

Abstract
The ability to recognize and repair abnormal DNA structures is common to all forms of life. Studies in a variety of species have identified an incredible diversity of DNA repair pathways. Documenting and characterizing the similarities and differences in repair between species has important value for understanding the origin and evolution of repair pathways as well as for improving our understanding of phenotypes affected by repair (e.g., mutation rates, lifespan, tumorigenesis, survival in extreme environments). Unfortunately, while repair processes have been studied in quite a few species, the ecological and evolutionary diversity of such studies has been limited. Complete genome sequences can provide potential sources of new information about repair in different species. In this paper we present a global comparative analysis of DNA repair proteins and processes based upon the analysis of available complete genome sequences. We use a new form of analysis that combines genome sequence information and phylogenetic studies into a composite analysis we refer to as phylogenomics. We use this phylogenomic analysis to study the evolution of repair proteins and processes and to predict the repair phenotypes of those species for which we now know the complete genome sequence.
Keywords: DNA repair, molecular evolution, phylogenomics, gene duplication and gene loss, orthology and paralogy
1. Introduction
Genomic integrity is under constant threat in all species. These threats come in many forms (e.g., agents that damage DNA, spontaneous chemical changes, and errors in DNA metabolism), lead to a variety of alterations in the normal DNA structure (e.g., single- and double-strand breaks, chemically modified bases, abasic sites, bulky adducts, inter- and intra-strand cross-links, and base-pairing mismatches) and have many direct and indirect effects on cells and organisms (e.g., mutations, genetic recombination, the inhibition or alteration of cellular processes, chromosomal aberrations, tumorigenesis, and cell death). Given this diversity of threats and their effects, it is not surprising that there is a corresponding diversity of DNA repair processes. Overall, repair pathways have been found that can repair just about any type of DNA abnormality. The cellular functions of all known repair pathways are also diverse. These functions include the correction of replication errors, resistance to killing by DNA damaging agents, chromosome duplication and segregation, cell cycle control, generation of antibody diversity in vertebrates, regulation of interspecies recombination, meiotic and mitotic recombination, transcription or replication elongation, and tumor suppression. The diversity of DNA repair pathways can be readily seen by comparing and contrasting different pathways. For example, some pathways are able to repair only a single type of abnormality, others are quite broad and are able to repair many abnormalities. Similarly, some pathways are simple, involving single enzymes and single steps; others are highly complex, involving many steps and dozens of enzymes working in concert. In addition, some pathways have single functions while others have roles in a variety of cellular processes.
The diversity of specificity, functions, and complexity of repair pathways is best understood by comparing mechanisms of action among pathways. Such comparisons are simplified by the division of repair processes into three major classes based on general mechanism of action: direct repair (in which abnormalities are chemically reversed), recombinational repair (in which homologous recombination is used to repair abnormalities) and excision repair (in which a section of the DNA strand containing an abnormality is removed and a repair patch is synthesized using the intact strand as a template). Within each of these classes there are multiple types and sometimes even subtypes of repair. For example, there are dozens of different subtypes of base excision repair (BER), which itself is one of three main types of excision repair (the other two being nucleotide excision repair (NER) and mismatch excision repair (MMR)).
The diversity of DNA repair pathways outlined above is the diversity of all known repair processes in all species. One aspect of this overall diversity is that found within species. For example, Escherichia coli, Saccharomyces cerevisiae, and humans each exhibit all the major classes of repair, and multiple types and even subtypes of each class. It is likely that most or even all species also have many classes and types of repair. The within species diversity allows a species to recognize and repair many types of abnormalities and also provides redundancy since there is overlap among many pathways. Another aspect of the diversity of DNA repair is that due to differences between species. These interspecific differences come in two forms. First, although all species have many types of repair, the exact repertoire of types differs between species. For example, although all species studied have BER, the particular types of abnormal bases that are repaired by BER differ greatly. Similarly, photoreactivation (PHR) is found in some species, such as E. coli and yeast, but not others, such as humans (1). There are also differences within particular types and subtypes of repair between species. For example, in those species that have been found to have MMR, the particular mismatches that are best repaired is highly species specific (2). Differences in specificity exist in almost every type of repair even between closely related species.
Differences in the specificity and types of repair such as those described above can have profound biological effects. For example, it has been suggested that the accelerated mutation rate in mycoplamsas may be due in part to deficiencies in DNA repair (3, 4). Examples of other phenotypes and features that may be variable between individuals, strains or species due to differences in repair include cancer rates (5), lifespan (6, 7), pathogenesis (8, 9, 10), codon usage and GC content (11, 12), evolutionary rates (13), survival in extreme environments (14), speciation (15, 16), and diurnal/nocturnal patterns (17). Thus, to understand differences in any of these phenotypes, it is useful to understand differences in repair.
Characterization of repair in different species is also of great use in understanding the evolution of repair proteins and processes. This is important not just because repair is a major cellular process but also because information about the evolution of repair provides a useful perspective for comparative repair studies. In general an evolutionary perspective is useful in any comparative study because it allows a focus on how and why similarities and differences arose rather than the simple identification and characterization of similarities and differences (18). For studies of DNA repair, we believe an evolutionary perspective is the key to understanding differences in repair between species, as well as the mechanisms and functions of particular repair processes (19, 20, 21).
Unfortunately, comparative and evolutionary studies of DNA repair processes have been limited because of the lack of detailed studies of repair in a wide ecological and evolutionary diversity of species (19). Recently, a potential new source of comparative repair data has emerged: complete genome sequences. In theory, complete genome sequences should enable the prediction of the phenotype of a particular strain or species, while also providing a wealth of data for comparative analysis. In practice, however, obtaining useful information from complete genome sequences is quite difficult. We have been developing a new approach that combines the analysis of complete genome sequences with evolutionary reconstructions into a composite analysis we refer to as phylogenomics (21, 22, 23). We present here a global phylogenomic analysis of DNA repair proteins and processes. We use this phylogenomic analysis to infer the evolutionary history of repair pathways and the respective proteins that comprise them and to make predictions about the repair phenotypes of species for which genomes have been sequenced. In addition, we discuss the uses of evolutionary analysis in studies of complete genome sequences, the uses of complete genome sequences in studies of evolution, and the advantages of the combined phylogenomic approach.
2. Methods
Our phylogenomic analysis can be divided into a series of steps, with feedback loops between some steps such that initial analyses are subsequently refined (see Table 2 for an outline of methods used). The steps are described below as well as in some previous papers (21, 22, 23).
Table 2.
Components of phylogenomic analysis
| Component | How is it Determined? | Uses of This Component |
|---|---|---|
| Gene Analysis | ||
| 1. Database of genes of interest. | Personal choice, characterized genes. | Similarity searches (2). |
| 2. Searching for homologs. | Blast, PSI-blast, BLOCKS. Set homology threshold. | Presence/absence (4); gene tree (7). |
| 3. Functional predictions. | Overlay known functions of genes onto gene tree. | Prediction of phenotypes (6); functional evolution. |
| Genome Analysis | ||
| 4. Gene presence/absence in species. | Searches (2) of complete genome sequences. Some refinement from evolutionary analysis (7, 10). | Evolutionary analysis (8, 10) |
| 5. Correlated presence/absence. | Analyze presence/absence (4) in different species. | Functional predictions (3), pathway evolution (11). |
| 6. Phenotype predictions. | Combine functional predictions (3), presence/absence (4) and pathway evolution (11). | Identify universal activities. |
| Evolutionary Analysis | ||
| 7. Gene trees. | Set homology threshold for searches (2) and use phylogenetic analysis of all homologs. | Presence/absence (4); identifying evolutionary events (10), functional predictions (3). |
| 8. Evolutionary distribution patterns. | Overlay gene presence/absence (4) onto species tree. | Identifying gene evolutionary events; pathway evolution. |
| 9. Congruence. | Compare gene tree (7) to species tree. | Distinguish lateral transfer from other events (8). |
| 10. Gene evolution events. | Analysis of gene tree (7), congruence (9) and evolutionary distribution patterns (8). | Pathway evolution (11), correlated and convergent events, presence/absence (4); functional predictions (3) |
| 11. Pathway evolution. | Integrate gene evolution (10), evolutionary distribution (8), correlated presence/absence (5). | Phenotype predictions (6); functional predictions (3). |
2. 1. Presence and Absence of Homologs
The first major step in phylogenomic analysis is the determination of the presence and absence of homologs of genes of interest in different species. For the analysis here, genes with established roles in DNA repair processes were identified by a comprehensive review of the literature. Likely homologs of these genes were identified by searching a variety of sequence databases using the blast and blast2 search algorithms (24). A conservative operational definition of homology (i.e., high threshold of sequence similarity) was used to limit the number of false positive results (i.e., identifying genes as homologs that do not share common ancestry). In some cases, this threshold was lowered if other evidence suggested that homologs were highly divergent (see Discussion). Since this conservative approach might lead to false negatives, iterative search methods (e.g., PSI-blast (24) and manual methods) were used to increase the likelihood of identifying highly divergent homologs of the reference protein. Presence and absence of homologs of genes in particular species was determined by searching (using the above methods) against complete genome sequences (Table 1). Homologs of repair genes that had been cloned from species for which complete genomes were not available were identified by searching against the nr and EST databases at the National Center for Biotechnology (www.ncbi.nlm.nih.gov). The amino-acid sequences of all putative homologs of a particular gene were aligned using the clustalw program (25). The alignments were examined by eye to assess the reliability of the homology assignments. In addition, block-motifs were made of alignments using the blocks web server (www.blocks.fhcrc.org). These were then used for additional database searches to identify sequences containing motifs similar to those that were aligned together.
Table 1.
Completely or nearly completely sequenced genomes analyzed
| Species | Classification | Size (mb) | # Orfs | Ref. |
|---|---|---|---|---|
| Bacteria | ||||
| Escherichia coli K-12 | Proteobacteria (γ) | 4.60 | 4288 | (172) |
| Haemophilus influenzae Rd KW20 | Proteobacteria (γ) | 1.83 | 1743 | (30) |
| Rickettsia prowazekii Madrid E | Proteobacteria (α) | 1.1 | ~834 | (173) |
| Helicobacter pylori 26695 | Proteobacteria (ε) | 1.67 | 1590 | (174) |
| Helicobacter pylori 26695 | Proteobacteria (ε) | 1.67 | 1590 | (175) |
| Campylobacter jejuni NCTC 1168 | Proteobacteria (ε) | 1.70 | n/a | (176) |
| Bacillus subtilis 169 | Low GC Gram + | 4.20 | 4100 | (177) |
| Mycoplasma genitalium G-37 | Low GC Gram + | 0.58 | 470 | (178) |
| Mycoplasma pneumoniae M129 | Low GC Gram + | 0.82 | 679 | (179) |
| Mycobacterium tuberculosis H37rV | High GC Gram + | 4.41 | ~4000 | (180) |
| Borrelia borgdorferi B31 | Spirochete | 1.44 | 1283 | (181) |
| Treponema pallidum Nichols | Spirochete | 1.14 | 1041 | (182) |
| Chlamydia trachomatis serovar D | Chlamydia | 1.05 | n/a | (183) |
| Synechocystis sp. PCC6803 | Cyanobacteria | 3.57 | 3168 | (184) |
| Deinococcus radiodurans R1 | Deinoccus/Thermus | 3.20 | 3193 | (185) |
| Thermotoga maritima MSB8 | Thermotogales | 1.80 | 1877 | (171) |
| Aquifex aeolicus VF5 | Aquificaceae | 1.55 | 1512 | (186) |
| Archaea | ||||
| Methanococcus jannaschii DSM 2661 | Euryarchaeota | 1.66 | 1738 | (187) |
| Methanobacterium thermoautotrophicum ΔH | Euryarchaeota | 1.75 | 1855 | (188) |
| Pyrococcus horikoshii OT3 | Euryarchaeota | 1.80 | ~2000 | (189) |
| Archaeoglobus fulgidus VC-16, DSM4304 | Euryarchaeota | 2.18 | 2436 | (190) |
| Eukaryote | ||||
| Saccharomyces cerevisiae S288C | Fungi | 13.0 | 5885 | (191) |
2.2. Evolutionary Relationships among Homologs
The second major step in phylogenomic analysis is the characterization of the evolutionary relationships among all homologs of each gene. To do this, phylogenetic trees were generated for each group of homologs from the sequence alignments (excluding poorly conserved regions) by the neighbor-joining and parsimony methods of the PAUP* program (26). The robustness of phylogenetic patterns was assessed using bootstrapping and by comparing phylogenetic trees generated with different algorithms.
2.3. Inference of Evolutionary Events
In the third major step in phylogenomic analysis, four main events in the history of each gene family (gene origin, gene duplication, lateral gene transfer and gene loss) are inferred. The first step in identifying these events involves determining evolutionary distribution patterns (EDPs) for each gene. EDPs, which are determined by overlaying gene presence/absence information onto an evolutionary tree of species, reveal a great deal about the evolutionary history of particular genes (see Table 3). For example, if a gene is present in only one subsection of the species tree, then it likely originated in that subsection. However, some EDPs do not have a single likely mechanism of generation and thus require further analysis before being used to identify specific evolutionary events. For example, an uneven distribution pattern (scattered presence and absence throughout the species tree) can be explained either by lateral transfer to the species with an unexpected presence of the gene or by gene loss in species with an unexpected absence. Ascertaining which event occurred can usually be accomplished by comparing the gene tree to the species tree and testing for congruence. If there has been a lateral transfer in the past, then the species tree and the gene tree should be incongruent (i.e., they should have different branching topology). In contrast, if there has been a gene loss in the past, then the gene and species trees should be congruent except that some species will not be represented in the gene tree. This comparison of gene tree, species tree, and EDPs was used to identify likely cases of gene duplication, loss, and lateral transfer in the history of every DNA repair gene. Then parsimony reconstruction methods were used to determine the likely timing of gene origin, loss, duplication and transfer events. In this analysis, we used the MacClade computer program (27) to attempt to identify the evolutionary scenario that requires the fewest events to arrive at the current distribution patterns. Since this type of analysis is not commonly used for molecular data, an example (for tracing gene loss) is presented in Figure 1.
Table 3.
Evolutionary distribution patterns
| Type of pattern1 | Description | Likely explanations | How resolve ambiguities? |
|---|---|---|---|
| Universal | All species have the gene. | Gene is ancient and probably universally required in all species. | n/a |
| Uniform presence | Gene is in only one evolutionary lineage. | Gene originated in that lineage. | n/a |
| Uniform absence | Gene is missing from one lineage. | Gene lost in that lineage. | n/a |
| Uneven | Presence/absence scattered through tree. | Gene loss or lateral transfer. | Compare gene tree vs. species tree. |
| Multicopy | Multiple homologs in some species. | Gene duplication or lateral transfer. | Compare gene tree vs. species tree. |
Determined by overlaying presence/absence of genes onto evolutionary tree of species
Figure 1. Demonstration of using evolutionary distribution patterns to trace gene gain and loss.

An evolutionary tree of the relationships among some representatives of the bacteria, Archaea, and eukaryotes is shown. Presence of genes in these species is indicated by a colored box at the tip of the terminal branches of the tree. Gain and loss of the gene is inferred through parsimony reconstruction techniques. Within the bacterial part of the tree, we divide the species into major phyla but have collapsed the branches joining the different phyla to indicate that the relationships among these phyla are ambiguous.
An essential component in the identification of gene loss, duplication, origin, and transfer is the species tree. Unfortunately, there is no general consensus concerning the relationships among all of the species analyzed here. For the analysis described here, a species tree based upon the Ribosomal Database Project trees (28) was used. In this tree, Archaea, bacteria and eukaryotes are each monophyletic and Archaea are a sister group to eukaryotes. In the sections on specific repair pathways, the possible effects of alternative species trees are discussed.
2. 4. Refining homology groups
In the fourth major step in phylogenomic analysis, evolutionary analysis is used to refine the list of presence and absence of homologs of particular genes. For the analysis reported here, this involved two types of refinement. First, if gene duplication events were identified, genes were divided into groups of orthologs and paralogs, and then presence and absence was determined only for orthologs of the query gene. In addition, in some cases, gene trees were used to subdivide gene families into evolutionarily distinct subfamilies, and then presence and absence was determined only for homologs in the same subfamily as the query gene.
2.5. Functional predictions and functional evolution
The fifth major step in phylogenomic analysis involves studies of functional evolution for individual gene families. Functional evolution was studied by overlaying information on gene functions onto the gene trees. Then parsimony reconstruction methods were used to trace changes of function over evolutionary time (this was done in much the same was as for presence and absence of genes described above). Tracing functional evolution is an important component of making functional predictions for both ancestral genes and uncharacterized genes as described (22) and phenotypic predictions for species. For example, if there have been many functional changes in the history of a particular gene or gene family, then the identification of the presence of homologs of such genes in a species is not sufficient information to predict the presence of a particular activity. Thus tracing functional changes helps prevent incorrect predictions of function. In addition, tracing functional changes can also greatly improve the chances of making correct functional predictions for ancestral genes and uncharacterized genes (22). Such functional predictions are made based on the position of the gene of interest in the gene tree relative to genes with known functions and based on identifying evolutionary events such as gene duplications that may identify groups of genes with similar functions (22). Specific functional predictions made for repair genes are discussed in the sections on the different repair pathways. It is important to note that all studies of functional evolution and predictions of gene function should use only experimental information on gene functions and not database annotation. Thus for our analysis we made extensive use of the primary literature on gene functions. We apologize for being unable to cite all sources here.
2.6 Pathway analysis
The last major step in phylogenomic analysis involves comparing and combining the results of analyses of different genes and pathways. One aspect of this is the comparison of the presence and absence of all the genes in a pathway in different species. If genes in a pathway are always present or absent as a unit (i.e., no gene in the pathway is ever present without the other genes), this suggests a conserved association among these genes. If the genes are not always present together, there are multiple possible explanations including that the pathway is found in the different species but that some genes have been replaced by non-orthologous genes (29)); that the pathway does not function the same way in all species; or that the genes do not work together as was thought. The presence and absence of genes in different species was studied for all repair pathways. Another important aspect of pathway analysis is determining if there are any correlated evolutionary events for different genes in a pathway (e.g., gene loss or duplication). Such correlated events lend extra support to a conserved association among genes, especially if correlated events occurred multiple times in different lineages. Correlated events were studied for all repair pathways. A third component of phylogenomic analysis of pathways involves comparing functional evolution between and within pathways. For example, if a particular activity evolved only once, then the presence or absence of the gene(s) required for that activity can be used as a good estimator of the presence and absence of the activity. If a particular activity evolved separately many times, then there may be many as of yet uncharacterized genes that can also provide that activity. Therefore, even if a species does not encode any of the genes known to have that activity, one should not conclude that the species does not have that activity.
3. Results and Discussion
The publication in 1995 of the first complete genome sequence of a free-living organism initiated a new phase of biology research (30). Currently, more than twenty complete genome sequences are publicly available (Table 1) and it is likely that there will be hundreds more available within a few years. These genome sequences provide an unprecedented window into the biology of the species that have been sequenced as well as into the evolution of life on the planet. To make the most out of genome sequences, both for studies of the biology of species and for studies of evolution, we believe that evolutionary reconstructions and genome analysis should be integrated into a single composite approach, which we refer to as phylogenomics (21, 22).
The first reason to combine evolutionary reconstructions and genomics is that evolutionary analysis can greatly improve what can be learned from genome sequences. In general, an evolutionary perspective is useful in any comparative biological study because it allows one to go beyond identifying what is similar or different between species and to focus instead on understanding how and why such similarities and differences may have arisen (18, 31). The benefits of an evolutionary perspective are well known in some aspects of comparative biology such as comparative physiology and ecology (18, 31). Although it is not well recognized, an evolutionary perspective has also been quite useful in many aspects of comparative molecular biology including making functional predictions (22), inferring mutation processes (32), determining secondary and tertiary structures of ribosomal RNA (33) and proteins (34), making motif-patterns for conserved proteins (35), and in sequence searching algorithms (36). All such methods can be of use in comparative genomic analysis as well.
Just as evolutionary methods can benefit comparative genomic studies, genome analysis is incredibly useful in studies of evolution. One aspect of this is the wealth of comparative data provided by genome sequences which allow studies of the evolutionary relationships among species in a way never before possible. It is the completeness of complete genome sequences that allows one to address questions never before possible in evolutionary studies. For example, one can now analyze codon usage of all genes in a genome and compare this between species (37). Complete genomes can also be used to compare and contrast the evolution of different pathways both within and between species.
The reason for a composite phylogenomic approach is that there are feedback loops between genome analysis and evolutionary reconstruction such that they are impossible to separate in some cases and in most other cases they can be combined for a mutual benefit. For example, in the inference of gene loss, complete genome information is required to show that a species does not encode any homologs of the gene thought to have been lost. Evolutionary analysis is then required to show that an ancestor of the species without the gene likely had the gene. Similarly, in the inference of gene duplication, genome analysis is required to determine the number of homologs of a particular gene in different species. Then evolutionary analysis is needed to divide the homologs into orthologs and paralogs. Finally genome analysis is required to determine the presence and absence of the different orthologs. There are many other areas in which genome and evolutionary analysis can be combined for mutual benefit including making functional predictions for individual genes (22), predicting species phenotpye, and tracing the evolution of pathways. We have incorporated many of these into our phylogenomic analysis (see Table 2 and Methods).
Here we apply our phylogenomic approach to the study of DNA repair processes. We have divided our analysis into two main sections. In the first section, we discuss our results on a pathway by pathway basis. For each pathway, we review what is known about the pathway and the proteins in that pathway in the species in which the pathway is best characterized. Then we discuss what is known about this pathway in other species. Finally we present the results of our phylogenomic analysis as well as results of other comparative or evolutionary studies of this pathway, such as the recently published comprehensive analysis of DNA repair domains (38). In the second section, we discuss our results from a broader perspective, looking at all repair pathways together. To simplify our discussion, we have summarized our results in a few ways. In Figure 3 we have traced the inferred gain and loss of repair genes onto an evolutionary tree of the species. In Table 6 we have sorted the repair genes by pathway and by the inferred timing of the origin of each gene.
Figure 3. Evolutionary gain and loss of DNA repair genes.

The gain and loss of repair genes is traced onto an evolutionary tree of the species for which complete genome sequences were analyzed. Gain and loss were inferred by methods described in the main text. Origins of repair genes (+) are indicated on the branches while loss of genes (−) is indicated along side the branches. Gene duplication events are indicated by a “d” while possible lateral transfers are indicated by a “t”.
Table 6.
Origin of DNA repair genes and pathways
| Pathway | Ancient | Evolved Within Bacteria |
Evolved in Arch-Euk Lineage |
Evolved Within Archaea |
Evolved Within Eukaryota |
Ambiguous Origin |
General Mechanisms Conserved? |
Comments |
|---|---|---|---|---|---|---|---|---|
| Photoreactivation | PhrI PhrII | - | - | - | - | - | Yes | Specificity varies between species. PhrI and PhrII genes lost many times. Also some lateral transfer and duplication. |
| Alkyltransfer | Ogt | Ada | - | - | - | - | Yes | Addition of Ada domain to Ada protein occurred in bacteria. |
| Base Excision Repair | Ung? MutY/Nth AlkA | Fpg/Nei TagI | Ogg | - | - | 3MG GT MMR | Yes | Ung may have originated in bacteria. Specificity varies greatly between species for MutY-Nth, AlkA, and others. Many cases of gene loss. |
| AP Endonucleases | Xth Nfo | - | - | - | - | - | Yes | Many cases of gene loss of Xth and Nfo. All species have one or the other. |
| Nucleotide Excision Repair | - | UvrABCD | Rad1 Rad2 | - | All other euk. NER proteins | Rad25 | Yes/No | UvrABCD in M. thermoautotrophicum (Archaea) probably by lateral transfer. |
| Transcription-Coupled Repair | - | Mfd | - | - | CSA, CSB | - | ? | Mfd missing from some bacteria. |
| General Mismatch Repair | MutLS? | MutH Dam Vsr | - | - | dup MutS dup MutL | - | Yes/No | Strand recognition systems and exonucleases differ between species. Many cases of loss of MutLS genes. Duplication in eukaryotes allows use of heterodimers. |
| Recombination Initiation | SbcCD | AddAB RecBCD RecFJNOR RecET SbcB | - | - | dup RecQ | RecQ | No | Many cases of gene loss in bacteria. RecF pathway genes not always present together. |
| Recombinase | RecA | RecT? | - | - | dup RecA | - | Yes | Lateral transfer from chloroplast to plant nucleus has occurred. RecT is of phage origin. |
| Branch Migration | - | RuvAB RecG | - | - | - | - | Yes/No | RuvAB and RecG missing from some bacteria. |
| Branch Resolution | - | RuvC Rus RecG | - | - | CCE1 | - | Yes/No | CCE1 may function in mitochondria. Rus is likely of phage origin and is only found in a few species. |
| Other Recombination | - | - | - | - | Rad52-59 XRS2 | - | - | - |
| Non-homologous end joining | - | - | - | - | XRCC4 Ku70, 86 DNA-PKcs | - | - | - |
| Ligation | - | LigI | LigII | - | - | - | Maybe | - |
| Induction | - | LexA | - | - | P53 | - | No | - |
| Other | MutT UmuC SMS? | SSB | - | - | RFAs | Dut | - | Eukaryotic SSB came from mitochondria. |
3.1. Direct Repair
3.1.1. Photoreactivation (PHR)
Photoreactivation (PHR) is a general term used to refer to the ability of cells to make use of visible light to reverse the toxic effects of UV irradiation. PHR has been found in bacteria, Archaea, and eukaryotes. Despite the highly general way that PHR is defined, all characterized enzymatic PHR processes involve a similar type of direct repair of UV irradiation induced DNA lesions (39). Therefore the term PHR is frequently used more narrowly to refer to this type of DNA repair. Two different types of PHR have been discovered – the most common one involving the reversal of cyclobutane pyrimidine dimers (CPDs) and the other involving the reversal of 6-4 pyrimidine-pyrimidone photoproducts (6-4s). In addition, PHR processes differ from each other in their action spectrum, the wavelength of light required for peak activity, and the particular cofactor used to facilitate energy transfer (39). Despite the different substrates, all PHR processes are quite similar - all are single step processes that have similar mechanisms and all enzymes that perform PHR (known as photolyases) are homologous.
The comparison of photolyases is somewhat complicated by the fact that some photolyase homologs do not repair any lesions but instead function as blue-light receptors (40). Comparative sequence analysis reveals that the photolyase gene family can be divided into two subfamilies, referred to as classI (or PhrI) and class II (or PhrII) (39). ClassI includes the photolyases of E. coli, H. halobium and yeast, as well as the blue-light receptors from plants and a human gene with no known function (1). ClassII includes the photolyases from M. xanthus, M. thermoautotrophicum, goldfish and marsupials.
Most species for which complete genome sequences are available do not encode any photolyase homolog, and those that do encode either a PhrI or a PhrII, but not both. It is important to note that some species for which the complete genomes are not available encode both PhrI and PhrII homologs (e.g., Arabidopsis thaliana). Phylogenetic trees of the photolyase gene family (ours and those of (39)), and the fact that both PhrI and PhrII are found in each of the major domains of life (Table 4), suggest that the two gene families are the result of an ancient duplication event. Thus we conclude that the last common ancestor of all life encoded both a PhrI and a PhrII and that the uneven distribution pattern of these genes is best explained by gene loss events in some lineages. For example, a PhrI gene loss likely occurred recently in the H. influenzae lineage since many other γ-Proteobacteria (including E. coli, N. gonorrhoeae, and S. typhimurium) encode a PhrI. It is possible that gene loss has occurred in humans as well. Marsupials encode a PhrII, but a PhrII has not yet been found in humans. The rampant loss of PhrI and PhrII genes is not particularly surprising since many species may have switched from high to low UV irradiation environments and thus may not have much use for PHR. In addition, since both 6-4s and CPDs can be repaired by other pathways such as NER, PHR is not absolutely necessary for repair of these lesions. Some gene duplication has also occurred in the photolyase gene family - for example, Synechocystis sp. encodes two PhrIs. In addition, it is likely that there have been some lateral gene transfers of Phr genes - A. thaliana encodes a PhrI that likely was transferred from the chloroplast genome (data not shown).
Table 4.
Presence and absence of repair gene homologs in complete genome sequences
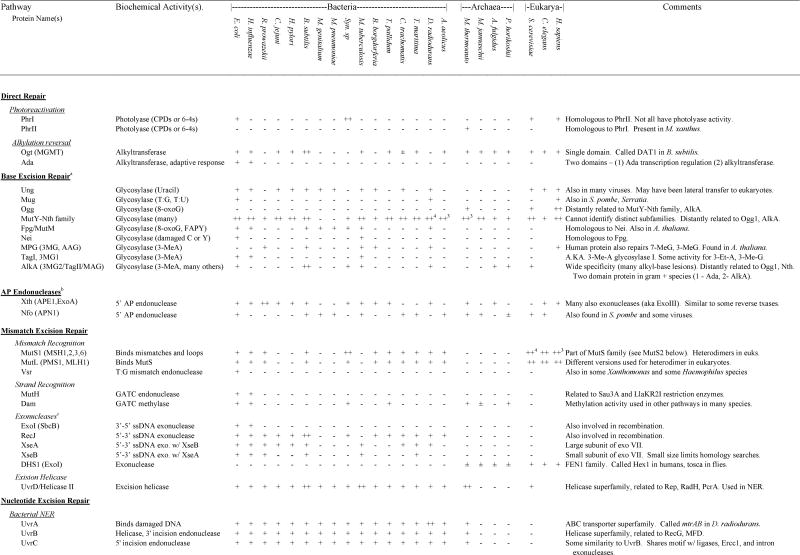
|

|

|
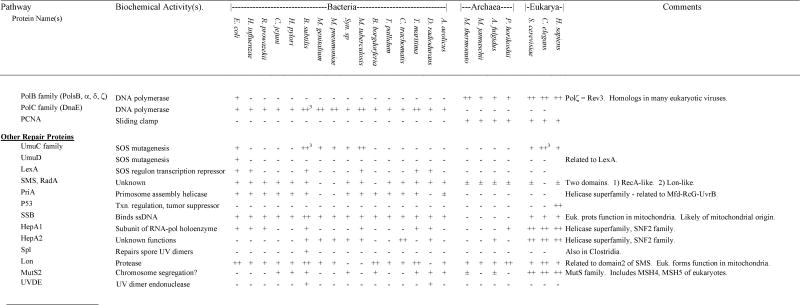
|
In those cases in which a species encoded a gene for which homology to the gene of interest was ambiguous, we indicated ±. If a gene was found in any other species within bacteria, Archaea or eukaryotes, this is listed in the “ANY” column. For those genes that were part of multigene families, we used phylogenetic analysis to divide the family into subfamilies and groups of orthologs and paralogs (see Results and Discussion). If subfamilies could be determined unambiguously, we only identify presence and absence of a homolog within the same subfamily as the search gene. If subfamilies could not be determined unambiguously, we listed the number of homologs of a particular gene (e.g., MutY-Nth). In cases of relatively recent gene duplications, presence of multiple homologs (++ for two and ++# for more) was indicated for a few species if a limited number of species encoded multiple orthologs of a gene. If lateral transfers were identified, this is indicated in the Comments column. Additional details can be found in the discussion.
The first step in BER involves glycosylases. See text for details on other steps. Some of these glycosylases also have AP lyase or dRPase activity.
Functions similarly to AP-Endonuclease but biochemical activity is AP lyase (in conjunction with role in base excision repair).
Many exonucleases can serve this role in mismatch repair.
RecBCD complex (ExoV) has many activities including dsDNA and ssDNA exonuclease and endonuclease, ATPase, helicase, and Chi-site recognition.
By tracing the evolution of functions of photolyase homologs, we conclude that the ancestral Phr protein was a photolyase and thus that the last common ancestor could perform PHR. Photolyase genes may have been more important in the early evolution of life since there was no ozone layer then to attenuate the intense solar UV flux (41). This analysis also shows that the blue-light receptors descended from photolyases and thus have lost PHR activity but retained the ability to absorb blue-light (39). Our analysis also shows that there have been multiple cases of change of function between CPD and 6-4 specificity. Because the history of photolyases is filled with functional changes and loss of function, we believe that the presence of a photolyase homolog in a species cannot be used to unambiguously predict the presence of PHR activity or its nature (e.g., CPD vs. 6-4).
The specific origin of photolyase enzymes is difficult to determine since the photolyase gene family does not show any obvious homology to any other proteins. However, it is useful to recognize that limited photolyase activity can be provided by a tripeptide sequence (Lys-Trp-Lys) (42, 43, 44), suggesting that a photolyase protein could have evolved relatively easily early in evolution.
3.1.2. Alkylation Reversal
A common form of damage to DNA bases occurs when alkyl groups (especially methyl and ethyl groups) are covalently linked to DNA bases. One way that cells repair this damage is by transferring the alkyl group off the DNA, a form of direct repair known as alkyltransfer repair (45, 46). Alkyltransfer repair has been found in bacteria, Archaea, and eukaryotes (47). All alkyltransfer repair processes are highly similar. First, all are catalyzed by a single protein which transfers O-6-alkyl guanine from the DNA to itself in a suicide process (the protein is never used again). In addition, comparative sequence analysis reveals that all alkyltransferases share a highly conserved core domain and thus are all homologs (48, 49). The comparison of alkyltransferase proteins is somewhat complicated because some contain additional domains (Figure 2). For example, in E. coli the Ogt protein contains only the alkyltransferase domain while the Ada protein contains the alkyltransferase domain and a transcriptional regulatory domain. Ada uses the second domain as part of an inducible response to alkylation damage.
Figure 2.

Schematic diagram of an alignment of alkyltransferase genes.
Our analysis shows that many but not all species encode alkyltransferase homologs (Table 4). Since alkyltransferase homologs are found in at least some species from each of the major domains of life (Table 4), we conclude that they are ancient proteins and were present in the last common ancestor of all organisms. Thus the absence of an alkyltransferase homolog from some species (e.g., D. radiodurans, the two mycoplasmas, Synechocystis sp., R. prowazekii, and Borrelia borgdorferi) is likely due to gene loss. The two alkyltransferases in E. coli are likely the result of gene duplication and domain shuffling. Specifically, we infer that in the γ-Proteobacteria there was a duplication into two alkyltransferase genes and subsequently the transcriptional regulatory domain was added onto the Ada protein. Interestingly, Gram positive bacteria also encode a two domain protein with an Ada transcriptional-regulatory domain, but in this case the Ada domain is fused to an alkyl glycosylase domain (see Figure 2).
Since all characterized members of this gene family function as alkyltransferases, we conclude that the presence of an alkyltransferase homolog in a species likely indicates the presence of alkyltransferase activity. Thus the last common ancestor was likely able to perform alkylation repair. In addition, since no other proteins have been found to have this activity, we conclude that the absence of an alkyltransferase homolog likely indicates the absence of alkyltransferase activity. However, the species without an alkyltransferase homolog likely are still able to repair alkylation damage - some encode alkylation glycosylases for BER and all encode genes for NER (see below).
3.1.3. DNA Ligation
DNA ligation (the process of joining together two separate DNA strands) is required for replication, recombination, and all forms of excision repair, and when used to repair DNA strand breaks, is a form of direct repair. DNA ligation is usually performed by a single ligase enzyme, although accessory proteins frequently aid in the process. The ligases that are used for DNA repair can be divided into two apparently unrelated families. Ligase-Is, which have been found and characterized in many bacterial species (e.g., DnlJ of E. coli), are all NAD-dependent (50). Ligase-IIs, which have been found in many viruses, Archaea, and eukaryotes (51), are all ATP-dependent. Multiple Ligase-IIs with similar but not completely overlapping functions have been found in many eukaroytes.
Our comparative analysis shows that, of the species analyzed here, all bacteria and only bacteria encode a Ligase-I, some bacteria encode a Ligase-II, all Archaea encode a Ligase-II, and all eukaryotes encode multiple Ligase-IIs. We therefore conclude that Ligase-Is originated early in bacterial evolution. We also conclude that Ligase-IIs originated in a common ancestor of Archaea and eukaryotes and that subsequently there were duplications in eukaryotes and lateral transfers to some bacteria and viruses. Although functional information is not available for any of the bacterial Ligase-IIs, given the functional conservation among members of this gene family in eukaryotes and Archaea, we suggest that they act as ligases. Perhaps they provide an alternative type of ligase function to the universal bacterial Ligase-Is that are found in these species. Since all species encode a homolog of one of the two ligase families and since all characterized members of these gene families are ligases, it is likely that all species have ligation activity.
3.2. Mismatch Excision Repair
The ability to recognize and repair mismatches in DNA has been well documented in many species. Since mismatches can be generated in many ways, processes that repair mismatches have many functions including the repair of some types of DNA damage, the regulation of recombination, and perhaps most importantly, the prevention of mutations due to replication errors (52). Mismatches can be repaired by three main mechanisms - by base excision repair glycosylases which recognize specific mismatches (discussed in the BER section); by a general mismatch excision repair process (referred to here as mismatch repair or MMR) that can repair many types of mismatches; and by a variant of the general MMR process that uses endonucleases specific for certain mismatches as well as many of the proteins involved in general MMR.
MMR has been found in a wide diversity of species and has been best characterized in E. coli in which it works in the following way (52). First, the MutS protein binds to a mismatch or a small unpaired loop and, with the cooperation of MutL, the region is targeted for excision repair. The newly replicated strand (and thus the strand containing the replication error) is targeted for repair by the fact that it will not yet have been methylated by the Dam protein. This lack of methylation makes the newly replicated strand the target of the MutH endonuclease which, when activated by the MutS-MutL complex, cuts the unmethlyated strand at GATC sites near the mismatch. Various exonucleases and the UvrD helicase complete the excision of the target strand and a very large repair patch is resynthesized using the intact strand as a template.
While the overall scheme of general MMR is similar between species, not all details are identical (5, 53, 54). For example, while all species exhibit strand specificity, the mechanism of strand recognition is different between species. In addition, there are many differences in the post-cleavage steps between species. However, there is a conserved core of general MMR: homologs of the E. coli MutS and MutL proteins are absolutely required for MMR in all species (5, 53). MutS (and its homologs) are always responsible for the recognition step and MutL (and its homologs) have an as of yet poorly characterized structural role. Some of the unusual features of MutS and MutL homologs in different species are what led us to explore phylogenomic methods (21). For example, eukaryotes encode multiple functionally distinct homologs of MutS and MutL, only some of which participate in MMR. In addition, some species encode MutS homologs but not MutL homologs. We showed that this is explained by the finding that the MutS family is composed of two major subfamilies (MutS1 and MutS2) and only those proteins in the MutS1 subfamily are involved in MMR. This functional information is supported by the finding that all species either encode homologs of both MutS1 and MutL or neither. Thus in the species with only a MutS homolog and no MutL, the MutS is always a MutS2.
The origins of the MutS1 and MutL proteins are difficult to determine with certainty from the currently available data. In particular, the presence of homologs of these two genes in bacteria and eukaryotes but not Archaea needs to be explained. Some evidence suggests that MutS1 and MutL were present in the last common ancestor of all species. The arguments for why MutS1 is likely ancient have been previously discussed (21). We propose that MutL is also ancient because we have isolated a clone of a portion of a MutL homolog from the Archaea Haloferax volcanii (19). Thus we conclude that the absence of MutS1 and MutL from the Archaea analyzed here is due to gene loss. An alternative theory suggests that all eukaryotic MutS homologs were transferred to the nucleus from the mitochondrial genome (38). We do not believe this is correct because the eukaryotic MutS and MutL homologs do not all branch in evolutionary trees close to the Rickettsia prowazekii MutS and MutL homologs. However, the eukaryotic MSH1 genes do branch in MutS family trees next to the Rickettsia prowazekii MutS homolog. Thus we propose that the MSH1 genes were transferred from the mitochondrial genome, which is consistent with experiments that show that these genes function in mitochondrial MMR. Interestingly, there is a MutS homolog encoded by the mitochondrial genome of a coral species but this is a gene in the MutS2 subfamily and likely does not function in MMR.
Whether or not MutS1 and MutL are ancient, since homologs of these genes are found in most bacteria, we conclude that the ancestor of all bacteria encoded MutS1 and MutL homologs. Thus we infer that the absence of these genes from some species is due to gene loss. Tracing gene loss events shows that loss of MutS1 and MutL has occurred many times in the history of bacteria including in the mycoplasmal lineage (they are absent from the mycoplasmas but present in other low-GC gram-positive species), the ε-Proteobacterial lineage (they are absent from C. jejuni and H. pylori but present in other Proteobacteria), and the M. tuberculosis lineage (21). Since the function of MutS1 and MutL homologs is highly conserved, we conclude that species with homologs of these likely have MMR. Since no other proteins are known to perform general MMR we conclude that species without homologs of these genes (H. pylori, C. jejuni, M. tuberculosis, the two mycoplasmas, and the Archaea) do not have MMR.
That there have been multiple parallel losses of the MutL and MutS1 genes suggests either that these genes are particularly unstable and are easily lost, or that there is some advantage to the loss of these genes. We believe that the latter explanation is more likely and that MMR genes might have been lost to increase the mutation rate. Such an increased mutation rate should allow a species/strain to more readily evolve in response to unstable changing environments (8, 10, 55, 56). In particular, absence of MMR would result in a very high mutation rate in microsatellite sequences, which in turn could contribute to generating diversity in antigen proteins of these species (57). In addition, since MMR plays a role in other processes such as the regulation of interspecies recombination, differences in MMR could also affect these processes (58).
The limited distribution of MutH homologs supports experimental evidence that only close relatives of E. coli use methyl-directed strand recognition. Interestingly, MutH is closely related to the restriction enzymes Sau3AI from Staphylococcus aureus (59) and LlaKR2I from Lactococcus lactis (60). We propose that the mutH methylation based system evolved from a restriction modification system. This suggests that other species may have co-opted separate restriction systems for strand recognition. This may explain why many species encode a Dam homolog but not a MutH homolog. In addition it may also explain the interaction of a methyl CpG binding endonuclease (MED1) with the MutL homolog MLH1 in humans (61).
Interestingly, the Vsr mismatch endonuclease, that is involved in specific mismatch repair of GT mismatches, also has many functional and structural similarities to restriction enzymes (62). As with MutH, the Vsr system also appears to be of recent origin in the Proteobacterial lineage.
3.3. Nucleotide Excision Repair
Nucleotide excision repair (NER) is a generalized repair process that allows cells to remove many types of bulky DNA lesions (63, 64). The overall scheme of NER, which is highly conserved between species, works in the following way: recognition of DNA damage; cleavage of the strand containing the damage (usually on both the 5′ and 3′ sides of the lesion); removal of an oligonucleotide containing the damage; resynthesis of a repair patch to fill the gap; and ligation to the contiguous strand at the end of the gap. Since the biochemical details of NER are quite different between bacteria and eukaryotes, we have divided the analysis into multiple sections, first summarizing NER studies in bacteria and eukaryotes, then comparing the origins of the eukaryotic and bacterial system, and finally analyzing what this analysis suggests about NER in Archaea.
3.3.1. Bacterial NER – UvrABCD pathway
NER in bacteria has been best characterized in E. coli, in which it works in the following way (65, 66). First, a homodimer of UvrA recognizes the putative lesion and recruits UvrB to aid in the verification that a lesion exists. UvrA leaves the site and UvrB then recruits UvrC, revealing a cryptic endonuclease activity to produce dual incisions 12–13 nucleotides apart bracketing the lesion. The UvrD helicase, in concert with DNA polymerase I, removes the damaged oligonucleotide, and a repair patch is synthesized by pol I that is then sealed into place by DNA ligase. An accessory protein, Mfd, is involved in targeting NER to the transcribed strand of actively transcribing genes – a subpathway known as transcription coupled repair (TCR) (67, 68). Homologs of UvrABCD are required for NER in all bacterial species studied. Although Mfd homologs have been found in many species, other than E. coli the function has only been studied in B. subtilis. As in E. coli, in B. subtilis, Mfd is involved in TCR. However, the B. subtilis gene may also be involved in recombination (69, 70).
3.3.2. Eukaryotic NER – XP pathways
NER in eukaryotes has been most thoroughly studied in yeast and humans. (63, 71). In humans, multiple proteins are involved in the initial damage recognition steps, including XPA, RPA, XPE and XPC. The helicase activities are provided by those of XPB and XPD in the basal transcription factor TFIIH, that interestingly serves dual functions in transcription and NER. In NER, TFIIH forms a bubble to enable separate flap endonucleases XPG and an XPF-ERCC1 heterodimer to produce incisions 3′ and 5′ of the lesion, respectively, about 30 nucleotides apart. Repair replication is then carried out by the same proteins required for genomic replication, including RPA, RFC, PCNA and DNA polymerase δ/ε.
Although NER is highly conserved among eukaryotes (the names of yeast homologs of the human proteins are given in Table 4), some major differences exist among eukaryotes in targeting NER to particular parts of the genome. For example, the CSA protein in humans is involved in TCR but its putative ortholog in yeast is not. Similarly, XPC in humans is required for global genome repair (GGR) but Rad4, the XPC ortholog in yeast, is not. Instead, in yeast Rad7 and Rad16 are required for GGR but orthologs of these have not yet been found in humans. There are even more subtle differences in targeting lesions between humans and rodents. In particular, humans and rodents are nearly identical in the repair of 6-4 photoproducts but rodents do not carry out efficient global repair of CPDs as well as humans, evidently because they lack inducible up-regulation of NER.
3.3.3. Comparison of bacterial and eukaryotic NER
One major difference between the bacterial and eukaryotic NER systems is that many more proteins are needed to carry out each step in eukaryotic compared to bacterial NER. However, even more striking is that, despite the overall similarity of biochemical mechanism of each of the steps, the bacterial and eukaryotic NER systems appear to be of completely separate origins. For example, UvrA has no homology to any of the damage recognition proteins of eukaryotes. Similarly, the early initiation steps in eukaryotes require many proteins yet none of these share a direct common ancestry with any of the bacterial NER proteins. Interestingly, in some cases, the eukaryotic and bacterial NER systems have separately recruited similar proteins for particular functions. For example, UvrC and Ercc1 have similar activities and share a similar motif, but are probably not homologs. In the early initiation steps eukaryotes use the 5′-3′ and 3′-5′ helicases encoded by XPB and XPD, respectively while bacteria use the distantly related helicase UvrB to carry out the analogous activity. In addition, for TCR, eukaryotes and bacteria each use proteins in the helicase family that are not helicases, but these proteins (CSB and MFD) are not particularly closely related to each other (20).
3.3.4. Origins of bacterial NER
Our comparative analysis shows that orthologs of the UvrABCD proteins are found in all the bacterial species analyzed (Table 4). Therefore we infer that these genes were present in the common ancestor of all bacteria. Surprisingly, orthologs of UvrA, UvrB, UvrC, and UvrD are also found in the Archaea M. thermoautotrophicum. Since the genes for these four proteins are located next to each other in the M. thermoautotrophicum genome Aravind et al. suggest that these were transferred to M. thermoautotrophicum as a single unit (38). However, as of yet these genes have not been found together in any bacterial species so we believe the alternative possibility is still possible - that UvrA, UvrB, UvrC, and UvrD were present in a common ancestor of bacteria and Archaea and were then lost in some Archaeal lineages. Orthologs of Mfd are found in all bacteria except the mycoplasmas and A. aeolicus. Therefore Mfd likely originated near the beginning of bacterial evolution and was then lost from the mycoplasmal and A. aeolicus lineages. Since the functions of UvrA, UvrB, UvrC, and UvrD are conserved in many bacteria we conclude that all the bacteria analyzed here as well as the bacterial common ancestor can/could perform NER in much the same way as does E. coli. Since Mfd is absolutely required for TCR in E. coli and B. subtilis it is likely that the species without Mfd cannot perform TCR.
The specific origins of these proteins help in understanding the origins of the bacterial NER process. UvrA is a member of the ABC transporter family of proteins (72). All proteins in this family for which functions are known (other than UvrA) are involved in transport across membranes (73), although it is important to note that for transport an additional membrane spanning domain is required. Based on this relationship to ABC transporters, we propose that bacterial NER evolved from a system that transported toxins out of the cell (a function that many of the ABC transporters such as the MDR proteins still have). We further propose that bacterial NER may still have a transport function - transporting DNA damage containing oligonucleotides out of the cell. Evidence for this includes that NER is associated with the bacterial membrane (74), that some UvrA homologs are possibly involved in transporting DNA damaging antibiotics out of the cell (75), and that some species are known to export DNA repair products out of the cell (14). The origins of UvrB are also revealing. UvrB is a member of the helicase superfamily of proteins, most closely related to Mfd and RecG. The relationship of UvrB and Mfd is of particular interest since both interact with UvrA. Maybe the original NER system only used one protein to interact with UvrA and a gene duplication event allowed Mfd and UvrB to diverge in function. Our analysis shows that UvrD and UvrC are also part of large multigene families and thus arose by gene duplication as well. UvrD is also a member of the helicase superfamily and is part of a subfamily that includes the RecB, rep, and helicase IV proteins of bacteria and RadH from yeast. UvrC likely shares a common ancestry with homing endonucleases from mitochondrial introns and with a family of uncharacterized proteins found in many bacteria (see also (38)). Thus, all the proteins involved in bacterial NER originated by gene duplication events rather than by invention of new proteins.
3.3.5. Origins of eukaryotic NER
Our comparative analysis reveals that most of the proteins involved in eukaryotic NER are only found in eukaryotes and thus likely evolved during eukaryotic history. However, some bacteria do encode likely orthologs of some of the eukaryotic NER proteins. For example, some homologs of Rad25 are found in two bacterial species, orthologs of CSB are found in many bacteria, and many bacteria encode a protein DinG that is probably an ortholog of XPD. The functions of these proteins in bacteria are unknown. In addition, homologs of these and some other genes are found in Archaea (see below).
3.3.5. Archaeal NER
While NER has been studied in detail in bacteria and eukaryotes there have been only very limited studies in Archaea (19, 76). Our comparative genomic analysis sheds little light on NER in Archaea. XPF/Rad1 and XPG/Rad2 homologs are found in all Archaea suggesting that these genes originated in a common ancestor to Archaea and Eukaryotes. However, the functions of these genes in Archaea are hard to predict and there are a few reasons to think that they may not function in NER. First, XPF works in concert with Ercc1 in NER, but no Ercc1 homologs are found in any of the Archaea. In addition some of the eukaryotic XPF homologs do not function only in NER. For example, the XPF homolog in yeast (RAD1) also functions in recombination. Different functions for the Archaeal XPF homologs are also suggested by the fact that the Archaeal XPF homologs have likely functional helicase motifs while the eukaryotic genes have degenerate helicase motifs (77). It is also not possible to predict conclusively the functions of the Archaeal XPG homologs since they are not much more similar to XPG than to other members of the FEN1 family with different functions (78). The Archaeal XPB/Rad25 and CSB/Rad26 orthologs are likely not involved in NER either. Interestingly, the one Archaea in which a NER-like process has been characterized in-vitro (79) is the one that encodes UvrABCD orthologs. With the separate origin of the bacterial and eukaryotic NER systems, we believe it is likely that the Archaea without UvrABCD homologs have an Archaeal specific NER system made up in part of genes yet to be characterized.
3.4. Alternative Excision Repair
A novel mechanism for the initiation of excision repair of UV induced photoproducts has been reported in Neurospora crassa and in Schizosaccharomyces pombe. In this process, the UV dimer endonuclease protein (UVDE) introduces an incision immediately 5′ of the lesion (80, 81, 82). Following incision, the subsequent steps of repair are thought to occur just as in the “normal” NER described above, although the specific details are not known. Homologs of UVDE are also found in the bacteria B. subtilis and D. radiodurans. In D. radiodurans, the UVDE homolog likely corresponds to the UV endonuclease b, a UV damage specific endonuclease active in uvrA mutants. Since the bacterial NER system is so different from the eukaryotic system (see above) it would be interesting to see if the UVDE homologs in bacteria also work in conjunction with NER. There is some recent evidence that the UVDE homologs of some species may also work on AP sites (83).
3.5. Base Excision Repair (BER)
In BER, damaged or altered bases are detached from the DNA backbone by DNA glycosylases that cleave the glycosylic bond (84). Subsequently the backbone of the DNA is incised by an abasic-site endonuclease, the sugar is removed, and a repair patch of a single or a few nucleotides is synthesized using the base opposite the excised base as a template. In this section, we discuss the evolution of different DNA base glycosylases.
3.5.1. Uracil DNA glycosylases (UDG or UNG)
Uracil can appear in DNA via two routes – incorporation during replication and by spontaneous deamination of cytosine. While the incorporation during replication can be limited by controlling the dUTP pools (such as with dUTPase), the deamination of cytosine is spontaneous and cannot be readily controlled. This deamination is potentially mutagenic because replication will lead to an adenine being incorporated opposite the uracil, rather than the guanine that should have been incorporated opposite the cytosine. A variety of proteins have been found to act as uracil DNA glycosylases including homologs of the E. coli Ung protein, glyceraldehyde-3-phosphate dehydrogenase (85), a cyclin-like protein (86), and the MUG protein (see below). We focus here on homologs of Ung since these apparently provide the major uracil DNA glycosylase activity for most species (87). Ung homologs have been characterized in many bacterial and eukaryotic species, as well as in many viruses (mostly herpes related viruses) and these proteins have strikingly similar structures and functions.
Our comparative analysis shows that Ung homologs are found in eukaryotes, many bacteria, but not in any of the Archaea analyzed. Since Ung is found in a wide diversity of bacteria, we conclude that the bacterial ancestor encoded a Ung homolog and that the absence of Ung homologs from some bacteria (T. pallidum, Syn. sp, R. prowazekii, and A. aeolicus) is due to gene loss. Our phylogenetic analysis suggests that the eukaryotic Ung homologs were transferred from the mitochondrial genome (they branch within the Proteobacterial Ung homologs). The possibility of a mitochondrial transfer is supported by the finding that an alternatively splice form of the human Ung functions in the mitochondria (88). However, since no Ung sequence is yet available from the α-Proteobacteria which are thought to be the closest living relatives of the mitochondria, we cannot conclusively resolve the origin of the eukaryotic Ung genes. Due to the high degree of functional conservation among characterized Ung homologs, it is likely that the species with Ung homologs have uracil glycosylase activity. However, the absence of an Ung homolog should not be used to imply the absence of uracil glycosylase activity, because many other proteins have some uracil glycosylase activity. The absence of uracil glycosylase activity would be particularly surprising in thermophiles like A. aeolicus and the Archaea since the deamination of cytosine increases with increasing temperature. One possibility is that these species have a novel means of preventing or limiting deamination. However, more likely, these species have an alternative protein that acts as a uracil DNA glycosylase. Although these species do not encode a MUG homolog (see below) some do encode a novel G:U glycosylase that was originally described in T. maritima (89). This enzyme may explain the uracil glycosylase activity found in many thermophiles (90)
3.5.2. G:U and G:T mismatch glycosylase (MUG)
The first protein in this family to be characterized was the thymine DNA glycosylase (TDG) of humans (91, 92). This protein was originally shown to cleave the glycosylic bond of thymine from G-T mismatches but was subsequently found to also cleave the uracil from G-U mismatches. Subsequently, homologs of this protein were found in other mammals as well as some bacteria. The E. coli protein is called the mismatch specific uracil DNA glycosylase (MUG) although is works on both G-U and G-T mismatches like the human protein. These proteins are likely used for the repair of deamination of cytosine and methyl-cytosine, which will lead to G-U and G-T mismatches, respectively. Since these proteins can cleave uracil from DNA, they can be confused with uracil DNA glycosylases. Until more sequences are available for these genes, the evolutionary history of this gene family cannot be determined accurately.
3.5.3. MutY-Nth family
The MutY and Nth proteins of E. coli are both DNA-glycosylases and, although they are homologs of each other, they have quite different substrate specificity and cellular functions (47, 93). MutY cleaves the glycosylic bond of adenine from G:A, C:A, 8-oxo-G:A or 8-oxo-A:A base pairs (94). Its primary role is protection against mutations due to oxidative damage of guanine (95). Nth has a very broad specificity and excises a variety of damaged pyrimidines. Homologs of MutY and Nth have been cloned from many species and all that have been characterized are DNA glycosylases. Some of these are clearly MutY-like or Nth-like in sequence and function (e.g., the MutY (96, 97) and Nth (98) of mammals). However, many have quite different specificity than the E. coli proteins including the pyrimidine dimer glycosylase of Micrococcus luteus (99), the yeast NTG1 and NTG2 (that excise similar substrates to the E. coli Nth as well as ring opened purines, the formamidopyrimidines (FAPY)), the GT mismatch repair enzyme of the Archaea M. thermoformicum (100), and a methyl-purine glycosylase from T. maritima (101).
Our comparative analysis shows that all species except the two mycoplasmal species encode at least one member of the MutY-Nth gene family. We attempted unsuccessfully to use phylogenetic analysis to divide this gene family into subfamilies of orthologs. Some proteins are clearly more related to MutY or to Nth than others are, but there is no obvious, well-supported subdivision. Therefore, we list the MutY-Nth gene family together without attempting to distinguish orthologs of these two proteins. Since this family is so widespread, we conclude that it is ancient, and thus that the last common ancestor encoded at least one MutY-Nth like protein. Thus the absence of a MutY-Nth like gene from the mycoplasmas is likely due to gene loss. However, since our phylogenetic analysis was ambiguous and since the activity is not conserved among these proteins, we cannot infer any activity other than a broad “glycosylase” activity for the ancestral protein. For similar reasons we also cannot reliably predict the functions of any of the MutY-Nth family members for which functions are not known. The MutY-Nth family is distantly related to the Ogg and AlkA glycosylases (see below). Thus all three of these gene families likely descended from a single ancestral glycosylase gene. Since some species encode three or four members of this gene family there must have been some more recent duplications in this gene family.
3.5.4. Fpg-Nei family
The Fpg protein in E. coli (also known as MutM) excises damaged purines (including 8-oxo-G and FAPY) from DNA (102). Its primary function is the protection against mutation due to oxidative DNA damage (103). Homologs of Fpg have been isolated from a variety of bacterial species and all that have been characterized have functions similar to that of the E. coli protein (104, 105). Somewhat surprisingly, when the Nei protein was cloned, it was found to be a homolog of Fpg (106, 107). Nei is a glycosylase that excises thymine glycol and dihydrothymine. Thus the Nei-Fpg family has a great deal of functional diversity, while exhibiting a common theme of the repair of DNA damage due to reactive oxygen species.
Our comparative analysis shows that although members of the Fpg-Nei family are found in many bacterial species, they are not found in Archaea and the only gene found in eukaryotes (that of A. thaliana) is likely derived from the chloroplast genome (108). Therefore this family is of bacterial origin. Our phylogenetic analysis of the members of this family has allowed us to divide it into clear Fpg and Nei orthologous groups (therefore they are listed separately in Table 4). Of the proteins in the family, most are orthologs of Fpg. The distribution of Fpg orthologs suggests that Fpg was present in the ancestor of most bacteria. Therefore, the absence of Fpg from some species (H. pylori, the spirochetes and A. aeolicus) is likely due to gene loss. Since Fpg proteins have similar activities between species, the presence of an Fpg homolog likely indicates the presence of FAPY-and 8-oxoG glycosylase activity. The origin of Nei is somewhat less clear. Only one species other than E. coli (M. tuberculosis) has been found to encode a likely ortholog of Nei. We do not find evidence for a Nei ortholog in cyanobacteria as found by Aravind et al. (38). It is not possible to determine if there was a lateral transfer between these two lineages or if there was a gene duplication in the common ancestor and subsequent gene loss of Nei from many species.
3.5.5. Ogg1 and 2
The Ogg1 and Ogg2 proteins of yeast are homologous and both act as 8-oxo-G glycosylases (109). Ogg1 excises 8-oxo-G if it is opposite cytosine or thymine and Ogg2 if opposite guanine or adenine. Although these proteins have similar substrate specificity to Fpg proteins, and both are β-lyases like Fpg, they are not homologs of Fpg despite initial reports. As mentioned above, they may be distantly related to the MutY-Nth family and to AlkA. Homologs of Ogg1 and Ogg2 have been cloned from humans (110, 111, 112). Some isoforms of these function in the nucleus and others in the mitochondria (113). Our comparative analysis reveals that a homolog of Ogg1 is present in M. thermoautotrophicum, but not in the other Archaea or any bacteria analyzed here. Aravind et al. suggest that Ogg orthologs are found in some bacterial species (38), but we cannot find evidence for this. We conclude that an Ogg homolog was present in the eukaryotic common ancestor. It is not possible to determine if M. thermoautotrophicum obtained its Ogg protein by lateral transfer, or if Ogg originated prior to the divergence of Archaeal and eukaryotic ancestors and then was subsequently lost from some Archaeal lineages.
3.5.6. Alkylation glycosylases
Alkylation glycosylases can be divided into three gene families (47, 114, 115). One includes AlkA of E. coli (also known as TagII) and MAG of yeast. AlkA can excise many alkyl-base lesions (e.g., 3-me-A, 3-me-G, 7-meG, and 7-me-A), and a variety of other damaged bases including hypoxanthine. The AlkA homolog in yeast, MAG, has a similar broad specificity. A second family includes TagI of E. coli and its homologs in other bacteria. TagI is highly specific for 3-methyl-adenine (3-me-A), although it can also remove 3-methyl-guanine (3-me-G), but with much lower efficiency. The third family includes the MPG proteins of mammals that, like AlkA and MAG, have a broad specificity.
Our comparative analysis shows that each of the three alkylation glycosylases families has an uneven distribution pattern. TagI is only found in a limited number of species and thus likely evolved within bacteria. AlkA homologs are found in many species of bacteria, Archaea, and eukaryotes. Thus, we conclude that the AlkA family is ancient and that its absence from some species is due to gene loss. MPG homologs are found in many eukaryotes (including many species not listed in Table 4) and some bacteria. The origins of the MPG family are not clear.
Since the functions of homologs of each of the three alkylation glycosylase families are highly conserved between species we conclude that the presence of one of these genes indicates the likely presence of alkylation glycosylase activity. It is likely that those species with AlkA or MPG homologs can repair many different types of alkylation damage. Many species (the mycoplasmas, the spirochetes, A. aeolicus, and M. jannaschii) do not encode a homolog of any of these glycosylases. Given that alkylation glycosylases have apparently evolved separately many times, it is possible that these species have novel alkylation glycosylases. However, since alkylation damage can be repaired by other pathways (e.g., NER and alkyltransferases) these species may still be adequately protected from alkylation damage.
3.5.7. T4 Endonuclease V
The DENV protein (also known endonuclease V) of T4 phage is a glycosylase that acts specifically on UV irradiation induced CPDs. This protein may serve as a back-up system for the host’s NER enzymes (it can functionally complement mutants in bacteria or eukaryotes with deficiencies in the early steps in NER). Homologs of DENV have been cloned in a paramecium virus and phage RB70 (116), but the activities of these are not known. DENV homologs are not present in any of the complete genome sequences.
3.6. AP Endonucleases (Abasic site endonucleases)
AP endonucleases, which cleave the DNA backbone at sites at which bases are missing, are required for BER and for the repair of base loss. (117). There are two distinct families of AP endonucleases. One includes the Xth protein of E. coli, RRP1 of D. melanogaster, and the APE1/BAP1/HAP1 proteins of mammals (118). The other includes the Nfo protein of E. coli and the APN1 protein of yeast. Some other proteins can serve as AP endonucleases, but usually these activities are part of base-glycosylase (e.g., Nth and DENV) that do not function as AP endonucleases on their own. In addition, Xth is distantly related to the p150 proteins of LINE elements, although it is not clear if these proteins have similar activities.
Our comparative analysis shows that members of the Xth/APE1 family are found in almost every species (with the exception of the two mycoplasmas and M. jannaschii). The Nfo/APN1 family have a more limited distribution, although representatives are found in all domains of life, suggesting that these proteins are also ancient. Since both gene families are likely ancient, the absence of either gene from a particular species is likely due to gene loss. Interestingly, although each gene has been lost many times in different lineages, all species encode a homolog of one of the two AP endonucleases. Thus the loss of one of the two is tolerable, but loss of both is not. Since all characterized members of these gene families function as AP endonucleases, we conclude that AP endonuclease activity is universal. This is not surprising in view of the high frequency of spontaneous depurination of DNA.
3.7. Recombination and Recombinational Repair
Homologous recombination is required for a variety of DNA repair and repair related activities (119, 120, 121). Before discussing the role of homologous recombination in repair, it is useful to review some of the details of homologous recombination in general. Homologous recombination can be divided into four main steps: (a) initiation (during which the substrate for recombination is generated); (b) strand pairing and exchange; and (c) branch migration and (d) branch resolution. Different pathways within a species often differ from each other in the first step (initiation) and the last steps (migration and resolution) but use the same mechanism and proteins for the pairing and exchange step. For example, in E. coli, there are at least four pathways for the initiation of recombination - the RecBCD, RecE, RecF, and SbcCD pathways. These pathways generate substrates that are used by RecA to catalyze the pairing and exchange steps. The branch migration and resolution steps are then carried out by either the RuvABC, RecG or Rus pathways.
One form of damage that can be repaired by homologous recombination is the double-strand break (DSB). DSBs can be created by many agents including reactive oxygen species, restriction enzymes and normal cellular processes like VDJ recombination. It is important to note that DSBs can also be repaired by non-homologous end joining (NHEJ) (discussed in more detail below). In E. coli and yeast, the majority of the repair of DSBs is carried out by homologous recombination pathways, although in yeast some DSBs are also repaired by NHEJ. In contrast, in humans, most of the repair of DSBs is carried out by NHEJ, although some homologous recombination based repair is also performed.
Homologous recombination is also used in many species to repair post-replication daughter strand gaps (DSGs). When DNA is being replicated, if the polymerase encounters a DNA lesion, it has three choices - replicate the DNA anyway, and risk that the lesion might be miscoding; stop replication and wait for repair; or leave a gap in the daughter strand and continue replication a little but further downstream. In E. coli, the choice depends on the type of lesion, but frequently gaps are left in the daughter strand. In such cases, it is no longer possible to perform excision repair on the lesion because there is no intact template to allow for the repair synthesis step. However, such gaps can be repaired by daughter-strand gap repair (DSGR) in which homologous recombination with an undamaged homologous section of DNA is used to provide a patch for the unreplicated daughter strand section (122). Thus, although DSGR does not remove the instigating DNA damage, it is still a form of DNA repair.
Homologous recombination can be used to repair a variety of other DNA abnormalities such as interstrand cross-links. Below we discuss different pathways for homologous recombination, focusing on those known to be involved in some type of DNA repair. In Table 4, and below, the proteins are categorized by the stage in which they participate in the recombination process.
3.7.1. Initiation Pathways
3.7.1.1. RecBCD pathway
The primary pathway for the initiation of homologous recombination in E. coli is the RecBCD pathway (see (123) for review). This pathway is used for the majority of chromosomal recombination (such as during Hfr mating) and for the repair of DSBs. The initiation steps for this pathway require primarily the RecB, RecC and RecD proteins, although other proteins such as PriA may also be required. Together, RecB, RecC and RecD make up an exonuclease/helicase complex that is used to assemble a substrate for RecA-mediated recombination.
Our comparative analysis shows a limited distribution of RecB, RecC and RecD orthologs (they are only found in some enterobacteria, M. tuberculosis, and possibly in B. borgdorferi). Based on this, we conclude that the RecBCD pathway evolved relatively recently within bacteria. The timing of the origin of the RecBCD pathway is somewhat ambiguous. The pathway could have evolved within the Proteobacteria and M. tuberculosis could have received it by lateral transfer. Alternatively, the pathway could have been present in the common ancestor of high-GC Gram-positive species and Proteobacteria, and its absence from many Proteobacteria and possibly the low-GC Gram-positive species would have to be due to gene loss.
The finding that most species either have orthologs of all three or of none of these proteins suggests that these proteins have a conserved affiliation with each other. Analysis of the individual proteins suggests that this complex may have an ancestry in other recombination and repair functions. RecB and RecD are both in the helicase superfamily of proteins and both are closely related to proteins with recombination or repair roles (RecB is related to UvrD and AddA, RecD is related to TraA which is involved in DNA transfer in Agrobacterium tumefaciens (124, 125). Functionally similar complexes that are composed of proteins that are not orthologs of RecBCD have been described and isolated from many bacterial species (126). Interestingly, the proteins in some of these complexes are related to proteins in the RecBCD complex, even though they are not orthologous (38).
3.7.1.2. RecF pathway - DSGR initiation in bacteria
In E. coli, the RecF pathway is responsible for most plasmid recombination, for daughter-strand gap-repair, for some replication related functions (127) and for a process known as thymineless death (128, 129). This pathway has only a limited role in “normal” homologous recombination accounting for less than 1% of the recombination in E. coli. The proteins involved in recombination initiation in this pathway are RecF, RecJ, RecN, RecO, RecR and RecQ (121). Not all of these proteins are required for every function of the pathway. For example, RecF, RecJ and RecQ are required for thymineless death while RecF, RecR, and RecO are evidently required for replication restart functions (127). In addition, some of the proteins in this pathway are involved in other repair pathways. For example, RecJ can be used as an exonuclease in MMR if other exonucleases are defective.
Homologs of some of the proteins in the RecF pathway have been characterized in a variety of species. RecF and RecJ homologs in many bacteria have similar functions to the E. coli proteins. RecQ homologs have been characterized in many eukaryotic species and many of these have been shown to be helicases (130, 131), like the E. coli RecQ. However, their cellular functions are not well understood and it is not clear if they use their helicase activity in similar ways as the E. coli RecQ. The yeast RecQ homolog SGS1 is involved in the maintenance of chromosome stability, possibly through interaction with topoisomerases during recombination (132). Humans encode at least three RecQ homologs and all that is known about their function is that Werner’s syndrome is caused by a defect in one of these (133) and Bloom’s syndrome is caused by a defect in another (134).
Our comparative analysis of proteins in the RecF pathway was somewhat limited by difficulty identifying orthologs of many of the proteins. In particular, orthologs of RecN and RecO were difficult to identify because they show only limited conservation between species. Using low stringency searches we were able to identify more distantly related homologs of RecO and RecN. This was helpful for identifying RecO orthologs but did not work well for RecN because the lower stringency searches pulled up many homologs in most species. In particular, we were unable to distinguish whether the RecN-like proteins of the mycoplasmas and B. borgdorferi were RecN orthologs or paralogs.
Our analysis shows that, in contrast to many other repair pathways, the proteins in the RecF pathway do not have strongly correlated distribution patterns between species. For example, orthologs of RecJNR are found in H. pylori while orthologs of RecFOQ are not. Eukaroytes encode orthologs of RecQ but not of any other proteins in this pathway. Thus if other species have a RecF-like pathway, it cannot work in the same way as in E. coli. One possibility is that other species have replaced some genes in the RecF pathway by non-orthologous gene displacement (see (29) for a description of non-orthologous gene displacement). It is known that some of the functions of RecJ in E. coli can be complemented by other 5′ exonucleases such as RecD. Alternatively, it is possible that the functions of the RecF pathway are specific for E. coli and that other species do not have a similar pathway. We also note that we do not find any evidence for multiple RecR orthologs in any species as suggested by Aravind et al. (38). It is possible that this suggestion was due to the fact that in some species RecR orthologs are referred to as RecM.
Since most of the genes in the RecF pathway are found in a wide diversity of bacteria, we conclude that they were present in the bacterial common ancestor. Since RecQ orthologs are found in eukaryotes and RecJ orthologs are found in some Archaeal species (see Table 3 and (38)), RecQ and RecJ may have originated somewhat before the origins of bacteria. It is not possible to determine the ultimate origins of many of the RecF pathway proteins; but at least three of the proteins originated by some type of gene duplication (RecQ within the Dead family of the helicase superfamily and RecN and RecF within the SMC superfamily (135, 136).
3.7.1.3. RecE pathway – alternative initiation pathway in bacteria
The RecE recombination pathway of E. coli is only activated in recBrecC, sbcA mutants. This pathway requires many of the proteins in the RecF pathway, as well as two additional proteins RecE and RecT (137, 138, 139). These additional proteins are both encoded by a cryptic lambda phage. RecE is an exonuclease that can generate substrates for recombination either by RecT or by RecA. RecT may be able to catalyze strand invasion without RecA (140). Our comparative analysis shows that the species distribution of these proteins is extremely limited. RecT is found in some lowGC gram-positive bacteria. RecE homologs are not found in any species other than E. coli. The presence of these genes on a cryptic phage may reflect a recent lateral transfer between species.
3.7.1.4. SbcBCD pathway
The SbcB, SbcC and SbcD proteins were all identified as genes that, when defective, led to the suppression of the phenotype of recBC mutants (121). SbcB is an exonuclease and is also known as exonuclease I, exoI, or Xon. When it is defective, the RecE and RecF pathways are revealed. SbcC and SbcD together make up an exonuclease that cleaves hairpin structures. The main function of the SbcCD complex is thought to be the elimination of long cruciform or palindromic sequences which would remove sequences that may interfere with DNA replication (141). Homologs of SbcC and SbcD have not been characterized in many bacteria. However, these proteins do share some sequence similarity to the yeast Rad50 and MRE11 and may be distant homologs of these proteins (discussed below) (142).
Our comparative analysis shows that SbcB homologs are found only in E. coli and H. influenzae and thus this protein apparently originated within the γ-Proteobacteria. Homologs of SbcC and SbcD are present in many bacteria and are always present together. Thus the interaction of these proteins appears to have been conserved over time. Given the likely homology of these proteins to MRE11/RAD50 (which are found in eukaryotes and Archaea) we believe SbcC and SbcD are ancient proteins. Thus, the absence of these genes from some species is likely due to gene loss.
3.7.1.5. Rad52 pathway - DSBR in eukaryotes
The primary pathway for homologous recombination in yeast is the Rad52 pathway. This pathway is used for mitotic and meiotic recombination as well as for double-strand break repair. Although the exact biochemical details of this pathway are not completely worked out, it is known that the initiation step depends on three proteins - MRE11, Rad50 and XRS2, which form a distinct exonucleolytic complex (143, 144). It is believed that this complex functions to induce DSBs for mitotic and meiotic recombination and that it may alter DSBs caused by DNA damage to allow them to be repaired by homologous recombination. Genetic studies have found that these genes are also involved in NHEJ (see below). A similar exonucleolytic complex that is also involved in homologous recombination and NHEJ has been identified in humans. This complex is composed of five proteins including homologs of MRE11 and Rad50 but not XRS2 (145). Defects in one of the other proteins in the human complex (NBS1) lead to the Nijmegen breakage syndrome (145).
Our comparative analysis shows that homologs of MRE11 and Rad50 are found in all the Archaea analyzed (although the Archaeal Rad50 homologs may not be orthologs of the eukaryotic Rad50s – our phylogenetic analysis was ambiguous). Since these genes are related to the SbcC and SbcD genes of bacteria (142), we conclude that the SbcC/Rad50 and SbcD/MRE11 proteins are ancient proteins. XRS2 appears to be of recent origin in yeast since homologs are not yet found in any other species.
3.7.2. Strand Pairing and Recombinases
The RecA protein catalyzes strand pairing and invasion, is required for all homologous recombination in E. coli. Comparative studies have shown that homologous recombination depends on RecA homologs in many other bacterial species as well as in Archaea (RadA) and eukaryotes (Rad51 and DMC1) (146, 147, 148). There is some divergence of function in eukaryotes. Rad51 is the recombinase for the majority of mitotic recombination and both Rad51 and DMC1 are used for aspects of meiotic recombination.
The comparative analysis of RecA and its homologs is somewhat complicated by the fact that RecA is part of a multigene family that includes the proteins mentioned above, as well as SMS in bacteria, RadB in Archaea, and Rad55 and Rad57 in eukaryotes. Our analysis shows that the proteins that act as recombinases (RecA, RadA, Rad51, DMC1) are all orthologs of RecA and thus we focus on these here. Our comparative analysis shows that all species for which complete genomes are available encode orthologs of RecA. RecA is the only repair gene with such a universal distribution. Since all characterized RecA orthologs are recombinases, this suggests that all these species have recombinase activity. The universal presence also suggests that recombinase activity is fundamental to life. However, there have been reports of some mycoplasma species encoding defective RecA proteins (and possibly thus being defective in all homologous recombination (149, 150)). The presence of multiple functionally distinct orthologs of RecA in eukaryotes is likely due to a duplication and functional divergence early in eukaryotic evolution (148). A few other features of RecA evolution are worth mentioning. Some bacteria also encode two RecA orthologs (e.g., Myxococcus xanthus), although it is not clear if both are functional (151). Phage T4 also encodes a RecA ortholog, UvsX, which also has recombinase activity (152, 153). In addition, Arabidopsis thaliana encodes two bacterial-like RecAs in its nuclear genome. One of these, which functions in the chloroplast, likely was transferred from the chloroplast genome (146). Perhaps the other functions in the mitochondria.
3.7.3. Branch Migration and Resolution
In E. coli, many pathways have been identified that can perform branch migration and/or resolution. In the RuvABC pathway, which is likely the main branch migration and resolution pathway, RuvA binds to Holliday junctions, RuvB is a helicase that catalyzes branch migration and RuvC is a resolvase. The RecG protein catalyzes branch migration and possibly Holliday junction resolution as well (154). The RusA protein, the expression of which is normally suppressed, also can serve as a junction resolvase (155, 156, 157). It is encoded by a defective prophage DLP12 and is similar to protein in phage82. Homologs of RuvABC and RecG have been found in many other bacteria and shown to function in similar ways (158). Little is known about the proteins required for migrations and resolution in eukaryotes. CCE1, appears to be involved in resolution in yeast mitochondria (159). It has been suggested that Rad54 may be involved in branch migration in the Rad52 pathway.
Our comparative analysis reveals that RuvABC, RecG and RusA orthologs are only found in bacteria. Thus these pathways all likely evolved within bacteria. Since Rus has a limited distribution, we conclude that it evolved recently. Since RuvABC and RecG are found in a wide diversity of bacterial species, we conclude that they evolved early in bacterial evolution. Since the functions of RuvABC and RecG are conserved across species, those species with either RuvABC or RecG likely have branch migration and resolution abilities. Interestingly, while RuvA and RuvB orthologs are found in all bacterial species, many of these species do not encode a RuvC ortholog. Two of these species that do not encode RuvC orthologs do encode RecG orthologs. Perhaps as in E. coli RecG can replace some of the functions of RuvC in these species (160). Three of the species that do not encode a RuvC ortholog (the two Mycoplasmas and B. burgdorferi) also do not encode RecG or any other resolvase homolog. Whether these species encode alternative resolvases remains to be determined. However, since resolvase activity has apparently evolved many times, it is possible that these species have novel resolvase proteins.
The origins of each of these proteins reveals some clues to the origins of branch migration and resolution activities. RuvC is somewhat similar in structure to RnaseH1 (158) so it is possible that these proteins share a common ancestor. The branch migration activities are a little more constrained in that all of them appear to use some DNA helicase protein. However, the particular helicases used are quite different. RecG is closely related to Mfd and UvrB (see NER section) while RuvB is particularly closely related to an uncharacterized group of RuvB-like proteins found in many bacterial species.
3.8. Non-Homologous End Joining
In mammals, most of the repair of double-strand breaks is carried out without homologous recombination by NHEJ (161, 162). In this process, DSBs are simply restitched back together. Thus NHEJ is in essence a form of direct repair although we discuss it here because many of the genes involved are also used in recombinational repair. Genetic studies have shown that at least four proteins (XRCC4-7) are specifically required for NHEJ in humans. Together XRCC5-7 make up the DNA-dependent protein kinase complex composed of Ku80/86 (XRCC5), Ku70 (XRCC6) and DNA-PKcs (XRCC7). This complex likely functions by binding to DNA ends and stimulating DNA ligase activity. Putative homologs of Ku70 and Ku86 have been identified in yeast and these have been found to be involved in the repair of DSBs by NHEJ (163). In addition, as mentioned above, MRE11 and Rad50 are also involved in NHEJ in humans and yeast (143). As discussed above, in yeast, most of the repair of DBSs is carried out by homologous recombination based pathways (164).
Our comparative analysis shows that there are no homologs of XRCC4 or any of the three subunits of DNA-PK in Archaea or bacteria. Therefore this pathway most likely evolved in eukaryotes (165). Our analysis also shows that the sequence similarity between the yeast and mammalian proteins is very limited. Although it is likely that these proteins are homologous, the low level of sequence similarity suggests that they also may have many functional differences. An ortholog of DNA-PKcs is not found in yeast.
3.9. DNA Replication
Most repair pathways require some DNA synthesis as part of the repair process. In some cases, specific polymerases are used only for repair. In other cases, the normal replication polymerases are used for repair synthesis. Since the evolution of polymerases has been reviewed elsewhere it will not be discussed in detail here (166). Obviously, all species are able to replicate their DNA in some way and thus should be able to perform repair synthesis. The specific types of polymerases used may help determine the accuracy of repair synthesis.
3.10. Inducible Responses
3.10.1. LexA and the SOS system in bacteria
The SOS system in E. coli is an inducible response to a variety of cellular stresses, including DNA damage (167). A key component of the SOS system is the LexA transcription repressor. In response to stresses such as DNA damage, the RecA protein is activated to become a coprotease and assists the autocatalytic cleavage of LexA. When LexA is cleaved, it no longer functions as a transcription repressor, and the genes that it normally represses are induced. The induction of these LexA-regulated SOS genes is a key component of the SOS system.
SOS-like processes have been documented in a wide variety of bacterial species. Those that have been characterized function like the E. coli system, with regulation of SOS genes by LexA homologs, although sometimes different sets of genes are repressed by LexA and different “SOS-boxes” are used (167). Our comparative analysis suggests that LexA appeared near the origin of bacteria since it is found in a wide diversity of bacterial species (Table 4). Thus the absence of LexA from some species is likely due to gene loss. Since the function of the characterized LexAs is highly conserved, we conclude that those species that encode LexA likely have an SOS system. However, since there are many ways to regulate responses to external stimuli, it is possible that the species without a LexA homolog have co-opted another type of transcription regulator to control an SOS-like response.
LexA is part of a large multigene family that includes many other proteins that are also cleaved when RecA is activated including many transcriptional repressors of phage and the SOS-mutagenesis protein UmuD. Since UmuD is found in only a few γ-Proteobacteria, we propose that it evolved from a LexA like protein.
3.10.2. p53 in animals
Inducible responses to DNA damage have also been found in some eukaryotes. One gene that is involved in inducible responses in animals is p53. One of p53’s activities is transcriptional activation, and this activity is stimulated by the presence of DNA damage. In fact it has been shown that the efficient NER of UV induced DNA damage in human cells requires activation of p53, and that the mode of action involves the regulated expression of the p48 gene that is a component of XPE (168). Homologs of p53 have only been found in animals, suggesting that this inducible system evolved after animals diverged from other eukaryotes. In addition, some species encode multiple p53 homologs suggesting that there was a duplication in this gene family early in the evolution of animals.
4. Discussion II - The Big Picture
In the preceding sections we have focused the discussion on what the phylogenomic analysis reveals about specific repair proteins and pathways. We believe it is also important to take a “big picture” approach and consider all of the pathways together. One reason to take such a global approach is that the different pathways overlap a great deal in their specificity. For example, CPDs can be repaired by PHR, NER, BER (by T4EV), and can be tolerated through recombinational repair. In fact, it is rare for a particular lesion to be repaired only by one pathway. Another reason for the big picture approach is that some repair genes function in multiple pathways. In addition, it is useful to compare and contrast the evolution of different genes and pathways to identify unusual features of any pathway.
4.1. Distribution patterns of particular genes
Examination of the distribution of all DNA repair genes reveals that only one DNA repair gene, RecA, is found in every species analyzed here. The universality of RecA suggests both that it is an ancient gene, and that its activity is irreplaceable (at least for these species). Since many DNA repair genes have important cellular functions, we were surprised that only one gene was present in all species. There are however, many genes that are found in all or most members of some of the major domains of life. For example, in Table 5a we list those repair genes found in all or most bacteria. Focusing only on genes found in all members of a particular group can be somewhat misleading because it does not reveal whether these genes are found in other groups. For example, it is important to realize that, although UvrABCD are found in all bacteria, they are also found in one Archaeal species. Therefore, we also present a comparison of genes that are found in bacteria but not eukaryotes and the converse, genes that are found in eukaryotes but not bacteria (Table 5b)
Table 5a.
DNA repair genes present in all or most bacteria
| Process | In all bacteria | In most bacteria |
|---|---|---|
| Nucleotide excision repair | UvrABCD | UvrABCD |
| Holliday junction resolution | - | RuvABC |
| Recombination | RecA | RecA; RecJ, RecG |
| Replication | PolA, C | PolA, C; PriA; SSB |
| Ligation | LigaseI | LigaseI |
| Transcription-coupled repair | - | Mfd |
| Base excision repair | - | Ung, MutY-Nth |
| AP endonuclease | - | Xth |
| Single-strand binding protein | SSB | SSB |
Table 5b.
DNA repair genes present in bacteria or eukaryotes but not both
| Process | Only in bacteria | Only in eukaryotes |
|---|---|---|
| Transcription-coupled repair | Mfd | CSB, CSA |
| Mismatch strand recognition | MutH | - |
| Nucleotide excision repair | UvrABCD1 | XPs, TFIIH, etc. |
| Recombination initiation | RecBCD, RecF | KU, DNA-PK |
| Holliday junction resolution | RuvABC | CCE1 |
| Base excision | Fpg-Nei, TagI | - |
| Inducible responses | LexA | P53 |
Also found in some Archaea
4.2. Timing of origin of repair genes – ancient, old, and recent
It is informative to compare and contrast the origins of different repair genes to look for general patterns as well as to attempt to infer the repair processes of common ancestors of particular groups or of all life. It is important to note that in all our analysis of repair gene origins we may be underestimating gene loss. For example, we have inferred that UvrA, UvrB, UvrC, and UvrD originated in bacteria or in a common ancestor of bacteria and Archaea, because these genes are found in all bacteria and one Archaea, but not in any eukaryote. However, it is possible that the common ancestor of all life encoded these genes and that they were lost early in eukaryotic evolution. Our estimates of gene origin are thus very conservative, many genes could have originated even earlier than we suggest. Nevertheless, with this caveat we still have inferred that many repair genes were present in the last common ancestor (Table 6). Based on our functional evolution studies for these genes, we conclude early in the evolution of life, many DNA repair activities were present including PHR, alkyltransfer, recombination, AP endonuclease, a few DNA glycosylases, and MMR. Interestingly, most of these ancient repair pathways have been lost from at least one evolutionary lineage (Fig. 3). Thus these genes are not absolutely required for survival in all species. The ancient origin of many repair helicases and the nature of the particular helicases being used led Aravind et al. to suggest that many of the DNA repair helicases evolved from RNA helicases that functioned in an RNA world (38). This theory is one of the first links made between repair pathways and the RNA world. Another possible link between repair and early evolution was suggested by Lewis and Hanawalt (169).
Our analysis suggests that many other repair genes originated at or near the origins of major evolutionary groups (Figure 3). Interestingly, in many cases, genes with similar functions originated separately in different groups (e.g., UvrABCD vs. XPs, RuvABC vs. CCE1, Ligase I vs. Ligase II). Perhaps most surprisingly to us, we found that many repair genes are of very recent origin (e.g., MutH, SbcB, Rus, RecBCD, RecE, and AddAB). Thus repair processes are continuing to be originated in different lineages.
4.3. Mechanism of origin of repair genes
DNA repair genes have originated by a variety of mechanisms. One common means is by gene duplication. Perhaps the best example of this comes from the helicase superfamily of proteins (170). Members of this superfamily are involved in almost every repair pathway and in many cases multiple distinct superfamily members are used in a single pathway (e.g., UvrB, UvrD, and Mfd in NER). It is important to note, however, that not all proteins in this superfamily have helicase activity and that the motifs are actually indicative of DNA-dependent ATPase activity. Since all helicase motif containing proteins are homologous, each distinct gene must have been created by a separate gene duplication events. Thus gene duplication has allowed repair pathways to copy helicases used for other functions and then use them in slightly different ways. Other examples of gene duplication in the history of repair genes are given in Table 7. The particular details of each duplication reveal a great deal about each protein. For example, many eukaryotic repair genes were created by gene duplication events within the SNF2 family (which itself is a member of the helicase superfamily). This suggests that this particular family has an activity very useful for repair in eukaryotes (20).
Table 7.
Gene duplications in the history of DNA repair genes
| When Duplication Occurred | Duplicated Genes |
|---|---|
| Ancient | SNF2 family |
| MutS1-MutS2 | |
| RecA-SMS | |
| PhrI-PhrII | |
| MutY-Nth | |
| Early helicase evolution | |
| In eukaryotes | Rad23a-Rad23b in animals |
| RecQL-Blooms-Werner’s in animals | |
| SNF2 family massive duplication | |
| Rad51-DMC1 | |
| MSH1-6 (MutS family) | |
| PMS1-MLH1-MLH2 (MutL family) | |
| Rad52-Rad59 | |
| polB family | |
| Ligase family II | |
| In bacteria | Fpg-Nei, |
| UvrB-Mfd-RecG | |
| UvrA | |
| LexA-UmuD | |
| Ada-Ogt in Proteobacteria | |
| Phr in some cyanobacteria | |
| UvrD-Rep-RecB | |
| RecA1-RecA2 in Myxococcus xanthus |
Another mechanism of origin of repair genes is by co-opting genes from other functions. For example, MutH may have descended from a restriction enzyme. New repair genes have also originated by gene fusion or fusion of domains (38). For example, SMS is a fusion between Lon and RecA domains and Ada is a fusion between alkyltransferase and transcription regulatory domains (Figure 2). A final way that a species can get new repair activities is by lateral gene transfer. Since this does not involve creation of new repair genes it is discussed in a separate section below.
4.4. Gene loss
Our analysis shows that gene loss is a frequent event in the history of DNA repair (Figure 3). As discussed above, it is important to remember that we may be underestimating the total amount of gene loss in the history of repair. In some cases, whole pathways have been lost as a unit (e.g., MutLS, SbcCD). Such correlated loss of multiple genes in a pathway suggests that the functional association among these genes is conserved between species. In other cases single genes or only parts of pathways are lost (e.g., components of the RecF pathway). The number of times a particular gene or pathway has been lost can also be informative. For example, that MutL and MutS1 have been lost many times in separate lineages supports the suggestion that there is sometimes a selective advantage to losing these genes. Limitations on gene loss can also be informative. For example, our analysis shows that the last common ancestor of life encoded two AP endonuclease – Nfo and Xth. Some lineages have lost Nfo, some have lost Xth, but none have lost both, supporting the suggesting that AP endonuclease activity is required for all species. Differences in gene loss between lineages are even more striking. For example, there has been extensive loss of repair genes in the mycoplasmal lineage (e.g., Table 8, Fig. 3). Not surprisingly, loss of repair genes is more common in species or lineages with small genomes. This points to a problem in drawing too many conclusions about the likelihood of gene loss in general from the analysis of currently available genome sequences. Many species have been picked for genome sequencing because their genomes are small and thus may have undergone more gene loss than an average species.
Table 8.
DNA repair genes that were lost in the mycoplasmal lineage
| Process | Protein |
|---|---|
| Base excision repair | MutY-Nth, AlkA |
| Recombination initiation | RecF pathway, SbcCD |
| Recombination resolution | RecG, RuvC |
| Mismatch repair | MutLS |
| Transcription coupled repair | MFD |
| Induction | LexA |
| Direct repair | PhrI, Ogt |
| AP endonuclease | Xth |
| Other | MutT, Dut, PriA, SMS |
4.5. Lateral gene transfer
New repair activities can be acquired by a particular lineage without the creation of a new gene by the process of lateral transfer. The best examples of this are genes transferred from the chloroplast to the plant nucleus (e.g., RecA, MutM, photolyase). Transfers of genes from the mitochondrial to the eukaryotic nucleus also seem likely (e.g., Ung, MSH1). These and other possible cases of lateral transfer (listed with a “t” in Figure 3). Given that lateral gene transfer appears to be quite common over evolutionary history (171), it is likely organisms could replace lost genes relatively easily by gene transfer.
4.6. Conservation of pathways
Comparisons of the evolution of the different classes of repair reveal a great deal of diversity in how well conserved the classes of repair are. In addition, the ways in which classes of repair differ between species are also variable. The conservation between species can be classified according to the level of homology of the pathways. Some pathways are completely homologous between species (they make use of homologous genes in all species). This is only the case for some of the single enzyme pathways (PHR and alkyltransfer). Interestingly, all the single enzyme pathways are direct repair pathways. Other pathways are partially homologous. For example, some of the proteins involved in MMR are homologous between E. coli and eukaryotes (e.g., MutS and MutL), but others are not (e.g., MutH and UvrD). Finally, there are some pathways that are not homologous at all between species despite performing the same functions. The best example of this is NER in bacteria compared to that in eukaryotes. These systems are clearly of completely separate origins. In addition to different levels of homology, pathways also differ between species is by functional divergence of homologs. Examples of this include the divergence of 6-4 and CPD photolyases and the divergence of MSH genes for MMR in eukaryotes.
4.4. Prediction of species phenotypes and universal DNA repair activities
We believe that the key to making functional and phenotypic predictions for any species is an understanding of the evolution of the functions of interest. For example, functional predictions for homologs of repair genes are improved by evolutionary analysis (see methods). Such evolutionary functional prediction has been particularly helpful in studies of a variety of repair genes such as MutS, photolyases, many of the base excision repair glycosylases and many of the large helicase-motif containing families. In addition, identifying how many times new genes have evolved with particular functions helps determine whether the absence of particular homologs is meaningful. For example, the fact that uracil-DNA glycosylase activity has evolved many times in non-homologous proteins suggests that the absence of homologs of these proteins cannot be used to predict the absence of uracil glycosylase activity. A similar case can be made for recombination initiation and resolution activities. In contrast, since alkyltransferase activity has apparently only evolved once, it is likely that those species without a homolog of the known alkyltransferase gene family do not have alkyltransferase activity. Similarly, the fact that all generalized MMR systems use homologs of MutS and MutL suggests that the absence of mutL and mutS genes means the absence of general MMR. Finally, it is important to realize that some species may have novel activities that have not been characterized in any species.
The difficulties in making functional and phenotypic predictions are exacerbated by the biased sampling of the evolutionary tree in studies of DNA repair. For example, there has been very little experimental work on DNA repair in Archaea and what has been done is usually the characterization of homologs of known repair genes. Thus any repair processes that evolved within Archaea will likely be missed by comparative genomic approaches. Given that many processes appeared to have evolved in bacteria or in eukaryotes it seems very likely that there are also many that have evolved in Archaea. One can easily see the “bias” of model systems by following the gain of repair genes in Figure 3. Essentially all of the gain events are in the lineages leading up to E. coli, B. subtilis, yeast, and humans. This is not surprising because almost all the repair genes we analyzed are from these species. These genes must have been present in some ancestor of these species and thus the only place they could have been “gained” is along the lineage leading up to these species. Clearly, repair genes must have originated in other lineages - especially given the evidence that new repair genes have originated relatively recently (see above). For similar reasons we have an underestimation of the amount of loss of repair genes in these model organisms. They could not have lost their own genes.
Despite all these potential problems, we have still tried to make phenotypic predictions (Table 9). It should be remembered that all predictions need to be confirmed by experimental studies. We believe such predictions are a useful starting point for designing experiments on these species and for determining if the predicted presence or absence of particular repair activities can be correlated with any interesting biological properties. For example, the predicted absence of many repair pathways from mycoplasmas is consistent with the high mutation and evolutionary rates of mycoplasmas. Thus we can use the absence of certain genes to make some predictions. For example, the presence of UvrABCD but the absence of Mfd from the two Mycoplasmas and A. aeolicus suggests that these species can perform NER but not the TCR component of it.
Table 9.
Predicted DNA repair phenotypes of selected species.1
| Pathway | Proteins with Activity | Bacteria | Archaea | Eukarya | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E. coli | H. influenzae | H. pylori | B. subtilis | M. genitlaium | M. pneumoniae | M. tuberculosis | Syn. sp | B. borgdorferia | T. pallidum | A. aeolicus | All | M. thermoauto | M. jannaschii | A. fulgidus | All | S. cerevisiae | H. sapiens | All | |||
| Photoreactivation | PhrI, PhrII | + | − | − | − | − | − | − | + | − | − | − | − | + | − | − | − | + | − | − | |
| Alkyltransfer | Ada/Ogt/MGMT | + | + | + | + | − | − | + | − | − | + | + | − | + | + | + | + | + | + | + | |
| Nucleotide excision repair | UvrABCD or XPs | + | + | + | + | + | + | + | + | + | + | + | + | + | ? | ? | ? | + | + | + | |
| Transcription-coupling | Mfd or CSA/CSB | + | + | + | + | − | − | + | + | + | + | − | + | ? | ? | ? | ? | + | + | + | |
| Base excision repair | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||
| Uracil glycosylase | Ung, GT | + | + | + | + | + | + | + | − | + | − | − | − | − | − | − | − | + | + | + | |
| Alkylation glycosylase | AlkA, TagI, MPG | + | + | + | + | − | − | + | + | − | − | − | − | − | − | + | − | + | + | + | |
| Misc. damaged bases | MutY/Nth, Fpg/Nei, Ogg | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| AP endonucleases | Xth/APE1, Nfo/APN1 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Mismatch repair | MutLS | + | + | − | + | − | − | − | + | + | + | + | − | − | − | − | − | + | + | + | |
| Recombination | |||||||||||||||||||||
| Initiation | |||||||||||||||||||||
| RecBCD pathway | RecBCD | + | + | − | − | − | − | + | − | ± | − | − | − | − | − | − | − | − | − | − | |
| RecF pathway | RecFJNORQ | + | + | ±− | + | − | − | ± | + | − | ± | ± | − | − | − | − | − | − | − | − | |
| AddAB pathway | AddAB | − | − | − | + | − | − | − | − | − | ± | − | − | − | − | − | − | − | − | − | |
| SbcCD pathway2 | SbcC/MRE11, SbcD/Rad50 | + | − | − | + | − | − | − | + | + | + | + | − | + | + | + | + | + | + | + | |
| Recombinase | RecA, RadA, Rad51 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Branch migration | RuvAB, RecG | + | + | + | + | + | + | + | + | + | + | + | + | ? | ? | ? | ? | ? | ? | ? | |
| Resolution | RuvC, Rus, RecG, CCE1 | + | + | + | + | − | − | + | + | − | + | + | − | ? | ? | ? | ? | + | ? | ? | |
| Non-homologous end joining | Ku, DNA-PK | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | + | + | + | |
| Ligation | LigI, LigII | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |
+ = likely present, ± = may be present, - = likely absent, ? = not able to predict well
Resolution is possible non-enzymatically so absence of genes does not mean resolution is not possible.
One generalization that can be made from our phenotypic predictions is that, despite the lack of many universal genes, it appears that there are many universal activities. For example, we predict that all species have AP endonuclease activity. However, no AP endonuclease gene is universal because there are two evolutionarily unrelated AP endonuclease families (Nfo or Xth). All species encode at least one of these genes. Similarly, all species encode at least one of the two ligase genes.
5. Summary and Conclusions
We believe that the analysis reported here can serve as a starting point for experimental studies of repair in species with complete genome sequences and for understanding the evolution of DNA repair proteins and processes. However, it is important to restate some of the caveats to this type of analysis. First, it should be remembered that all functional and phenotypic predictions are just that - predictions. They need to be followed up by experimental analysis. In addition, the species for which complete genome sequences are available is not a random sampling of ecological and evolutionary diversity. In particular, many have small genomes (this is the reason they were sequenced) and have likely undergone large-scale gene loss events in the recent past. This is one of the reasons they were sequenced. Thus this may give a misleading picture about what an average bacterium or Archaeon is like.
Despite these limitations, the phylogenomic analysis of DNA repair proteins presented here reveals many interesting details about DNA repair proteins and processes and the species for which complete genome sequences were analyzed. We have identified many examples of gene loss, gene duplication, functional divergence and recent origin of new pathways. All of this information helps us to understand the evolution of DNA repair as well as to predict phenotypes of species based upon their genome sequences. In addition, our analysis helps identify the origins of the different repair genes and has provided a great deal of information about the origins of whole pathways. We believe our analysis also helps identify potentially rewarding areas of future research. There are some unusual patterns that require further exploration such as the presence of UvrABCD in some Archaea and the only limited number of homologs of known repair genes in any of the three Archaea. In addition, the areas with empty spaces in the tree tracing the origin of repair genes may be of interest to determine if novel pathways exist in such lineages. In summary, we believe that this composite phylogenomic approach is an important tool in making sense out of genome sequence data and in understanding the evolution of whole pathways and genomes. Combining genomics and evolutionary analysis into phylogenomics is useful because genome information is useful in inferring evolutionary events and evolutionary information is useful in understanding genomes.
Acknowledgments
We would like to thank M.B. Eisen, M-I Benito, J. H. Miller, A. J. Clark, J. Laval, B. van Houten, I. Mellon, P. Warren, R. Woodgate, W.F. Dollittle, J. Hays, S. Henikoff, P. Forterre, D. Botstein, R. Myers, A. Ganesan, J. DiRuggiero, H. Ochman, F. Robb, M. Riley, S. Suzen, M. White, V. Mizrahi, A. Villeneuve, M. Galperin, C. M. Fraser, and J. C. Venter for helpful comments and encouragement. In addition we would like to thank the two anonymous reviewers for helpful comments and criticisms. This paper was supported by Outstanding Investigator Grant CA44349 from the National Cancer Institute to P. Hanawalt. Supplemental material related to this paper can be found at http://www.tigr.org/~jeisen/Repair/Repair.html.
References
- 1.Li YF, Kim ST, Sancar A. Evidence for lack of DNA photoreactivating enzyme in humans. Proc Natl Acad Sci U S A. 1993;90:4389–4393. doi: 10.1073/pnas.90.10.4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisen JA. Mechanistic basis of microsatellite instability. In: Goldstein DB, Schlotterer C, editors. Microsatellites: Evolution and Applications. Oxford University Press; Oxford: 1999. pp. 34–48. [Google Scholar]
- 3.Labarére J. DNA replication and repair. In: Maniloff J, editor. Mycoplasmas: Molecular Biology And Pathogenesis. American Society For Microbiology; Washington, D. C: 1992. pp. 309–323. [Google Scholar]
- 4.Dybvig K, Voelker LL. Molecular biology of mycoplasmas. Annu Rev Microbiol. 1996;50:25–57. doi: 10.1146/annurev.micro.50.1.25. [DOI] [PubMed] [Google Scholar]
- 5.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 6.Cortopassi GA, Wang E. There is substantial agreement among interspecies estimates of DNA repair activity. Mech Ageing Dev. 1996;91:211–218. doi: 10.1016/s0047-6374(96)01788-5. [DOI] [PubMed] [Google Scholar]
- 7.Promislow DE. DNA repair and the evolution of longevity: a critical analysis. J Theor Biol. 1994;170:291–300. doi: 10.1006/jtbi.1994.1190. [DOI] [PubMed] [Google Scholar]
- 8.LeClerc JE, Li B, Payne WL, Cebula TA. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science. 1996;274:1208–1211. doi: 10.1126/science.274.5290.1208. [DOI] [PubMed] [Google Scholar]
- 9.Matic I, Radman M, Taddei F, Picard B, Doit C, Bingen E, Denamur E, Elion J. Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science. 1997;277:1833–1834. doi: 10.1126/science.277.5333.1833. [DOI] [PubMed] [Google Scholar]
- 10.Taddei F, Matic I, Godelle B, Radman M. To be a mutator, or how pathogenic and commensal bacteria can evolve rapidly. Trends Microbiol. 1997;5:427–428. doi: 10.1016/S0966-842X(97)01157-8. [DOI] [PubMed] [Google Scholar]
- 11.Sueoka N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J Mol Evol. 1995;40:318–325. doi: 10.1007/BF00163236. [DOI] [PubMed] [Google Scholar]
- 12.Eyre-Walker A. DNA mismatch repair and synonymous codon evolution in mammals. Mol Biol Evol. 1994;11:88–98. doi: 10.1093/oxfordjournals.molbev.a040095. [DOI] [PubMed] [Google Scholar]
- 13.Sharp PM, Shields DC, Wolfe KH, Li WH. Chromosomal location and evolutionary rate variation in enterobacterial genes. Science. 1989;246:808–810. doi: 10.1126/science.2683084. [DOI] [PubMed] [Google Scholar]
- 14.Battista JR. Against all odds: the survival strategies of Deinococcus radiodurans. Annu Rev Microbiol. 1997;51:203–224. doi: 10.1146/annurev.micro.51.1.203. [DOI] [PubMed] [Google Scholar]
- 15.Matic I, Rayssiguier C, Radman M. Interspecies gene exchange in bacteria: the role of SOS and mismatch repair systems in evolution of species. Cell. 1995;80:507–515. doi: 10.1016/0092-8674(95)90501-4. [DOI] [PubMed] [Google Scholar]
- 16.Sniegowski P. Mismatch repair: origin of species? Curr Biol. 1998;8:R59–61. doi: 10.1016/s0960-9822(98)70035-1. [DOI] [PubMed] [Google Scholar]
- 17.Cleaver JE, Speakman JR, Volpe JP. Nucleotide excision repair: variations associated with cancer development and speciation. Cancer Surv. 1995;25:125–142. [PubMed] [Google Scholar]
- 18.Felsenstein J. Phylogenies and the comparative method. Am Nat. 1985;125:1–15. doi: 10.1086/703055. [DOI] [PubMed] [Google Scholar]
- 19.Eisen JA. PhD Thesis. Stanford University; 1999. [Google Scholar]
- 20.Eisen JA, Sweder KS, Hanawalt PC. Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res. 1995;23:2715–2723. doi: 10.1093/nar/23.14.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisen JA. A phylogenomic study of the MutS family of proteins. Nucleic Acids Res. 1998;26:4291–4300. doi: 10.1093/nar/26.18.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisen JA. Phylogenomics: improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 1998;8:163–167. doi: 10.1101/gr.8.3.163. [DOI] [PubMed] [Google Scholar]
- 23.Eisen JA, Kaiser D, Myers RM. Gastrogenomic delights: a movable feast. Nature (Medicine) 1997;3:1076–1078. doi: 10.1038/nm1097-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson JD, Higgins DG, Gibson TJ. Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swofford D. Phylogenetic Analysis Using Parsimony (PAUP) 3.0d. Illinois Natural History Survey; 1991. [Google Scholar]
- 27.Maddison WP, Maddison DR. MacClade 3. Sinauer Associates, Inc; 1992. [Google Scholar]
- 28.Maidak BL, Larsen N, McCaughey MJ, Overbeek R, Olsen GJ, Fogel K, Blandy J, Woese CR. The ribosomal database project. Nucleic Acids Res. 1994;22:3485–3487. doi: 10.1093/nar/22.17.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koonin EV, Mushegian A, Bork P. Non-orthologous gene displacement. Trends Genet. 1996;12:334–336. [PubMed] [Google Scholar]
- 30.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–498. 507–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 31.Dobzhansky T. Nothing in biology makes sense except in thelight of evolution. American Biology Teacher. 1973;35:125–129. [Google Scholar]
- 32.Li WH, Gojobori T, Nei M. Pseudogenes as a paradigm of neutral evolution. Nature. 1981;292:237–9. doi: 10.1038/292237a0. [DOI] [PubMed] [Google Scholar]
- 33.Gutell RR, Larsen N, Woese CR. Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol Rev. 1994;58:10–26. doi: 10.1128/mr.58.1.10-26.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldman N, Thorne JL, Jones DT. Using evolutionary trees in protein secondary structure prediction and other comparative sequence analysis. J Mol Biol. 1996;263:196–208. doi: 10.1006/jmbi.1996.0569. [DOI] [PubMed] [Google Scholar]
- 35.Henikoff S, Henikoff J. Protein family classification based on searching a database of blocks. Genomics. 1994;19:97–107. doi: 10.1006/geno.1994.1018. [DOI] [PubMed] [Google Scholar]
- 36.Dayhoff MO. Atlas of protein sequence and structure. 3. Vol. 5. National Biomedical Research Foundation; Washington, D.C: 1978. [Google Scholar]
- 37.Lafay B, Lloyd AT, McLean MJ, Devine KM, Sharp PM, Wolfe KH. Proteome composition and codon usage in spirochaetes: species-specific and DNA strand-specific mutational biases. Nucleic Acids Res. 1999;27:1642–1649. doi: 10.1093/nar/27.7.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aravind L, Walker DR, Koonin EV. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999;27:1223–1242. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanai S, Kikuno R, Toh H, Ryo H, Todo T. Molecular evolution of the photolyase-blue- light photoreceptor family. J Mol Evol. 1997;45:535–548. doi: 10.1007/pl00006258. [DOI] [PubMed] [Google Scholar]
- 40.Zhao S, Sancar A. Human blue-light photoreceptor hCRY2 specifically interacts with protein serine/threonine phosphatase 5 and modulates its activity. Photochem Photobiol. 1997;66:727–731. doi: 10.1111/j.1751-1097.1997.tb03214.x. [DOI] [PubMed] [Google Scholar]
- 41.Cockell CS. Biological effects of high ultraviolet radiation on early earth--a theoretical evaluation. J Theor Biol. 1998;193:717–729. doi: 10.1006/jtbi.1998.0738. [DOI] [PubMed] [Google Scholar]
- 42.Sutherland JC, Griffin KP. Monomerization of pyrimidine dimers in DNA by tryptophan- containing peptides: wavelength dependence. Radiat Res. 1980;83:529–536. [PubMed] [Google Scholar]
- 43.Chen J, Huang CW, Hinman L, Gordon MP, Deranleau DA. Photomonomerization of pyrimidine dimers by indoles and proteins. J Theor Biol. 1976;62:53–67. doi: 10.1016/0022-5193(76)90051-5. [DOI] [PubMed] [Google Scholar]
- 44.Helene C, Toulme F, Charlier M, Yaniv M. Photosensitized splitting of thymine dimers in DNA by gene 32 protein from phage T 4. Biochem Biophys Res Commun. 1976;71:91–98. doi: 10.1016/0006-291x(76)90253-9. [DOI] [PubMed] [Google Scholar]
- 45.Pegg AE, Byers TL. Repair of DNA containing O6-alkylguanine. Faseb J. 1992;6:2302–2310. doi: 10.1096/fasebj.6.6.1544541. [DOI] [PubMed] [Google Scholar]
- 46.Pegg AE, Dolan ME, Moschel RC. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase. Prog Nucleic Acid Res Mol Biol. 1995;51:167–223. doi: 10.1016/s0079-6603(08)60879-x. [DOI] [PubMed] [Google Scholar]
- 47.Labahn J, Scharer OD, Long A, Ezaz-Nikpay K, Verdine GL, Ellenberger TE. Structural basis for the excision repair of alkylation-damaged DNA. Cell. 1996;86:321–329. doi: 10.1016/s0092-8674(00)80103-8. [DOI] [PubMed] [Google Scholar]
- 48.Leclere MM, Nishioka M, Yuasa T, Fujiwara S, Takagi M, Imanaka T. The O6-methylguanine-DNA methyltransferase from the hyperthermophilic archaeon Pyrococcus sp. KOD1: a thermostable repair enzyme. Mol Gen Genet. 1998;258:69–77. doi: 10.1007/s004380050708. [DOI] [PubMed] [Google Scholar]
- 49.Skorvaga M, Raven NDH, Margison GP. Thermostable archaeal O6-alkylguanine-DNA alkyltransferases. Proc Natl Acad Sci U S A. 1998;95:6711–6715. doi: 10.1073/pnas.95.12.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo J, Barany F. Identification of essential residues in Thermus thermophilus DNA ligase. Nucleic Acids Res. 1996;24:3079–3085. doi: 10.1093/nar/24.15.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tomkinson AE, Mackey ZB. Structure and function of mammalian DNA ligases. Mutat Res. 1998;407:1–9. doi: 10.1016/s0921-8777(97)00050-5. [DOI] [PubMed] [Google Scholar]
- 52.Modrich P. Mechanisms and biological effects of mismatch repair. Annu Rev Genet. 1991;25:229–253. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]
- 53.Kolodner R. Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev. 1996;10:1433–1442. doi: 10.1101/gad.10.12.1433. [DOI] [PubMed] [Google Scholar]
- 54.Miller JH. Spontaneous mutators in bacteria: insights into pathways of mutagenesis and repair. Annu Rev Microbiol. 1996;50:625–643. doi: 10.1146/annurev.micro.50.1.625. [DOI] [PubMed] [Google Scholar]
- 55.Taddei F, Vulic M, Radman M, Matic I. Genetic variability and adaptation to stress. Experientia. 1997;83:271–290. doi: 10.1007/978-3-0348-8882-0_15. [DOI] [PubMed] [Google Scholar]
- 56.Sniegowski PD, Gerrish PJ, Lenski RE. Evolution of high mutation rates in experimental populations of E. coli. Nature. 1997;387:703–705. doi: 10.1038/42701. [DOI] [PubMed] [Google Scholar]
- 57.Mizrahi V, Andersen SJ. DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence? Mol Microbiol. 1998;29:1331–1340. doi: 10.1046/j.1365-2958.1998.01038.x. [DOI] [PubMed] [Google Scholar]
- 58.Matic I, Taddei F, Radman M. Genetic barriers among bacteria. Trends Microbiol. 1996;4:69–72. doi: 10.1016/0966-842X(96)81514-9. [DOI] [PubMed] [Google Scholar]
- 59.Ban C, Yang W. Structural basis for MutH activation in E.coli mismatch repair and relationship of MutH to restriction endonucleases. EMBO J. 1998;17:1526–1534. doi: 10.1093/emboj/17.5.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Twomey DP, McKay LL, O’Sullivan DJ. Molecular characterization of the Lactococcus lactis LlaKR2I restriction-modification system and effect of an IS982 element positioned between the restriction and modification genes. J Bacteriol. 1998;180:5844–5854. doi: 10.1128/jb.180.22.5844-5854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, Golemis EA, Genuardi M, Neri G. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci U S A. 1999;96:3969–3974. doi: 10.1073/pnas.96.7.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Glasner W, Merkl R, Schellenberger V, Fritz HJ. Substrate preferences of Vsr DNA mismatch endonuclease and their consequences for the evolution of the Escherichia coli K-12 genome. J Mol Biol. 1995;245:1–7. doi: 10.1016/s0022-2836(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 63.Hoeijmakers JH. Nucleotide excision repair. II: From yeast to mammals. Trends Genet. 1993;9:211–217. doi: 10.1016/0168-9525(93)90121-w. [DOI] [PubMed] [Google Scholar]
- 64.Hoeijmakers JH. Nucleotide excision repair I: from E. coli to yeast. Trends Genet. 1993;9:173–177. doi: 10.1016/0168-9525(93)90164-d. [DOI] [PubMed] [Google Scholar]
- 65.Sancar A. DNA excision repair. Annu Rev Biochem. 1996;65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- 66.Van Houten B, Snowden A. Mechanism of action of the Escherichia coli UvrABC nuclease: clues to the damage recognition problem. Bioessays. 1993;15:51–59. doi: 10.1002/bies.950150108. [DOI] [PubMed] [Google Scholar]
- 67.Selby CP, Sancar A. Structure and function of transcription-repair coupling factor. I. Structural domains and binding properties. J Biol Chem. 1995;270:4882–4889. doi: 10.1074/jbc.270.9.4882. [DOI] [PubMed] [Google Scholar]
- 68.Mellon I, Hanawalt PC. Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature. 1989;342:95–98. doi: 10.1038/342095a0. [DOI] [PubMed] [Google Scholar]
- 69.Ayora S, Rojo F, Ogasawara N, Nakai S, Alonso JC. The Mfd protein of Bacillus subtilis 168 is involved in both transcription-coupled DNA repair and DNA recombination. J Mol Biol. 1996;256:301–318. doi: 10.1006/jmbi.1996.0087. [DOI] [PubMed] [Google Scholar]
- 70.Zalieckas JM, Wray LV, Jr, Ferson AE, Fisher SH. Transcription-repair coupling factor is involved in carbon catabolite repression of the Bacillus subtilis hut and gnt operons. Mol Microbiol. 1998;27:1031–1038. doi: 10.1046/j.1365-2958.1998.00751.x. [DOI] [PubMed] [Google Scholar]
- 71.Wood RD. Nucleotide excision repair in mammalian cells. J Biol Chem. 1997;272:23465–23468. doi: 10.1074/jbc.272.38.23465. [DOI] [PubMed] [Google Scholar]
- 72.Doolittle RF, Johnson MS, Husain I, Van Houten B, Thomas DC, Sancar A. Domainal evolution of a prokaryotic DNA repair protein and its relationship to active-transport proteins. Nature. 1986;323:451–453. doi: 10.1038/323451a0. [DOI] [PubMed] [Google Scholar]
- 73.Linton KJ, Higgins CF. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol Microbiol. 1998;28:5–13. doi: 10.1046/j.1365-2958.1998.00764.x. [DOI] [PubMed] [Google Scholar]
- 74.Lin CG, Kovalsky O, Grossman L. DNA damage-dependent recruitment of nucleotide excision repair and transcription proteins to Escherichia coli inner membranes. Nucleic Acids Res. 1997;25:3151–3158. doi: 10.1093/nar/25.15.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lomovskaya N, Hong SK, Kim SU, Fonstein L, Furuya K, Hutchinson RC. The Streptomyces peucetius drrC gene encodes a UvrA-like protein involved in daunorubicin resistance and production. J Bacteriol. 1996;178:3238–3225. doi: 10.1128/jb.178.11.3238-3245.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCready S. The repair of ultraviolet light-induced DNA damage in the halophilic archaebacteria, Halobacterium cutirubrum, Halobacterium halobium and Haloferax volcanii. Mutat Res. 1996;364:25–32. doi: 10.1016/0921-8777(96)00018-3. [DOI] [PubMed] [Google Scholar]
- 77.Sgouros J, Gaillard PH, Wood RD. A relationship betweena DNA-repair/recombination nuclease family and archaeal helicases. Trends Biochem Sci. 1999;24:95–97. doi: 10.1016/s0968-0004(99)01355-9. [DOI] [PubMed] [Google Scholar]
- 78.Lieber MR. The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombinatino and repair. Bioessays. 1997;19:233–240. doi: 10.1002/bies.950190309. [DOI] [PubMed] [Google Scholar]
- 79.Ogrunc M, Becker DF, Ragsdale SW, Sancar A. Nucleotide excision repair in the third kingdom. J Bacteriol. 1998;180:5796–5798. doi: 10.1128/jb.180.21.5796-5798.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Doetsch PW. What’s old is new: an alternative DNA excision repair pathway. Trends Biochem Sci. 1995;20:384–386. doi: 10.1016/s0968-0004(00)89084-2. [DOI] [PubMed] [Google Scholar]
- 81.Bowman KK, Sidik K, Smith CA, Taylor JS, Doetsch PW, Freyer GA. A new ATP-independent DNA endonuclease from Schizosaccharomyces pombe that recognizes cyclobutane pyrimidine dimers and 6–4 photoproducts. Nucleic Acids Res. 1994;22:3026–32. doi: 10.1093/nar/22.15.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yasui A, McCready SJ. Alternative repair pathways for UV-induced DNA damage. Bioessays. 1998;20:291–297. doi: 10.1002/(SICI)1521-1878(199804)20:4<291::AID-BIES5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 83.Kanno S, Iwai S, Takao M, Yasui A. Repair of apurinic/apyrimidinic sites by UV damage endonuclease; a repair protein for UV and oxidative damage. Nucleic Acids Res. 1999;27:3096–3103. doi: 10.1093/nar/27.15.3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krokan HE, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochem J. 1997;325:1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meyer-Siegler K, Mauro DJ, Seal G, Wurzer J, deRiel JK, Sirover MA. A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceraldehyde-3-phosphate dehydrogenase. Proc Natl Acad Sci U S A. 1991;88:8460–8464. doi: 10.1073/pnas.88.19.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Muller SJ, Caradonna S. Cell cycle regulation of a human cyclin-like gene encoding uracil- DNA glycosylase. J Biol Chem. 1993;268:1310–1319. [PubMed] [Google Scholar]
- 87.Slupphaug G, Eftedal I, Kavli B, Bharati S, Helle NM, Haug T, Levine DW, Krokan HE. Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry. 1995;34:128–138. doi: 10.1021/bi00001a016. [DOI] [PubMed] [Google Scholar]
- 88.Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, Krokan HE. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997;25:750–755. doi: 10.1093/nar/25.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sandigursky M, Franklin WA. Thermostable uracil-DNA glycosylase from Thermotoga maritima a member of a novel class of DNA repair enzymes. Curr Biol. 1999;9:531–534. doi: 10.1016/s0960-9822(99)80237-1. [DOI] [PubMed] [Google Scholar]
- 90.Koulis A, Cowan DA, Pearl LH, Savva R. Uracil-DNA glycosylase activities in hyperthermophilic micro-organisms. FEMS Microbiol Lett. 1996;143:267–271. doi: 10.1111/j.1574-6968.1996.tb08491.x. [DOI] [PubMed] [Google Scholar]
- 91.Gallinari P, Jiricny J. A new class of uracil-DNA glycosylases related to human thymine- DNA glycosylase. Nature. 1996;383:735–738. doi: 10.1038/383735a0. [DOI] [PubMed] [Google Scholar]
- 92.Neddermann P, Jiricny J. Efficient removal of uracil from G.U mispairs by the mismatch-specific thymine DNA glycosylase from HeLa cells. Proc Natl Acad Sci U S A. 1994;91:1642–1646. doi: 10.1073/pnas.91.5.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Manuel RC, Czerwinski EW, Lloyd RS. Identification of the structural and functional domains of MutY, an Escherichia coli DNA mismatch repair enzyme. J Biol Chem. 1996;271:16218–16226. doi: 10.1074/jbc.271.27.16218. [DOI] [PubMed] [Google Scholar]
- 94.Au KG, Cabrera M, Miller JH, Modrich P. Escherichia coli mutY gene product is required for specific A-G----C.G mismatch correction. Proc Natl Acad Sci U S A. 1988;85:9163–9166. doi: 10.1073/pnas.85.23.9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nghiem Y, Cabrera M, Cupples CG, Miller JH. The mutY gene: a mutator locus in Escherichia coli that generates G.C--- -T.A transversions. Proc Natl Acad Sci U S A. 1988;85:2709–2713. doi: 10.1073/pnas.85.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McGoldrick JP, Yeh YC, Solomon M, Essigmann JM, Lu AL. Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Mol Cell Biol. 1995;15:989–996. doi: 10.1128/mcb.15.2.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Slupska MM, Baikalov C, Luther WM, Chiang JH, Wei YF, Miller JH. Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J Bacteriol. 1996;178:3885–3892. doi: 10.1128/jb.178.13.3885-3892.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aspinwall R, Rothwell DG, Roldan-Arjona T, Anselmino C, Ward CJ, Cheadle JP, Sampson JR, Lindahl T, Harris PC, Hickson ID. Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc Natl Acad Sci U S A. 1997;94:109–114. doi: 10.1073/pnas.94.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Piersen CE, Prince MA, Augustine ML, Dodson ML, Lloyd RS. Purification and cloning of Micrococcus luteus ultraviolet endonuclease, an N-glycosylase/abasic lyase that proceeds via an imino enzyme-DNA intermediate. J Biol Chem. 1995;270:23475–23484. doi: 10.1074/jbc.270.40.23475. [DOI] [PubMed] [Google Scholar]
- 100.Horst JP, Fritz HJ. Counteracting the mutagenic effect of hydrolytic deamination of DNA 5- methylcytosine residues at high temperature: DNA mismatch N-glycosylase Mig.Mth of the thermophilic archaeon Methanobacterium thermoautotrophicum. THF EMBO J. 1996;15:5459–5469. [PMC free article] [PubMed] [Google Scholar]
- 101.Begley TJ, Haas BJ, Noel J, Shekhtman A, Williams WA, Cunningham RP. A new member of the endonuclease III family of DNA repair enzymes that removes methylated purines from DNA. Curr Biol. 1999;9:653–656. doi: 10.1016/s0960-9822(99)80288-7. [DOI] [PubMed] [Google Scholar]
- 102.Michaels ML, Pham L, Cruz C, Miller JH. MutM, a protein that prevents G.C----T.A transversions, is formamidopyrimidine-DNA glycosylase. Nucleic Acids Res. 1991;19:3629–3632. doi: 10.1093/nar/19.13.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cabrera M, Nghiem Y, Miller JH. mutM, a second mutator locus in Escherichia coli that generates G.C---- T.A transversions. J Bacteriol. 1988;170:5405–5407. doi: 10.1128/jb.170.11.5405-5407.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duwat P, de Oliveira R, Ehrlich SD, Boiteux S. Repair of oxidative DNA damage in gram-positive bacteria: the Lactococcus lactis Fpg protein. Microbiol. 1995;141:411–417. doi: 10.1099/13500872-141-2-411. [DOI] [PubMed] [Google Scholar]
- 105.Mikawa T, Kato R, Sugahara M, Kuramitsu S. Thermostable repair enzyme for oxidative DNA damage from extremely thermophilic bacterium, Thermus thermophilus HB8. Nucleic Acids Res. 1998;26:903–910. doi: 10.1093/nar/26.4.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jiang D, Hatahet Z, Melamede RJ, Kow YW, Wallace SS. Characterization of Escherichia coli endonuclease VIII. J Biol Chem. 1997;272:32230–32239. doi: 10.1074/jbc.272.51.32230. [DOI] [PubMed] [Google Scholar]
- 107.Jiang D, Hatahet Z, Blaisdell JO, Melamede RJ, Wallace SS. Escherichia coli endonuclease VIII: cloning, sequencing, and overexpression of the nei structural gene and characterization of nei and nei nth mutants. J Bacteriol. 1997;179:3773–3782. doi: 10.1128/jb.179.11.3773-3782.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ohtsubo T, Matsuda O, Iba K, Terashima I, Sekiguchi M, Nakabeppu Y. Molecular cloning of AtMMH, an Arabidopsis thaliana ortholog of the Escherichia coli mutM gene, and analysis of functional domains of its product. Mol Gen Genet. 1998;259:577–90. doi: 10.1007/s004380050851. [DOI] [PubMed] [Google Scholar]
- 109.van der Kemp PA, Thomas D, Barbey R, de Oliveira R, Boiteux S. Cloning and expression in Escherichia coli of the OGG1 gene of Saccharomyces cerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N- methylformamidopyrimidine. Proc Natl Acad Sci U S A. 1996;93:5197–5202. doi: 10.1073/pnas.93.11.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Arai K, Morishita K, Shinmura K, Kohno T, Kim SR, Nohmi T, Taniwaki M, Ohwada S, Yokota J. Cloning of a human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene. 1997;14:2857–2861. doi: 10.1038/sj.onc.1201139. [DOI] [PubMed] [Google Scholar]
- 111.Radicella JP, Dherin C, Desmaze C, Fox MS, Boiteux S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1997;94:8010–8015. doi: 10.1073/pnas.94.15.8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosenquist TA, Zharkov DO, Grollman AP. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc Natl Acad Sci U S A. 1997;94:7429–7434. doi: 10.1073/pnas.94.14.7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Takao M, Aburatani H, Kobayashi K, Yasui A. Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res. 1998;26:2917–2922. doi: 10.1093/nar/26.12.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xiao W, Chow BL, Rathgeber L. The repair of DNA methylation damage in Saccharomyces cerevisiae. Curr Genet. 1996;30:461–648. doi: 10.1007/s002940050157. [DOI] [PubMed] [Google Scholar]
- 115.Laval J, Jurado J, Saparbaev M, Sidorkina O. Antimutagenic role of base-excision repair enzymes upon free radical-induced DNA damage. Mutat Res. 1998;402:93–102. doi: 10.1016/s0027-5107(97)00286-8. [DOI] [PubMed] [Google Scholar]
- 116.Furuta M, Schrader JO, Schrader HS, Kokjohn TA, Nyaga S, McCullough AK, Lloyd RS, Burbank DE, Landstein D, Lane L, et al. Chlorella virus PBCV-1 encodes a homolog of the bacteriophage T4 UV damage repair gene denV. Appl Environ Microbiol. 1997;63:1551–1556. doi: 10.1128/aem.63.4.1551-1556.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barzilay G, Hickson ID. Structure and function of apurinic/apyrimidinic endonucleases. Bioessays. 1995;17:713–719. doi: 10.1002/bies.950170808. [DOI] [PubMed] [Google Scholar]
- 118.Kuo CF, Mol CD, Thayer MM, Cunningham RP, Tainer JA. Structure and function of the DNA repair enzyme exonuclease III from E. coli. Ann N Y Acad Sci. 1994;726:223–234. doi: 10.1111/j.1749-6632.1994.tb52820.x. [DOI] [PubMed] [Google Scholar]
- 119.Camerini-Otero RD, Hsieh P. Homologous recombination proteins in prokaryotes and eukaryotes. Annu Rev Genet. 1995;29:509–552. doi: 10.1146/annurev.ge.29.120195.002453. [DOI] [PubMed] [Google Scholar]
- 120.Clark AJ, Sandler SJ. Homologous genetic recombination: the pieces begin to fall into place. Crit Rev Microbiol. 1994;20:125–142. doi: 10.3109/10408419409113552. [DOI] [PubMed] [Google Scholar]
- 121.Kowalczykowski S, Dixon D, Eggleston A, Lauder S, Rehrauer W. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hanawalt PC, Cooper PK, Ganesan AK, Smith CA. DNA repair in bacteria and mammalian cells. Annu Rev Biochem. 1979;48:783–836. doi: 10.1146/annurev.bi.48.070179.004031. [DOI] [PubMed] [Google Scholar]
- 123.Eggleston AK, West SC. Recombination initiation: easy as A, B, C, D... chi? Curr Biol. 1997;7:R745–749. doi: 10.1016/s0960-9822(06)00394-0. [DOI] [PubMed] [Google Scholar]
- 124.Farrand SK, Hwang I, Cook DM. The tra region of the nopaline-type Ti plasmid is a chimera with elements related to the transfer systems of RSF1010, RP4, and F. J Bacteriol. 1996;178:4233–4247. doi: 10.1128/jb.178.14.4233-4247.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Alt-Morbe J, Stryker JL, Fuqua C, Li PL, Farrand SK, Winans SC. The conjugal transfer system of Agrobacterium tumefaciens octopine-type Ti plasmids is closely related to the transfer system of an IncP plasmid and distantly related to Ti plasmid vir genes. J Bacteriol. 1996;178:4248–4257. doi: 10.1128/jb.178.14.4248-4257.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.el Karoui M, Ehrlich D, Gruss A. Identification of the lactococcal exonuclease/recombinase and its modulation by the putative Chi sequence. Proc Natl Acad Sci U S A. 1998;95:626–631. doi: 10.1073/pnas.95.2.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Courcelle J, Carswell-Crumpton C, Hanawalt PC. recF and recR are required for the resumption of replication at DNA replication forks in Escherichia coli. Proc Natl Acad Sci U S A. 1997;94:3714–3719. doi: 10.1073/pnas.94.8.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nakayama K, Shiota S, Nakayama H. Thymineless death in Escherichia coli mutants deficient in the RecF recombination pathway. Can J Microbiol. 1988;34:905–907. doi: 10.1139/m88-157. [DOI] [PubMed] [Google Scholar]
- 129.Nakayama H, Nakayama K, Nakayama R, Irino N, Nakayama Y, Hanawalt P. Isolation and genetic characterization of a thymineless death-resistant mutant of Escherichia coli K12: identification of a new mutation (recQ1) that blocks the RecF recombination pathway. Mol Gen Genet. 1984;195:474–480. doi: 10.1007/BF00341449. [DOI] [PubMed] [Google Scholar]
- 130.Gray MD, Shen JC, Kamath-Loeb AS, Blank A, Sopher BL, Martin GM, Oshima J, Loeb LA. The Werner syndrome protein is a DNA helicase. Nat Genet. 1997;17:100–103. doi: 10.1038/ng0997-100. [DOI] [PubMed] [Google Scholar]
- 131.Karow JK, Chakraverty RK, Hickson ID. The Bloom’s syndrome gene product is a 3′-5′ DNA helicase. J Biol Chem. 1997;272:30611–30614. doi: 10.1074/jbc.272.49.30611. [DOI] [PubMed] [Google Scholar]
- 132.Watt PM, Hickson ID, Borts RH, Louis EJ. SGS1, a homologue of the Bloom’s and Werner’s syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics. 1996;144:935–945. doi: 10.1093/genetics/144.3.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, et al. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 134.Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 135.Hirano T. SMC protein complexes and higher-order chromosome dynamics. Curr Opin Cell Biol. 1998;10:317–322. doi: 10.1016/s0955-0674(98)80006-9. [DOI] [PubMed] [Google Scholar]
- 136.Jessberger R, Frei C, Gasser SM. Chromosome dynamics: the SMC protein family. Curr Opin Genet Dev. 1998;8:254–259. doi: 10.1016/s0959-437x(98)80149-4. [DOI] [PubMed] [Google Scholar]
- 137.Clark AJ, Sharma V, Brenowitz S, Chu CC, Sandler S, Satin L, Templin A, Berger I, Cohen A. Genetic and molecular analyses of the C-terminal region of the recE gene from the Rac prophage of Escherichia coli K-12 reveal the recT gene. J Bacteriol. 1993;175:7673–7682. doi: 10.1128/jb.175.23.7673-7682.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kolodner R, Hall SD, Luisi-DeLuca C. Homologous pairing proteins encoded by the Escherichia coli recE and recT genes. Mol Microbiol. 1994;11:23–30. doi: 10.1111/j.1365-2958.1994.tb00286.x. [DOI] [PubMed] [Google Scholar]
- 139.Kusano K, Takahashi NK, Yoshikura H, Kobayashi I. Involvement of RecE exonuclease and RecT annealing protein in DNA double-strand break repair by homologous recombination. Gene. 1994;138:17–25. doi: 10.1016/0378-1119(94)90778-1. [DOI] [PubMed] [Google Scholar]
- 140.Noirot P, Kolodner RD. DNA strand invasion promoted by Escherichia coli RecT protein. J Biol Chem. 1998;273:12274–12280. doi: 10.1074/jbc.273.20.12274. [DOI] [PubMed] [Google Scholar]
- 141.Connelly JC, Kirkham LA, Leach DRF. The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc Natl Acad Sci U S A. 1998;95:7969–7974. doi: 10.1073/pnas.95.14.7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sharples GJ, Leach DRF. Structural and functional similarities between the SbcCD proteins of Escherichia coli and the Rad50 and Mre11 (Rad32) recombination and repair proteins of yeast. Mol Microbiol. 1995;17:1215–1217. doi: 10.1111/j.1365-2958.1995.mmi_17061215_1.x. [DOI] [PubMed] [Google Scholar]
- 143.Paul TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 144.Petrini JHJ, Walsh ME, Dimare C, Chen XN, Korenberg JR, Weaver DT. Isolation and characterization of the human MRE11 homologue. Genomics. 1995;29:80–86. doi: 10.1006/geno.1995.1217. [DOI] [PubMed] [Google Scholar]
- 145.Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, Hays L, Morgan WF, Petrini JHJ. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 146.Eisen JA. The RecA protein as a model molecule for molecular systematic studies of bacteria: comparison of trees of RecAs and 16s rRNAs from the same species. J Mol Evol. 1995;41:1105–1123. doi: 10.1007/BF00173192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gruber TM, Eisen JA, Gish K, Bryant DA. The phylogenetic relationships of Chlorobium tepidum and Chloroflexus aurantiacus based upon their RecA sequences. FEMS Microbiol Lett. 1998;162:53–60. doi: 10.1111/j.1574-6968.1998.tb12978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stassen NY, Logsdon JM, Jr, Vora GJ, Offenberg HH, Palmer JD, Zolan ME. Isolation and characterization of rad51 orthologs from Coprinus cinereus and Lycopersicon esculentum, and phylogenetic analysis of eukaryotic recA homologs. Curr Genet. 1997;31:144–157. doi: 10.1007/s002940050189. [DOI] [PubMed] [Google Scholar]
- 149.King KW, Woodard A, Dybvig K. Cloning and characterization of the recA genes from Mycoplasma pulmonis and M. mycoides subsp. mycoides. Gene. 1994;139:111–115. doi: 10.1016/0378-1119(94)90532-0. [DOI] [PubMed] [Google Scholar]
- 150.Marais A, Bove JM, Renaudin J. Spiroplasma citri virus SpV1-derived cloning vector: deletion formation by illegitimate and homologous recombination in a spiroplasmal host strain which probably lacks a functional recA gene. J Bacteriol. 1996;178:862–870. doi: 10.1128/jb.178.3.862-870.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Norioka N, Hsu MY, Inouye S, Inouye M. Two recA genes in Myxococcus xanthus. J Bacteriol. 1995;177:4179–4182. doi: 10.1128/jb.177.14.4179-4182.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bianco PR, Tracy RB, Kowalczykowski SC. DNA strand exchange proteins: a biochemical and physical comparison. Front Biosci. 1998;3:d570–603. doi: 10.2741/a304. [DOI] [PubMed] [Google Scholar]
- 153.Griffith JD, Harris LD. DNA strand exchanges. CRC Crit Rev Biochem. 1988;23:S43–86. [PubMed] [Google Scholar]
- 154.Muller B, West SC. Processing of Holliday junctions by the Escherichia coli RuvA, RuvB, RuvC and RecG proteins. Experientia. 1994;50:216–222. doi: 10.1007/BF01924004. [DOI] [PubMed] [Google Scholar]
- 155.Mandal TN, Mahdi AA, Sharples GJ, Lloyd RG. Resolution of Holliday intermediates in recombination and DNA repair: indirect suppression of ruvA, ruvB, and ruvC mutations. J Bacteriol. 1993;175:4325–4334. doi: 10.1128/jb.175.14.4325-4334.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sharples GJ, Chan SN, Mahdi AA, Whitby MC, Lloyd RG. Processing of intermediates in recombination and DNA repair: identification of a new endonuclease that specifically cleaves Holliday junctions. EMBO J. 1994;13:6133–6142. doi: 10.1002/j.1460-2075.1994.tb06960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chan SN, Vincent SD, Lloyd RG. Recognition and manipulation of branched DNA by the RusA Holliday junction resolvase of Escherichia coli. Nucleic Acids Res. 1998;26:1560–1566. doi: 10.1093/nar/26.7.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.West SC. Processing of recombination intermediates by the RuvABC proteins. Annu Rev Genet. 1997;31:213–244. doi: 10.1146/annurev.genet.31.1.213. [DOI] [PubMed] [Google Scholar]
- 159.Schofield MJ, Lilley DM, White MF. Dissection of the sequence specificity of the Holliday junction endonuclease CCE1. Biochemistry. 1998;37:7733–7740. doi: 10.1021/bi980399s. [DOI] [PubMed] [Google Scholar]
- 160.Ishioka K, Iwasaki H, Shinagawa H. Roles of the recG gene product of Escherichia coli in recombination repair: effects of the delta recG mutation on cell division and chromosome partition. Genes Genet Syst. 1997;72:91–99. doi: 10.1266/ggs.72.91. [DOI] [PubMed] [Google Scholar]
- 161.Jeggo PA, Taccioli GE, Jackson SP. Menage a trois: double strand break repair, V(D)J recombination and DNA-PK. Bioessays. 1995;17:949–957. doi: 10.1002/bies.950171108. [DOI] [PubMed] [Google Scholar]
- 162.Ramsden DA, Gellert M. Ku protein stimulates DNA end joining by mammalian DNA ligases: a direct role for Ku in repair of dna double-strand breaks. EMBO Journal. 1998;17:609–614. doi: 10.1093/emboj/17.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wilson TE, Grawunder U, Lieber MR. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature. 1997;388:495–498. doi: 10.1038/41365. [DOI] [PubMed] [Google Scholar]
- 164.Chu G. Double strand break repair. J Biol Chem. 1997;272:24097–24100. doi: 10.1074/jbc.272.39.24097. [DOI] [PubMed] [Google Scholar]
- 165.Keith CT, Schreiber SL. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science. 1995;270:50–51. doi: 10.1126/science.270.5233.50. [DOI] [PubMed] [Google Scholar]
- 166.Edgell DR, Doolittle WF. Archaea and the origin(s) of DNA replication proteins. Cell. 1997;89:995–998. doi: 10.1016/s0092-8674(00)80285-8. [DOI] [PubMed] [Google Scholar]
- 167.Shinagawa H. SOS response as an adaptive response to DNA damage in prokaryotes. Experientia. 1996;77:221–235. doi: 10.1007/978-3-0348-9088-5_14. [DOI] [PubMed] [Google Scholar]
- 168.Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci U S A. 1999;96:424–428. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lewis RJ, Hanawalt PC. Ligation of oligonucleotides by pyrimidine dimers--a missing ‘link’ in the origin of life? Nature. 1982;298:393–396. doi: 10.1038/298393a0. [DOI] [PubMed] [Google Scholar]
- 170.Gorbalenya AE, Koonin EV, Donchenko AP, Blinov VM. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989;17:4713–4730. doi: 10.1093/nar/17.12.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- 172.Blattner FR, Plunkett GI, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 173.Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, Podowski RM, Naslund AK, Eriksson AS, Winkler HH, Kurland CG. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- 174.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 175.Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 176.Sanger-Centre. personal communication.
- 177.Kunst A, Ogasawara N, Moszer I, Albertini A, Alloni G, Azevedo V, Bertero M, Bessieres P, Bolotin A, Borchert S, et al. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 178.Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, Bult CJ, Kerlavage AR, Sutton G, Kelley JM, et al. The minimal gene complement of Mycoplasma genitalium [see comments] Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- 179.Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, et al. Deciphiring the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 181.Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, Gwinn M, Hickey EK, Clayton R, Ketchum KA, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 182.Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, Gwinn M, Hickey EK, Clayton R, Ketchum KA, et al. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281:375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- 183.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 184.Kaneko T, Sato S, Kotani H, Tanaka A, Asamizu E, Nakamura Y, Miyajima N, Hirosawa M, Sugiura M, Sasamoto S, et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996;3:109–136. doi: 10.1093/dnares/3.3.109. [DOI] [PubMed] [Google Scholar]
- 185.White O, Eisen JA, Heidelberg JF, Hickey EK, Peterson JD, Dodson RJ, Haft DH, Gwinn ML, Nelson WC, Richardson DL, et al. Complete genome sequencing of the radioresistant bacterium, Deinococcus radiodurans. 1999 doi: 10.1126/science.286.5444.1571. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Deckert G, Warren PV, Gaasterland T, Young WG, Lenox AL, Grahams DE, Overbeek R, Snead MA, Keller M, Aujay M, et al. The complete genome of the hyperthemophilic bacterium Aquifex aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- 187.Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, Fitzgerald LM, Clayton RA, Gocayne JD, et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 188.Smith DR, Doucette-Stamm LA, Deloughery C, Lee H, Dubois J, Aldredge T, Bashirzadeh R, Blakely D, Cook R, Gilbert K, et al. Complete genome sequence of Methanobacterium thermoautotrophicumΔH: functional analysis and comparative genomics. J Bacteriol. 1996;179:7135–7155. doi: 10.1128/jb.179.22.7135-7155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kawarabayasi Y, Sawada M, Horikawa H, Haikawa Y, Hino Y, Yamamoto S, Sekine M, Baba S, Kosugi H, Hosoyama A, et al. Complete sequence and gene organization of the genome of a hyper-thermophilic archaebacterium, Pyrococcus horikoshii OT3. DNA Res. 1998;5:55–76. doi: 10.1093/dnares/5.2.55. [DOI] [PubMed] [Google Scholar]
- 190.Klenk HP, Clayton RA, Tomb JF, White O, Nelsen KE, Ketchum KA, Dodson RJ, Gwinn M, Hickey EK, Peterson JD, et al. The complete genomic sequence of the hyperthermophilic, sulfate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- 191.Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, et al. Life with 6000 genes. Science. 1996;274:546, 563–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]