give 44 (150 mg, 28%). 1H NMR (DMSO-d6): δ 0.08 (s, 6H), 0.91 (s, 9H), 4.63 (s, 2H), 6.76 (d, 1H, J = 8.0 Hz), 6.99 (s, 1H), 7.18 (d, 1H, J = 8.0 Hz), 10.07 (s, 1H).
3-Methoxy-4-(trifluoromethoxy)benzaldehyde (47a)
To the solution of 44 (3 g, 9.31 mmol) in dry DMF (10 mL) was added K2CO3 (2.57 g, 18.63 mmol) followed by MeI (1.16 mL, 18.63 mmol) and stirred for 3 h at room temperature. Reaction mixture was diluted with ether (50 mL) and washed with water (2 × 10 mL), brine, dried (anh.Na2SO4) and concentrated under reduced pressure to afford 3 g (8.928 mmol) of product which was dissolved in dry THF (12 mL) and treated with TBAF.3H2O (2.812 g, 8.928 mmol) at room temperature. After stirring the reaction mixture for 1.5 h, it was diluted with ether (30 mL). The organic layer was washed with water (2 × 5 mL) and brine solution, dried (anh.Na2SO4) and concentrated under reduced pressure. The residue upon purification by column chromatography over silica gel (100 – 200 mesh) using a solvent gradient of 20 – 23% EtOAc in hexane as eluent afforded 3-methoxy-4-(trifluoromethoxy)benzyl alcohol (1.0 g, 52%). 1H NMR (DMSO-d6): δ 3.84 (s, 3H), 4.50 (d, 2H, J = 5.6 Hz), 5.28 (t, 1H, J = 5.6 Hz), 6.95 (d, 1H, J = 8.4 Hz), 7.17 (s, 1H), 7.27 (d, 1H, J = 8.4 Hz). A solution of the above benzylic alcohol (900 mg, 4.484 mmol) in CH2Cl2 (10 mL) was added PCC (966 mg, 4.484 mmol) at room temperature. After stirring the reaction mixture for 1 h, it was diluted with CH2Cl2 (25 mL), washed with water (2 × 5 mL), brine and concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel using a solvent gradient of 10 – 15% EtOAc – petroleum ether as eluent to afford 47a (700 mg, 91%) of 3-methoxy-4-(trifluoromethoxy)benzaldehyde. 1H NMR (DMSO-d6): δ 3.95 (s, 3H), 7.62 (s, 2H), 7.71 (s, 1H), 10.01 (s, 1H).
Tert-butyl-(3-(methoxymethoxy)-4-(trifluoromethoxybenzyloxy)) dimethylsilane (46)
To a solution of 44 (150 mg, 0.46 mmol) in CH2Cl2 (2 mL) at 0 °C was added diisopropylethylamine (0.09 mL, 0.56 mmol) followed by methoxymethylchloride (0.04 mL, 0.56 mmol). After stirring at room temperature for 16 h, the reaction mixture was diluted with CH2Cl2, washed with water, brine, dried (anh. Na2SO4) and concentrated under reduced pressure. The residue upon purification by column chromatography over silica gel using a solvent gradient of 10% EtOAc in hexane as eluent afforded 46 (140 mg, 82%). 1H NMR (DMSO-d6): δ 0.00 (s, 6H), 0.82 (s, 9H), 3.30 (s, 3H), 4.63 (s, 2H), 5.17 (s, 2H), 6.92 (d, 1H, J = 8.00 Hz), 7.19 (s, 1H), 7.25 (d, 1H, J = 8.0 Hz).
3-(methoxymethoxy)-4-(trifluoromethoxy)benzaldehyde (47b)
To a solution of 46 (280 mg, 0.82 mmol) in THF (2 mL) was added tetra-n-butyl ammonium fluoride trihydrate (250 mg, 0.92 mmol). After stirring at room temperature for 30 min, it was diluted with water and extracted with CH2Cl2. The organic layer was washed with brine, dried (anhyd Na2SO4) and concentrated to give (3-(methoxymethoxy)-4-(trifluoromethoxyphenyl)methanol (160 mg, 0.65 mol) which was dissolved in CH2Cl2 (4 mL) and treated with pyridinium chlorochromate (240 mg, 1.1 mmol). After 1 h at room temperature, the reaction mixture was diluted with ethyl acetate. The organic layer was washed with water, brine, dried (anh. Na2SO4) and evaporated under reduced pressure. The residue was purified over silica gel eluting with 10% EtOAc in hexane to give 47b (140 mg, 73%). 1H NMR (CDCl3): δ 3.51 (s, 3H), 5.30 (s, 2H), 7.41 (d, 1H, J = 8.0 Hz), 7.55 (dd, 1H, J = 1.2, 8.0 Hz), 7.75 (d, 1H, J = 1.2 Hz), 9.96 (s, 1H).
Methyl 5-formyl-2-(trifluoromethoxy)benzoate (48)
To a solution of 43 (3.0 g, 9.8 mmol) in dry THF (45 mL) at −78 °C was added sec-BuLi (1.4M in cyclohexane, 14.7 mL, 20.56 mmol). After 1h at −78 °C, a solution of methyl chloroformate (1.13 mL, 14.75 mmol) in dry THF (5 mL) was added and the reaction mixture allowed to warm to room temperature over 1h. After 3h at room temperature, the reaction was quenched with sat. aq. NH4Cl solution and extracted with diethyl ether (2 × 30 mL). The organic layer was washed with brine (50 mL), dried (anh.Na2SO4) and concentrated under reduced pressure to give 4 g of crude product which was dissolved in dry THF (40 mL) and treated with TBAF.3H2O (4.16 g, 13.1 mmol) for 1h. The reaction mixture was diluted with water (50 mL), extracted with diethyl ether (2 × 50 mL), washed with water (3 × 50 mL) and brine (50 mL) dried (anh.Na2SO4) and concentrated under reduced pressure. The crude product (3 g, 12.0 mmol) was dissolved in dry CH2Cl2 (30 mL) and treated with PCC (3.0 g, 13.9 mmol). After 30 min, the reaction mixture was diluted with diethyl ether (50 mL), filtered through Celite and concentrated. The residue was purified by column chromatography over silica gel by using a solvent gradient mixture of 2 – 3% EtOAc in hexane as eluent to afford 48 (500 mg, 21%). 1H NMR (CDCl3): δ 3.98 (s, 3H), 7.51 (d, 1H, J = 8.4 Hz), 8.10 (dd, 1H, J = 1.6, 8.4 Hz), 8.47 (d, 1H, J =1.6 Hz), 10.06 (s, 1H).
Tert-butyldimethyl(3-(4-nitrophenoxy)-4-(trifluoromethoxy)benzyloxy)silane (50)
To a stirred solution of 44 (250 mg, 0.776 mmol) in DMF (3 mL) at 0 °C was added NaH (60% in oil, 40 mg, 0.931 mmol) and followed by 4-nitro-1-fluorobenzene (0.25 mL, 2.33 mmol) and stirred at 100 °C for 1h. The reaction mixture was diluted with water and extracted with ethyl acetate (2 × 20 mL). The organic layer was washed with water, brine, dried (Na2SO4) and concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel (100–200mesh) by using gradient mixture of 0–10% ethyl acetate in hexane as eluent to afford 50 (100 mg, 30%). 1H NMR (CDCl3): δ 0.09 (s, 6H), 0.91 (s, 9H), 4.73 (s, 2H), 7.02 (d, 2H, J = 9.0 Hz), 7.15 (br s, 1H), 7.22 (dd, 1H, J = 1.2, 8.8Hz), 7.35 (d, 1H, J = 8.4 Hz), 8.22 (d, 2H, J = 8.8 Hz).
(3-(4-Phenoxy)-4-(trifluoromethoxy)phenyl)methanol (51)
To stirred solution of 50 (300 mg, 0.677 mmol) in ethyl acetate (5 mL) was added Iron powder (380 mg, 6.77 mmol), NH4Cl (109 mg, 2.031 mmol) and H2O (1.5 mL) at room temperature. The reaction mixture was heated at reflux for 16h. Reaction mixture was filtered through celite and the filtrate diluted with ethyl acetate. The organic layer was washed with water, brine dried (Na2SO4) and concentrated under reduced pressure to afford 200 mg (0.48 mmol) of 4-(5-((tert-butyldimethylsilyloxy)methyl)-2-(trifluoromethoxy)phenoxy)aniline which was dissolved in 6N aqueous HCl (3 mL) and treated with aqueous NaNO2 (43 mg, 0.62 mmol). After stirring at 0° C for 30 Min, a cold solution of 50% H3PO2 (50%, 0.290 mL, 2.672 mmol) was added and stirred at 50° C for 1h. The reaction mixture was diluted with water (5 mL) and extracted with ethyl acetate (2 × 10 mL). The combined organic layer was washed with aq. NaHCO3, water, brine and dried (Na2SO4) and concentrated under reduced pressure to afford 51 (50 mg, 52%). 1H NMR (CDCl3): δ 4.63 (s, 2H), 7.00 (m, 3H), 7.10–7.14 (m, 2H), 7.31–7.37 (m, 3H).
3-Phenoxy-4-(trifluoromethoxy)benzaldehyde (52)
To a stirred solution of 51 (25 mg, 0.088 mmol) in CH2Cl2 (2 mL) was added PCC (19 mg, 0.088 mmol). After stirring for 1h at room temperature, the reaction mixture was filtered through celite. The filtrate was evaporated under reduced pressure and the residue purified by column chromatography over silica gel using a gradient mixture of 0–5% EtOAc in hexane as eluent to afford 52 (15 mg, 62%). 1H NMR (CDCl3): δ 7.04 (d, 2H, J = 8.4 Hz), 7.21 (t, 1H, J = 7.2 Hz), 7.42 (t, 2H, J = 7.2 Hz), 7.45 (s, 1H), 7.50 (d, 1H, J = 8.4 Hz), 7.63 (d, 1H, J = 8.4 Hz), 9.89 (s, 1H).
(S)-N-(2-Chloro-4-trifluoromethoxybenzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (41b)
A mixture of 34b (483 mg, 2.16 mmol) and amine 7 (200 mg 1.08, mmol) in DMF (5 mL) containing AcOH (1 mL) was stirred at room temperature for 1 h. NaBH3CN (165 mg, 2.52 mmol) was added to the reaction mixture and stirring continued for 24 h. The reaction mixture was diluted with water and extracted with EtOAc. The combined organic layer was washed with water, brine solution, dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by column chromatography over silica gel using a gradient mixture of 0–5 % MeOH-CHCl3 to afford 41b (110 mg, 26%). 1H NMR (DMSO-d6): δ 2.81–2.83 (m, 1H), 3.88 (d, 2H, J = 6.8 Hz), 4.01–4.04 (br d, 1H, J = 12.0 Hz), 4.118 (dd, 1H, J = 4.0, 12.0 Hz), 4.39–4.48 (m, 2H), 7.36 (d, 1H, J = 8.0 Hz), 7.52–7.53 (m, 1H), 7.61 (d, 1H, J = 8.0 Hz), 8.03 (s, 1H). HRMS calcd for C14H12ClF3N4O4 [M+H+] 393.0577, found 393.0572.
Adopting similar procedure, the following compounds were synthesized:
(S)-N-(2-Fluoro-4-(trifluoromethoxy)benzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (41a)
15% yield. 1H NMR (CDCl3): δ 3.34 (br s, 1H), 3.97 (m, 3H), 4.19 (dd, 1H, J = 4.0, 12.0 Hz), 4.39–4.43 (m, 2H), 6.97–6.98 (m, 1H), 7.02 (s, 1H, J = 8.0 Hz), 7.38 (d, 1H, J = 8.0 Hz), 7.40 (s, 1H). HRMS calcd for C14H12F4N4O4 [M+H+] 377.0873, found 377.0878. HPLC purity: 95.86%.
(S)-N-(2-Bromo-4-(trifluoromethoxy)benzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (41c)
46% yield. 1H NMR (DMSO-d6): δ 2.81–2.85 (m, 1H), 3.32–3.34 (m, 1H), 3.85 (d, 2H, J = 7.0 Hz), 4.03 (dd, 1H, J = 2.4, 120 Hz), 4.20 (dd, 1H, J = 4.0, 12.0 Hz), 4.38–4.48 (m, 2H), 7.40 (dd, 1H, J = 1.2, 7.5 Hz), 7.0 (d, 1H, J = 7.5 Hz), 7.66 (d, 1H, J = 1.6 Hz), 8.04 (s, 1H); 13C NMR (CDCl3): δ 47.5, 47.9, 49.9, 69.4, 115.6, 120.1, 121.5, 123.5, 125.3, 130.7, 136.9, 143.3, 147.2, 148.4. HRMS calcd for C14H12BrF3N4O4 [M+H+] 437.0072, found 437.0064. HPLC purity: 94.5%.
(S)-2-Nitro-N-(2-(piperidin-1-yl)-4-(trifluoromethoxy)benzyl)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (41d)
13% yield. 1H NMR (DMSO-d6): δ 1.50–1.65 (m, 6H), 2.69–2.84 (m, 5H), 3.24 (m, 1H), 3.77 (d, 2H, J = 7.2 Hz), 4.00 (dd, 1H, J = 3.2, 13.0 Hz), 4.17 (dd, 1H, J = 3.2, 13.0 Hz), 4.42–4.45 (m, 2H), 6.93 (br s, 1H), 6.98 (d, 1H, J = 8.4 Hz), 7.48 (d, 1H, J = 8.4 Hz), 8.03 (s, 1H). FT-ICR MS calcd for C19H22F3N5O4 [M+H+] 442.1696, found 442.1698.
(S)-N-(2-Morpholino-4-(trifluoromethoxy)benzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (41e)
24% yield. 1H NMR (DMSO-d6):. δ 2.67 (br s, 1H), 2.80–2.95 (m, 4H), 3.26–3.37 (m, 1H), 3.70–3.82 (m, 6H), 4.01 (dd, 1H, J = 3.6, 12.4 Hz), 4.18 (dd, 1H, J = 3.6, 12.4 Hz), 4.44 (d, 2H, J = 2.4 Hz), 6.97 (s, 1H), 7.03 (d, 1H, J = 8.0 Hz), 7.49 (d, 1H, J = 8.0 Hz), 8.05 (s, 1H); 13C NMR (CDCl3): δ 46.17, 47.9, 52.9, 67.1, 69.1, 112.9, 115.3, 116.2, 130.7, 132.0, 143.4, 147.2, 149.1, 152.5. HRMS calcd for C18H20F3N5O5 [M+H+] 444.1495, found 444.1490. HPLC purity: 95.4%.
(S)-N-(2-(4-Methylpiperazin-1-yl)-4-(trifluoromethoxy)benzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (41f)
32% yield. 1H NMR (DMSO-d6): δ 2.24 (s, 3H), 2.40–2.60 (m, 4H), 2.67 (br s, 1H), 2.80–3.00 (m, 4H), 3.20–3.40 (br s, 1H), 3.77 (br s, 2H), 3.98–4.02 (m, 1H), 4.16–4.20 (m, 1H), 4.43 (br s, 2H), 6.94 (s, 1H), 7.00 (d, 1H, J = 8.4 Hz), 7.48 (d, 1H, J = 8.4 Hz), 8.04 (s, 1H). FT-ICR MS calcd for C19H23F3N6O4 [M+H+] 457.1805, found 457.1804.
(S)-N-(3-Morpholino-4-(trifluoromethoxybenzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (56g)
31 % yield. 1H NMR (DMSO-d6): δ 2.80–2.90 Hz), 4.15 (dd, 1H, (m, 1H), 2.90–3.00 (m, 4H), 3.20–3.30 (m, 1H), 3.65–3.85 (m, 6H), 4.00 (dd, 1H, J = 2.8, 12.8 J = 4.0, 12.8 Hz), 4.35–4.45 (m, 2H), 7.00–7.10 (m, 2H), 7.21 (d, 1H, J = 8.4 Hz), 8.01 (s, 1H); 13C NMR (DMSO-d6): δ 38.6, 39.8, 41.5, 42.9 (2C), 58.6 (2C), 61.2, 108.3, 111.2, 113.2, 113.7, 132.0, 133.0, 134.4, 136.9, 139.9. HRMS calcd for C18H20F3N5O5 [M+H+] 444.1495, found 444.1506. HPLC purity: 95.3%.
(S)-N-(3-(4-Methylpiperazin-1-yl)-4-(trifluoromethoxy)benzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (56h)
20 % yield. 1H NMR (CDCl3): δ 2.39 (s, 3H), 2.60–2.70 (t, 4H, = 10.0 Hz), 3.94 (dd, 1H, J = 4.0 Hz), 3.05–3.15 (t, 4H, J = 4.0 Hz), 3.35–3.45 (m, 1H), 3.88 (d, 2H, J = 10.0 Hz), 3.94 (dd, 1H, J = 4.4, 12.0 Hz), 4.16 (dd, 1H, J = 4.8, 12.0 Hz), 4.35–4.45 (m, 2H), 6.92 (dd, 1H, J = 2.0, 8.0 Hz), 6.96 (d, 1H, J = 2.0 Hz), 7.13 (dd, 1H, J = 1.2, 8.0 Hz), 7.38 (s, 1H). HRMS calcd for C19H23F3N6O4 [M+H+] 457.1811, found 457.1798. HPLC purity: 93.4%.
(S)-Methyl 5-((2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-ylamino)methyl)-2-(trifluoromethoxy)benzoate (56f)
55% yield. 1H NMR (DMSO-d6): δ 2.93–2.94 (m, 1H), 3.26–3.29 (m, 1H), 3.84–3.87 (m, 5H), 3.97–4.00 (m, 1H), 4.15 (dd, 1H, J = 3.6, 12.4 Hz), 4.37–4.45 (m, 2H), 7.46 (d, 1H, J = 8.4 Hz), 7.67 (d, 1H, J = 8.4 Hz), 7.88 (s, 1H), 7.99 (s, 1H). ESI MS: m/z 417.1 (M+H). HRMS calcd for C16H15F3N4O6 [M+H+] 417.1022, found 417.1014. HPLC purity: 95.3%.
(S)-N-(3-Methoxy-4-(trifluoromethoxy)benzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (56c)
13% yield. 1H NMR (DMSO-d6): δ 2.80–2.90 (m, 1H), 3.24 (br s, 1H), 3.65–3.88 (m, 5H), 3.90–4.00 (m, 1H), 4.10–4.20 (m, 1H), 4.38–4.45 (m, 2H), 6.95 (d, 1H, J = 11.0 Hz), 7.19 (s, 1H), 7.25 (d, 1H, J = 11.0 Hz), 8.00 (s, 1H). FT-ICR HRMS calcd for C15H14F3N4O5 [M+H+] 389.1067, found 389.1069.
(S)-N-(3-(Methoxymethoxy)-4-(trifluoromethoxy)benzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (56d)
12% yield. 1H NMR (CDCl3): δ 3.41 (m, 1H), 3.45 (s, 3H), 3.90 (d, 2H, J = 10 Hz), 3.94 (m, 1H), 4.15 (dd, 1H, J = 4.0, 12.0 Hz), 4.35–4.45 (m, 2H), 5.21 (s, 2H), 6.95 (d, 1H, J = 8.4 Hz), 7.19–7.21 (m, 2H), 7.38 (s, 1H). ESI MS: m/z 419.2 (M+H).
(S)-N-(3-Fluoro-4-trifluoromethoxybenzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (56a)
35% yield. 1H NMR (CDCl3): δ 3.40 (br s, 1H), 3.88–3.97 (m, 4H), 4.20 (dd, 1H, J = 4.0, 12.0 Hz), 4.38–4.46 (m, 2H), 7.11 (d, 1H, J = 8.0 Hz), 7.22 (dd, 1H, J = 1.6, 10.0 Hz), 7.28 (d, 1H, J = 8.0 Hz), 7.39 (s, 1H); 13C NMR (CDCl3): δ 47.5, 47.8, 49.7, 69.3, 115.5, 116.4, 116.6, 119.1, 121.6, 123.7, 135.4, 140.3, 143.3, 147.3, 153.2, 155.7. HRMS calcd for C14H12F4N4O5 [M+H+] 378.0742, found 378.0729.
(S)-N-(3-Chloro-4-trifluoromethoxybenzyl)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (56b)
37% yield. 1H NMR (CDCl3): δ 3.40 (br s, 1H), 3.87–3.97 (m, 4H), 4.19 (dd, 1H, J = 4.4, 12.0 Hz), 4.18–4.46 (m, 2H), 7.26–7.30 (m, 2H), 7.40 (s, 1H), 7.46 (s, 1H); 13C NMR (CDCl3): δ 47.5, 47.9, 49.6, 69.3, 115.4, 119.1, 121.7, 122.6, 127.2, 127.5, 130.1, 139.6, 143.4, 144.2, 147.3. HRMS calcd for C14H12ClF3N4O4 [M+H+] 393.0577, found 393.0574.
(S)-2-Nitro-N-(3-phenoxy-4-(trifluoromethoxy)benzyl)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-amine (56e)
11% yield. 1H NMR (CDCl3): δ 3.35 (br s, 1H), 3.80–3.90 (m, 3H), 4.14 (dd, 1H, J = 4.0, 12.0 Hz), 4.30–4.41 (m, 2H), 6.95 (d, 1H, J = 1.6 Hz), 6.98 (d, 2H, J = 8.0 Hz), 7.07 (dd, 1H, J = 1.6, 8.0 Hz), 7.16 (t, 1H, J = 8.0 Hz), 7.30 (d, 1H, J = 8.4 Hz), 7.34–7.38 (m, 3H); 13C NMR (CDCl3): δ 47.6, 47.7, 50.0, 69.2, 115.2, 118.5, 119.5, 122.9, 123.5, 123.8, 129.8, 139.4, 143.4, 147.2, 149.5, 156.4. HRMS calcd for C20H17F3N4O5 [M+H+] 451.1229, found 451.1228.
(S)-5-((2-Nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-ylamino)methyl)-2-(trifluoromethoxy)-phenol (56i)
To a solution of 56d (20 mg, 0.047 mmol) in THF (1 mL) was added 6N HCl (1 mL). After 1h, the reaction mixture was neutralized with aq. NaHCO3 solution and extracted with ethyl acetate. The organic layer was washed with water, brine and dried (anh. Na2SO4) and concentrated under reduced pressure. The residue was triturated with ether/pentane to obtain 56i (8.0 mg 47%). 1H NMR (CDCl3): δ 3.40 (m, 1H), 3.91 (d, 2H, J = 10 Hz), 3.95 (m, 1H), 4.16 (dd, 1H, J = 4.0, 12.0 Hz), 4.39–4.44 (m, 2H), 5.50 (br s, 1H), 6.86 (d, 1H, J = 8.4 Hz), 7.02 (br s, 1H), 7.19 (d, 1H, J = 8.4 Hz), 7.35 (s, 1H). HRMS calcd for C14H13F3N4O5 [M+H+] 375.0916, found 375.0929. HPLC purity: 95.7%.
4-(Bromomethyl)-2-(methoxymethoxy)-1-(trifluoromethoxy)benzene(58a)
To a solution of 46 (1.1 g, 3.0 mmol) in THF (10 mL) was added TBAF (1.0 g, 3.6 mmol). After stirring at room temperature for 1 h, the reaction mixture was diluted with water and extracted with CH2Cl2. The organic layer was washed with brine, dried (anh. Na2SO4) and concentrated to give (3-(methoxymethoxy)-4-(trifluoromethoxyphenyl)methanol (600 mg, 1.039 mmol) which was dissolved in CH2Cl2 (20 mL) and treated with triphenylphosphine (644 mg, 2.46 mmol) and NBS (437 mg, 2.458 mmol) at 0 °C. After stirring for 30 min at room temperature, the reaction mixture was poured into water (5 mL) and extracted with diethyl ether (2 × 15 mL). The organic layer was washed with water, NaHCO3 solution, brine and dried (Na2SO4). The solvent was evaporated under reduced pressure and the residue passed through a short silica pad to afford 58a (500 mg, 66%) which was contaminated with 15% triphenylphosphine oxide. This material was used as such for next reaction, without further purification. 1H NMR (DMSO-d6): δ 3.40 (s, 3H), 4.69 (s, 2H), 5.29 (s, 2H), 7.17 (dd, 1H, J = 2.0, 8.4 Hz), 7.37 (d, 1H, J = 8.4 Hz), 7.41 (d, 1H, J = 2.0 Hz).
In a similar manner, 4-(bromomethyl)-2-methoxy-1-(trifluoromethoxy)benzene (58c) was prepared from (3-methoxy-4-(trifluoromethoxy)phenyl)methanol in 46% yield. 1H NMR (CDCl3): δ 3.89 (s, 3H), 4.46 (s, 2H), 6.96 (dd, 1H, J = 2.8, 10.8 Hz), 7.02 (d, 1H, J = 2.8 Hz), 7.18 (dd, 1H, J = 1.2, 10.8 Hz).
(S)-6-(3-(Methoxymethoxy)-4-(trifluoromethoxy)benzyloxy)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (59a)
To a solution of 4-(bromomethyl)-2-(methoxymethoxy)-1-(trifluoromethoxy)benzene (0.3 g, 0.9 mmol) (S)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-ol (57, 0.15 g, 0.81 mmol) in DMF (10 mL) was added NaH (60% in mineral oil, 36 mg, 0.9 mmol) at −78 °C and warmed to room temperature over 1 h. Water (5 mL) was added and the reaction mixture extracted with EtOAc (2 × 30 mL). The organic layer was washed with water, brine, dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by column chromatography over silica gel using a solvent gradient of 0 – 50% EtOAc in petroleum ether to afford 59a (115 mg, 34%). 1H NMR (DMSO-d6): δ 3.37 (s, 3H), 4.25 (m, 3H), 4.46 (d, 1H, J = 12.4 Hz), 4.60–4.70 (m, 3H), 5.26 (s, 2H), 7.02 (d, 1H, J = 8.4 Hz), 7.22 (s, 1H), 7.34 (d, 1H, J = 8.4 Hz), 8.03 (s, 1H). ESI MS: m/z 420.3 (M+H).
In a similar manner, (S)-6-(3-methoxy-4-(trifluoromethoxy)benzyloxy)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (59c) was synthesized in 30% yield. 1H NMR (DMSO-d6): δ 3.80 (s, 3H), 4.20–4.30 (m, 3H), 4.48 (m, 1H), 4.60–4.70 (m, 3H), 6.96 (d, 1H, J = 8.4 Hz), 7.14 (s, 1H), 7.31 (d, 1H, J = 8.4 Hz), 8.03 (s, 1H). FT-ICR HRMS calcd for C15H14F3N3O6 [M+H+] 390.0907, found 390.0909.
(S)-6-(3-Fluoro-4-(trifluoromethoxy)benzyloxy)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (59b)
To a stirred solution of 49b (7.0 g, 33.65 mmol) in methanol (50 mL) was added NaBH4 (1.9 g, 50.48 mmol) at room temperature under the N2 atmosphere. After 1h, ice water (100 mL) was added and the reaction mixture extracted with EtOAc (3 × 60 mL). The combined organic layer was washed with water (50 mL), brine, dried over (anh. Na2SO4) and concentrated under reduced pressure to afford 3-fluoro4-trifluoromethoxybenzyl alcohol (7 g, quant.). 1H NMR (DMSO-d6): δ 4.53 (d, 2H, J = 5.6 Hz), 5.44 (t, 1H, J = 5.6 Hz), 7.26 (d, 1H, J = 8.4 Hz), 7.40 (d, 1H, J = 11.2 Hz), 7.48–7.54 (t, 1H, J = 8.4 Hz). ESI MS: m/z 211.0 (M+H). To a solution of 3-fluoro-4-trifluoromethoxybenzyl alcohol (7 g, 33.33 mmol) in CH2Cl2 (70 mL) at 0 °C was added triphenylphosphene (13.0 g, 49.99 mmol) followed by NBS (8.9 g, 49.99 mmol) in portion-wise at room temperature under the N2 atmosphere. After 1h, water (100 mL) was added and the reaction mixture extracted with EtOAc (3 × 40 mL). The combined organic layer was washed with water (30 mL), brine, dried over (anh. Na2SO4) and concentrated under reduced pressure. The crude compound was purified by column chromatography over silica gel using a gradient of 0–5% EtOAc-hexane as eluent to afford 4-(bromomethyl)-2-fluoro-1-(trifluoromethoxy)benzene (58b, 6.0 g, 66%). 1H NMR (DMSO-d6): 4.73 (s, 2H), 7.42 (d, 1H, J = 8.4 Hz), 7.55–7.71 (m, 2H). Alkylation of 57 using 58b was carried out as described previously during the synthesis of 59a to afford 59b (32% yield). 1H NMR (DMSO-d6): δ 4.20–4.35 (m, 3H), 4.47 (d, 1H, J = 11.6 Hz), 4.65–4.75 (m, 3H), 7.26 (d, 1H, J = 8.0 Hz), 7.44 (d, 1H, J = 10.8 Hz), 7.55 (t, 1H, J = 8.0 Hz), 8.04 (s, 1H). 13C NMR (CD3OD): δ 58.3, 68.6, 69.2, 70.1, 117.1, 117.3, 117.7, 120.6, 123.1, 124.8, 124.9, 136.8, 141.0, 143.9, 149.1, 154.4, 156.9. HRMS calcd for C14H11F4N3O5 [M+H+] 378.0742, found 378.0729.
(S)-5-((2-Nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-yloxy)methyl)-2-(trifluoromethoxy) phenol (59d)
To a solution of 59a (150 mg, 0.357 mmol) in THF (5 mL) was added 6N HCl (10 mL) and stirred for 1h at room temperature. The reaction mixture was neutralized with sat. NaHCO3 solution, extracted with EtOAc (2 × 20 mL). The organic layer was washed with water, brine solution, dried (Na2SO4) and evaporated under reduced pressure. The residue was triturated with diethyl ether/pentane (1:1) mixture to afford 59d (90 mg, 67%). 1H NMR (DMSO-d6): δ 4.21–4.27 (m, 3H), 4.47 (d, 1H, J = 12.0 Hz), 4.55–4.67 (m, 3H), 6.78 (dd, 1H, J = 1.6, 8.4 Hz), 6.94 (d, 1H, J = 1.2 Hz), 7.20 (d, 1H, J = 8.0 Hz), 8.03 (s, 1H), 10.16 (s, 1H). 13C NMR (DMSO-d6): δ 46.7, 66.5, 67.8, 68.9, 116.3, 118.0, 119.0, 121.0, 122.8, 135.3, 138.3, 142.1, 147.0, 149.7. HRMS calcd for C14H12F3N3O6 [M+H+] 376.0756, found 376.0750.
In vivo Pharmacokinetic Studies
The Novartis Institute for Tropical Disease’s Animal Care and Use Committee approved all animal experimental protocols and animals use. The compounds were formulated in 10% w/v hydroxypropyl-β-cyclodextrin (Acros) and 10% w/v lecithin (Acros/Organics, New Jersey, USA). The formulation was centrifuged and the supernatant was injected into animals via the intravenous route at 5mg/kg. Blood samples were collected at various time points between 1min and 24h post dose. Plasma samples were extracted and analyzed using optimum LC/MS/MS conditions. Pharmacokinetic parameters were determined using WinNonLin Professional, version 5.0.1 (Pharsight, CA, USA), by non-compartmental analysis. The Extraction Ratio (ER) was calculated using the formula as previously described21.
Quantum Chemistry and Superposition Study
In our modeling studies, both pseudo-equatorial and pseudo-axial forms of 1 were used as reference structures22. The pseudo-equatorial conformer of 1 was built by replacing the methyl of 7-(R)-methyl-1 (X-ray structure) with a hydrogen atom while the X-ray structure of 1 was used for the pseudo-axial conformer without further modification. The geometries of these two conformers of 1 were then energy minimized with the density functional theory at the level of B3LYP/6-31G*23. The two resulting conformers were subsequently modified to build structures of the corresponding conformers of 32c, 32d, 32f, 41b, and 41f. These compounds (each with two conformations) were then energy minimized at the level of B3LYP/6-31G* in cyclohexane utilzing the polarized continuum solvent model with the UAKS parameters set. The resulting structures were taken as the assumed geometries of these compounds. Alternative conformers were considered by varying the hydrogen position of the linker group (NH), which can H-bond with the corresponding ortho-substituent. This was done by first changing the dihedral angle of H-N-C6-C5, and then after another cycle of full geometry optimization, the respective low energy conformers were chosen for comparison (see Fig. 2). For the superposition study, the optimized structures of 32c, 32d, 32f, 41b, and 41f were overlaid onto the geometry optimized 1 with the rigid fit of Quanta 2008 (Accelrys) using the nine heavy atoms of the imidazo-oxazine as a common alignment site. The root mean square deviations of such fit for each compound were all less than 0.01 Å and 0.03 Å for the pseudo-equatorial and the pseudo-axial conformers, respectively. The energy barrier for the conversion of pseudo-axial 1 to the pseudo-equatorial form was calculated by varying the dihedral angle of O8-C7-C6-O, and then further optimizing the geometry with the highest enegy on the potential energy surface with the keyword, opt=(ts,calcfs,noeigentest) as implemented in Gausssian 09 software23.
Acknowledgments
This work was funded, in part, by the intramural research program of NIH, NIAID and by a grant from the Bill and Melinda Gates Foundation and the Wellcome Trust through the Grand Challenges in Global Health Initiative. We thank Anne Goh and the bioanalytical team of NITD for the determination of compound concentrations in microsomal and plasma samples and Dr. Noel Whittaker, NIDDK for HRMS of some of the compounds. The quantum chemical study utilized PC/LINUX clusters at the Center for Molecular Modeling of the NIH (http://cit.nih.gov), and this research was also supported in part by the NIH Intramural Research Program through the Center for Information Technology.
Abbreviations
- AcOH
acetic acid
- DIPEA
N,N-diiospropylethylamine
- DMF
dimethylformamide
- ER
extraction ratio
- MAC
minimum anaerobicidal concentration
- MIC
minimum inhibitory concentration
- MsCl
methanesulfonyl chloride
- Mtb
Mycobacterium tuberculosis
- Mtz
metronidazole
- NBS
N-bromosuccinimide
- SAR
structure-activity relationship
- TB
tuberculosis
- TBAF
tetra-n-butylammonium fluoride
- TBS or TBDMS
tert-butyldimethylsilyl
- Tf
triflate
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TMEDA
tetramethlyethylenediamine
- TMSCN
trimethylsilyl cyanide
References
- 1.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 2.Matsumoto M, Hashizume H, Tomishige T, Kawasaki M, Tsubouchi H, Sasaki H, Shimokawa Y, Komatsu M. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006;3:e466. doi: 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sasaki H, Haraguchi Y, Itotani M, Kuroda H, Hashizume H, Tomishige T, Kawasaki M, Matsumoto M, Komatsu M, Tsubouchi H. Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles. J Med Chem. 2006;49:7854–7860. doi: 10.1021/jm060957y. [DOI] [PubMed] [Google Scholar]
- 4.Diacon AH, Dawson R, Hanekom M, Narunsky K, Maritz SJ, Venter A, Donald PR, van Niekerk C, Whitney K, Rouse DJ, Laurenzi MW, Ginsberg AM, Spigelman MK. Early bactericidal activity and pharmacokinetics of PA-824 in smear-positive tuberculosis patients. Antimicrob Agents Chemother. 2010;54:3402–3407. doi: 10.1128/AAC.01354-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tasneen R, Tyagi S, Williams K, Grosset J, Nuermberger E. Enhanced bactericidal activity of rifampin and/or pyrazinamide when combined with PA-824 in a murine model of tuberculosis. Antimicrob Agents Chemother. 2008;52:3664–3668. doi: 10.1128/AAC.00686-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nuermberger E, Tyagi S, Tasneen R, Williams KN, Almeida D, Rosenthal I, Grosset JH. Powerful bactericidal and sterilizing activity of a regimen containing PA-824, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob Agents Chemother. 2008;52:1522–1524. doi: 10.1128/AAC.00074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nuermberger E, Rosenthal I, Tyagi S, Williams KN, Almeida D, Peloquin CA, Bishai WR, Grosset JH. Combination chemotherapy with the nitroimidazopyran PA-824 and first-line drugs in a murine model of tuberculosis. Antimicrob Agents Chemother. 2006;50:2621–2625. doi: 10.1128/AAC.00451-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyagi S, Nuermberger E, Yoshimatsu T, Williams K, Rosenthal I, Lounis N, Bishai W, Grosset J. Bactericidal activity of the nitroimidazopyran PA-824 in a murine model of tuberculosis. Antimicrob Agents Chemother. 2005;49:2289–2293. doi: 10.1128/AAC.49.6.2289-2293.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Eum SY, Taylor K, Klein E, Manjunatha U, Gonzales J, Lee EG, Park SK, Raleigh JA, Cho SN, McMurray DN, Flynn JL, Barry CE., 3rd Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun. 2008;76:2333–2340. doi: 10.1128/IAI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barry CE, 3rd, Boshoff HI, Dowd CS. Prospects for clinical introduction of nitroimidazole antibiotics for the treatment of tuberculosis. Curr Pharm Des. 2004;10:3239–3262. doi: 10.2174/1381612043383214. [DOI] [PubMed] [Google Scholar]
- 11.Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, Daniels L, Dick T, Pang SS, Barry CE., 3rd Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103:431–436. doi: 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh R, Manjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, Kang S, Keller TH, Jiricek J, Barry CE., 3rd PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008;322:1392–1395. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim P, Zhang L, Manjunatha UH, Singh R, Patel S, Jiricek J, Keller TH, Boshoff HI, Barry CE, 3rd, Dowd CS. Structure-activity relationships of antitubercular nitroimidazoles. 1. Structural features associated with aerobic and anaerobic activities of 4- and 5-nitroimidazoles. J Med Chem. 2009;52:1317–1328. doi: 10.1021/jm801246z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim P, Kang S, Boshoff HI, Jiricek J, Collins M, Singh R, Manjunatha UH, Niyomrattanakit P, Zhang L, Goodwin M, Dick T, Keller TH, Dowd CS, Barry CE., 3rd Structure-activity relationships of antitubercular nitroimidazoles. 2. Determinants of aerobic activity and quantitative structure-activity relationships. J Med Chem. 2009;52:1329–1344. doi: 10.1021/jm801374t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baker WRSC, Keeler EL. Nitro-[2,1-b]imidazopyran compounds and antibacterial uses thereof. 6,087,358. US Patent. 2000
- 16.Baker WRSC, Keeler EL. Nitroimidazole antibacterial compounds and methods of use thereof. 5,668,127. US Patent. 1997
- 17.Palmer BD, Thompson AM, Sutherland HS, Blaser A, Kmentova I, Franzblau SG, Wan B, Wang Y, Ma Z, Denny WA. Synthesis and structure-activity studies of biphenyl analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2, 1-b][1,3]oxazine (PA-824) J Med Chem. 2010;53:282–294. doi: 10.1021/jm901207n. [DOI] [PubMed] [Google Scholar]
- 18.Harn NK, Gramer CJ, Anderson BA. Acylation of oxazoles by the copper-mediated reaction of oxazol-2-ylzinc chloride derivatives. Tetrahedron Letters. 1995;36:9453–9456. [Google Scholar]
- 19.Marzi E, Schlosser M. The site-selective functionalization of halogen-bearing phenols: an exercise in diversity-oriented organometallic synthesis. Tetrahedron. 2005;61:3393–3401. [Google Scholar]
- 20.Frey LF, Marcantonio KM, Chen CY, Wallace DJ, Murry JA, Tan LS, Chen WR, Dolling UH, Grabowski EJJ. Practical synthesis of a highly functionalized thiazole ketone. Tetrahedron. 2003;59:6363–6373. [Google Scholar]
- 21.Lau YY, Krishna G, Yumibe NP, Grotz DE, Sapidou E, Norton L, Chu I, Chen C, Soares AD, Lin CC. The use of in vitro metabolic stability for rapid selection of compounds in early discovery based on their expected hepatic extraction ratios. Pharm Res. 2002;19:1606–1610. doi: 10.1023/a:1020765025857. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Manjunatha UH, Goodwin MB, Knox JE, Lipinski CA, Keller TH, Barry CE, 3rd, Dowd CS. Synthesis and antitubercular activity of 7-(R)- and 7-(S)-methyl-2-nitro-6-(S)-(4-(trifluoromethoxy)benzyloxy)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazines, analogues of PA-824. Bioorg Med Chem Lett. 2008;18:2256–2262. doi: 10.1016/j.bmcl.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.2. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 24.Sinnokrot MO, Sherrill CD. High-Accuracy Quantum Mechanical Studies of p-p Interactions in Benzene Dimers. J Phys Chem A. 2006;110:10656–10668. doi: 10.1021/jp0610416. [DOI] [PubMed] [Google Scholar]
- 25.Grimme S. Do Special Noncovalent p–p Stacking Interactions Really Exist? Angew Chem Int Ed. 2008;47:3430 –3434. doi: 10.1002/anie.200705157. [DOI] [PubMed] [Google Scholar]