

Abstract
Genome-wide association (GWA) studies have identified a number of loci underlying variation in human serum uric acid (SUA) levels with the SLC2A9 gene having the largest effect identified so far. Gene-gene interactions (epistasis) are largely unexplored in these GWA studies. We performed a full pair-wise genome scan in the Italian MICROS population (n = 1201) to characterise epistasis signals in SUA levels. In the resultant epistasis profile, no SNP pairs reached the Bonferroni adjusted threshold for the pair-wise genome-wide significance. However, SLC2A9 was found interacting with multiple loci across the genome, with NFIA - SLC2A9 and SLC2A9 - ESRRAP2 being significant based on a threshold derived for interactions between GWA significant SNPs and the genome and jointly explaining 8.0% of the phenotypic variance in SUA levels (3.4% by interaction components). Epistasis signal replication in a CROATIAN population (n = 1772) was limited at the SNP level but improved dramatically at the gene ontology level. In addition, gene ontology terms enriched by the epistasis signals in each population support links between SUA levels and neurological disorders. We conclude that GWA epistasis analysis is useful despite relatively low power in small isolated populations.
Introduction
Serum uric acid is the final oxidation product of purine metabolism in humans. High serum uric acid (SUA) levels can lead to gout and is associated with cardiovascular diseases and diabetes [1], whereas low SUA levels may be associated with multiple sclerosis [2], [3]. High SUA levels are increasingly prevalent (reaching 15–20%) in many human populations and caused mainly by impaired renal excretion of urate [4]. Elevated urate is associated with insulin resistance [5] and neurological disorders such as Parkinson's disease [6], [7]. About 70% of SUA is excreted via the kidneys and the remainder is eliminated into the biliary tract and intestine, part of which is subsequently converted by colonic bacterial uricase to allantoin [4].
SUA level is a complex trait that is affected by environmental (e.g. diet and excessive body weight) and genetic factors with heritability estimates of 60–87% [8], [9]. Genome-wide association (GWA) studies so far have identified nine loci underlying SUA levels. Seven of these loci are membrane transporters suggesting that the genetic variation in urate transport proteins plays an important role [10], [11], [12]. The variants identified within the SLC2A9 gene are the most significant genetic risk factors associating with low fractional excretion of SUA and explain 5.3% of the SUA level variance in women and 1.7% of the SUA level variance in men in the VIS population [12]. However, because each of the eight remaining loci carries moderate marginal effects, the nine identified loci together only explain 5.22% of the SUA level variation [11], suggesting there may be more genetic loci to be detected.
One possible source of the unexplained variation in SUA levels is gene-gene interaction (epistasis) [13], [14]. Epistasis remains largely unexplored in previous GWA studies of SUA levels due to computational and statistical challenges, e.g. the lack of widely accepted algorithms that are fast enough to effectively handle high density SNPs and map different forms of epistasis while keeping false positive rates under control [15]. With the advances in computing technologies (e.g. GRID computing), full pair-wise genome scans are beginning to be applied in GWA data analyses [16], [17]. Nonetheless, fundamental questions about the potential values of GWA epistasis studies of normal GWA populations remain to be answered. Therefore, we performed a full pair-wise genome association scan in the Italian MICROS study cohort [18] to generate a profile of epistasis signals in SUA levels. The pair-wise genome scan used a regression-based comprehensive search algorithm that can detect epistasis signals with and without main effects [15], [19]. Epistasis signals detected in MICROS were tested for replication in a CROATIAN population combining the VIS [20] and KORCULA [21] study cohorts as well as the SOCCS (Phase 1) cohort [22]. Furthermore, we examined the gene ontology (GO) terms [23] enriched by the epistasis signals in both discovery and replication populations for any new insights into the genetic regulation of SUA levels.
Materials and Methods
Study cohorts and Ethics statement
The Italian MICROS cohort was recruited from the villages in South Tyrol [18]. This study was approved by the ethical committee of the Autonomous Province of Bolzano. The VIS [20] and KORCULA [21] cohorts were recruited from the islands of Vis and Korcula in Croatia respectively. This study was approved by the Ethical Committee of the Medical School, University of Zagreb and the Multi-Centre Research Ethics Committee for Scotland. The SOCCS (Phase 1) cohort was recruited in Scotland to study colorectal cancer [22]. This study was approved by the MultiCentre Research Ethics committee for Scotland. All participants gave written informed consent and were measured for a number of traits including SUA level, weight and height from which body mass index (BMI) values were calculated.
DNA samples were genotyped with Illumina Infinium HumanHap300v1/v2 or HumanCNV370v1 SNP bead microarrays and analyzed using the BeadStudio software. Quality control of the genotype data was performed for each cohort using the R/GenABEL package (Version 1.4.3) [24] based on a common set of criteria: individual call rate at 95%, SNP call rate at 98%, P value for deviation from Hardy-Weinberg equilibrium at 1.0e-10, minor allele frequency at 2%. The sample size and number of SNPs after the quality control were listed in Table 1 for each cohort.
Table 1. Summary information of each study cohort.* .
| MICROS | VIS | KORCULA | CROATIAN | SOCCS | |
| N | 1201 | 895 | 877 | 1772 | 1097 |
| #SNP | 293913 | 300265 | 307712 | 283971 | 305449 |
| SUA_median | 5.17 | 5.09 | 4.84 | NA | 4.41 |
| SUA_mean ± SD | 5.32±1.42 | 5.24±1.59 | 4.91±1.29 | NA | 4.60±1.25 |
| SUA and BMI correlation | 0.38 | 0.35 | 0.38 | NA | 0.24 |
| SUA polygenic heritability | 0.325 | 0.288 | 0.228 | 0.287 | NA |
*: SUA level in mg/dL; CROATIAN combined VIS and KORCULA; NA: not available; the phenotypic correlation between SUA level and BMI was significant (P<2.2E-16) across MICROS, VIS and KORCULA.
Statistical analysis
In each individual cohort the raw SUA levels were corrected for age, sex and BMI and normalised using the rntransform function that is implemented in the GenABEL package performing quantile normalisation of residuals from a generalized linear model analysis. The normalised SUA levels were then analysed using a linear mixed model to correct for polygenic effects and relatedness using the polygenic function in the GenABEL package and the resultant environmental residuals (i.e. pgresidualY) were used as the trait to test for association [25]. Polygenic heritability was estimated at the mixed model step. The CROATIAN combined population was created by merging the VIS and KORCULA study cohorts and the normalised SUA levels were corrected for study cohort, polygenic effects and relatedness as above and the resultant residuals were used as the trait for association tests.
A single SNP based GWA scan was performed in each population using a score test method (based on the additive model) implemented in the mmscore function in the GenABEL package. The consensus GWA threshold of 7.3 (−log10(5.0E-08)) was applied to identify GWA significant SNPs [26]. A full pair-wise genome scan was followed using regression models. Considering a pair of SNPs denoted as SNP1 and SNP2, the following genetic models are used to detect epistasis where genotypes of each SNP (i.e. homozygote of the minor allele, homozygote of the major allele and heterozygote) were fitted as fixed factors [19], [27]:
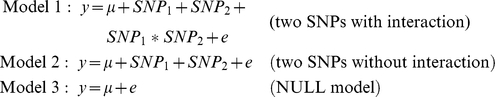 |
where y is the trait of interest, μ is the model constant, SNP1 (or SNP2) is a fixed factor with three levels, SNP1*SNP2 is the interaction term, e is the random error term. The F ratio test of Model 1 against Model 3 is for the whole pair effect including interaction (i.e. Fpair, 8 degrees of freedom). The F ratio test of Model 1 against Model 2 is for the interaction between the two SNPs (i.e. Fint, 4 degrees of freedom). SNP pairs with missing joint genotype classes (i.e. considering three genotypes per SNP, at least one of the nine joint genotype classes of a SNP pair had no individuals) were not evaluated to reduced the risk of inflation of the type I error rate. P values were calculated based on the F distribution with relevant degrees of freedom and transformed in the −log10 scale (i.e. −log10Ppair for the Fpair test, −log10Pint for the Fint test). We applied the same −log10 scaled thresholds for both the Fpair and Fint tests to control the type I error rate [15].
Genome-wide significance thresholds (all in the −log10 scale) were derived based on Bonferroni correction for multiple tests, i.e. the 5% nominal P value corrected by the number of pair-wise tests. Considering 300,000 SNPs, a SNP-genome scan and a full pair-wise genome scan perform 3.0E+05 and 4.5E+10 association tests respectively, thus the genome-wide threshold is 11.95 (−log10(0.05/4.5E+10)) for the pair-wise genome scan. SNP-genome scans have been used to test epistasis for genome-wide significant signals specifically to increase the power of detection [15], [19], [28], [29] and thus are applied here to examine the interactions between each GWA significant SNP and all other SNPs genotyped. The actual GWA threshold for SNP-genome scans is calculated as −log10(0.05/3.0E+05/N) if there are N GWA significant SNPs.
A full pair-wise genome scan was performed in the MICROS population and SNP pairs with a certain interaction signal (i.e. −log10Ppair>4.7 and −log10Pint>3.2) were retained. The retained results were evaluated using the predefined thresholds to identify genome-wide significant epistatic signals. Each SNP in the retained results was annotated to the nearest gene within a window of 20 kilobases flanking the SNP based on the physical distances to either the start or end of transcription of genes (the distance is set to zero if the SNP is within a gene) without considering linkage disequilibrium (LD). A full pair-wise genome scan was also performed in the CROATIAN population as above to prepare input for GO enrichment analyses (see below).
Replication, variance explained, and GO enrichment analysis
Epistatic pairs detected in the MICROS population were tested for replication in the CROATIAN population following the same procedures as above. The nominal threshold of 0.05 (or 0.05/K, if K epistatic pairs were tested) was used to claim significant replication for an epistatic pair (i.e. both replicated SNPs were exactly the same as the epistatic SNPs) because only one test was performed. When an epistatic pair was not replicated (e.g. due to missing genotype classes), we also tested the interactions between each of the adjacent SNPs of the first epistatic SNP and that of the second. In that case, the nominal threshold corrected by the actual number of tests was used to claim significant replication.
The polygenic function was also used to calculate the proportion of the phenotypic variance in SUA levels explained by epistatic pairs in the MICROS population. Because the quantile normalised SUA levels were based on the quantiles of the raw SUA levels, we used standardized SUA levels for variance calculation (i.e. the raw SUA levels were corrected for age, sex and BMI and then standardized (mean of 0 and variance of 1)). The standardized SUA levels were fitted into the full mixed model including polygenic effects and the identified SNP pairs. The difference of residual variance from a value of 1 is the proportion of phenotypic variance explained by the SNP pairs included.
A GO enrichment analysis was conducted for each of the MICROS and CROATIAN populations using the running mode of “Two unranked lists of genes” in GOrilla [23] where the full list of human genes was used as the background. We chose SNP pairs with a moderately high interaction signal (i.e. −log10Ppair>6.5 and −log10Pint>6.5) in each population and used the epistatic genes annotated from them as the target for the GO enrichment analysis to mine biological meanings from epistatic signals that were less significant. The GO terms enriched (P<1.0E-03) by the epistatic genes in each population were compared to identify replicated GO terms and then epistatic genes shared by each pair of replicated GO terms. The shared epistatic genes in the replicated GO terms were investigated further for a) associated biological functions or diseases and b) epistatic SNP pairs involved and their replication.
Results
The mean SUA level and phenotypic correlation between SUA level and BMI were similar across the MICROS, VIS and KORCULA cohorts (Table 1). The polygenic heritability estimates varied from 0.228 (KORCULA) to 0.325 (MICROS), suggesting a different genetic background in each individual cohort. Using the normalised SUA residuals as the trait, a conventional GWA scan identified seven genome-wide significant SNPs in MICROS: rs737267, rs13129697, rs13131257, rs6449213, rs1014290, rs10805346 and rs733175 that were all annotated to the SLC2A9 gene. The GWA scan for the CROATIAN population also identified seven genome-wide significant SNPs which were exactly the same as those in MICROS. Thus the genome-wide threshold of 7.62 (−log10(0.05/3.0E+05/7) was used for SNP-genome scans. The inflation factor λ (computed by regression in a quantile-quantile (QQ) plot, Figure S1) was 1.007 in both GWA scans, suggesting the family relatedness was well accounted for. In the SOCCS cohort 1097 unrelated individuals were measured for SUA and BMI where the mean SUA level (4.60±1.25) was lower than those in the three cohorts above (Table 1).
The full pair-wise genome scan in the MICROS population tested 43 billion pair-wise SNP combinations where 11 billion pairs (26.3%) had missing joint genotype classes and hence were ignored in this study. We plotted the P values of all the Fpair and Fint tests performed on chromosomes 3 and 4 (the total number of pair-wise combinations is too large to plot at the genome level) to illustrate why the two tests are needed (Figure 1). Using only the Fpair test (e.g. −log10Ppair>11.95 may pick up SNP pairs with very weak interactions (e.g. −log10Pint<3), whereas using only the Fint test (e.g. −log10Pint>7.62) could pick up those with weak whole pair effects when both SNPs had small marginal effects (e.g. −log10Ppair<6) (Figure 1a). The QQ plot for the Fpair tests (Figure 1b) showed an earlier departure (near the value of 3) from the expected line and that for the Fint tests (Figure 1c) showed a late departure from the expected line. The QQ plots suggested that many pairs of SNPs on chromosomes 3 and 4 had strong whole pair effects attributing to the marginal effects in the SLC2A9 region but only a few of them with interactions greater than expected under the null hypothesis. To further check the distributions of the test statistics, we randomly sampled 5000 SNPs from the MICROS genome and tested and stored all their pair-wise interactions with and without permutation. The two- sample Kolmogorov-Smirnov tests [30] found no significant difference (D = 0.0044, P>0.05) between the real (without permutation) −log10Pint distribution and the −log10Pint distribution under the null hypothesis.
Figure 1. Plots of the P values of the pair-wise tests performed on chromosomes 3 and 4.

SNP combinations with missing joint genotype classes were excluded. Left: scatter plot of the −log10Ppair against −log10Pint values of each SNP pair. Upper-right: QQ plot of the whole pair tests. Lower-right: QQ plot of the interaction tests.
We plotted the pair-wise tests for all the SNP pairs (−log10Ppair>4.7 and −log10Pint>3.2, 212933 in total) retained from the full pair-wise genome scan (Figure 2). No SNP pairs reached the pair-wise genome scan threshold (i.e. −log10Pint>11.95 along the x axis). However, we found a big cluster of SNP pairs with strong whole pair effects (i.e. −log10Ppair>11.95 along the y axis) but weak to high interactions (i.e. 3.2<−log10Pint<8.5 along the x axis). This cluster of SNP pairs all involved the SLC2A9 gene as one would expect. Two pairs: rs12130085 (NFIA) – rs737267 (SLC2A9) and rs737267 (SLC2A9) – rs9316212 (ESRRAP2) were significant based on the threshold of 7.62 for SNP-genome scans (Table 2). These two pairs showed different interaction patterns (Figure S2) and jointly explained 8.0% (4.4% by rs727367 alone and 3.4% by interactions) of the variance of the standardised SUA levels in MICROS.
Figure 2. Profile of all retained epistatic pairs in the MICROS population.

Each epistatic pair in the figure with −log10Ppair>4.7 and −log10Pint>3.2.
Table 2. SLC2A9 involved epistatic pairs and replication.1 .
| SNP1 | chr1 | gene1 | SNP2 | chr2 | gene2 | Ppair | Pint | MGC | CROATIAN replication3 | SOCCS replication3 |
| rs12130085 | 1 | NFIA | rs737267 | 4 | SLC2A9 | 4.1e-17 (16.38) | 6.6e-09 (8.20)2 | 2 | 0.045 | 0.178 |
| rs737267 | 4 | SLC2A9 | rs9316212 | 13 | ESRRAP2 | 6.2e-17 (16.21) | 1.1e-08 (7.97)2 | 2 | 0.048 | 0.238 |
| rs733175 | 4 | SLC2A9 | rs1818116 | 5 | (-) | 3.8e-12 (11.43) | 5.5e-08 (7.27) | 7 | 0.130 | 0.227 |
| rs733175 | 4 | SLC2A9 | rs137180 | 22 | SEZ6L | 8.0e-11 (10.09) | 2.6e-07 (6.58) | 4 | 0.035 | 0.155 |
: SNP1 (SNP2) – the first (second) SNP name; chr1 (chr2) – the chromosome where SNP1 (SNP2) locates; gene1 (gene2) – symbol of the gene annotated by SNP1 (SNP2); Ppair – P value of the whole pair test (−log10 P value in brackets); Pint – P value of the interaction test (−log10 P value in brackets); MGC – count of number of individuals in the minor joint genotype class; (-): no gene annotation.
: genome-wide significant.
: Best replication including adjacent SNPs: the SLC2A9 SNP is fixed and the other SNP is to replicate as itself or the first or second neighbour.
Considering only those with a moderately high interaction signal (−log10Ppair>6.5 and −log10Pint>6.5), in total 1326 SNP pairs involved 2063 unique SNPs of which 1148 (55.6%) were annotated to 910 unique genes, i.e. 1.5 pairs per gene. In contrast, 17 out of the 1326 SNP pairs involved the SLC2A9 gene including 4 GWA significant SNPs rs733175, rs737267, rs13131257 and rs13129697 (Tables 2 and S1). Epistatic pairs listed in Table 2 were tested for statistical replication in the CROATIAN population. The two genome-wide significant epistatic pairs missed the SNP level replication: the −log10Pint values of the best replicated pairs of rs737267 – rs1779851 (adjacent to rs12130085) and rs737267 – rs17064136 (adjacent to rs9316212) were 1.35 and 1.32 respectively. Combining the MICROS and CROATIAN data did not make the two significant pairs stronger (−log10Pint was 2.32 and 2.19 respectively) which is in line with the replication results. The two less significant SLC2A9 epistatic pairs also missed the SNP level replication in the CROATIAN population (Table 2). The four epistatic pairs in Table 2 also failed to achieve exact replication in the SOCCS cohort.
Epistatic genes annotated from the less significant SNP pairs (i.e. −log10Ppair>6.5 and −log10Pint>6.5) in MICROS (910 genes from 1326 pairs) and CROATIAN (984 genes from 1260 pairs) were tested for GO enrichment. The GO terms enriched by epistatic genes in MICROS showed a complicated relationship among biological functions (e.g. calmodulin binding, transporter activity) and highlighted the importance of glutamate receptor activity (Figure 3). Clearly, more than 50% of the GO terms enriched (P<1.0E-05) in MICROS were also enriched in CROATIAN (Table 3, Figures S3 and S4) which suggested GO terms regarding nervous system, synapse, glutamate receptors and plasma membrane were important in both populations. Comparing the epistatic genes enriched the 13 replicated GO terms (Table 3), we found 82 genes shared by both populations including SLC2A9 and a number of glutamate receptor genes (GRID1, GRIK1, GRIK2, GRM7 and GRIN2A) (Table S2). Surprisingly, 53 out of the 82 shared epistatic genes are previously published GWA loci [31] associated with phenotypes such as multiple sclerosis, Alzheimer's disease, bipolar disorder and schizophrenia, cognition and diabetes. Among SNP pairs where both SNPs were gene annotated and with at least one of the 82 shared epistatic genes from the retained results, we found 49 epistatic gene pairs (correspondingly to 120 SNP pairs) in MICROS were replicated in CROATIAN including SLC2A9 – LRRC16A (interaction between two SUA candidate genes) (Table S3). Two gene pairs were replicated exactly at the SNP level: rs737267 (SLC2A9) – rs4085921 (GPC6) and rs737267 (SLC2A9) – rs2302558 (CLEC16A). All the 49 epistatic gene pairs involved SLC2A9 and of these 5 involved interactions with other shared epistatic genes: ERC2, GPC6, CTNND2, CNTNAP2 and PTPRD. However, most of the 49 replicated gene pairs had a relatively weak interaction signal (−log10Pint<5).
Figure 3. Gene function ontology enrichment by epistatic signals in the MICROS population.

Table 3. Gene ontology terms enriched by epistatic genes in the MICROS population and their replication in the CROATIAN population.* .
| GO Term | Description | MICROS | CROATIAN |
| GO:0048731a | system development | 2.88E-10 | 1.50E-08 |
| GO:0007166a | cell surface receptor linked signaling pathway | 6.26E-09 | (-) |
| GO:0007268a | synaptic transmission | 1.85E-08 | 1.68E-05 |
| GO:0007169a | transmembrane receptor protein tyrosine kinase signaling pathway | 3.27E-07 | (-) |
| GO:0030224a | monocyte differentiation | 1.09E-06 | (-) |
| GO:0007611a | learning or memory | 2.40E-06 | (-) |
| GO:0007399a | nervous system development | 4.53E-06 | 4.39E-06 |
| GO:0023052a | signalling | 6.22E-06 | 5.78E-04 |
| GO:0031644a | regulation of neurological system process | 6.32E-06 | 5.63E-04 |
| GO:0050890a | cognition | 8.41E-06 | (-) |
| GO:0007267a | cell-cell signalling | 9.46E-06 | (-) |
| GO:0007165a | signal transduction | 9.53E-06 | 6.35E-07 |
| GO:0008066b | glutamate receptor activity | 3.39E-06 | 5.35E-05 |
| GO:0004970b | ionotropic glutamate receptor activity | 7.82E-06 | (-) |
| GO:0005516b | calmodulin binding | 9.68E-06 | 9.05E-05 |
| GO:0044459c | plasma membrane part | 1.49E-08 | 1.38E-07 |
| GO:0030054c | cell junction | 1.72E-08 | 2.25E-07 |
| GO:0044456c | synapse part | 2.11E-07 | 4.95E-08 |
| GO:0045202c | synapse | 2.96E-06 | 3.07E-08 |
| GO:0005886c | plasma membrane | 7.09E-06 | 5.28E-07 |
*: displayed only GO terms with P<1.0E-05; 910 and 984 epistatic genes used in MICROS and CROATIAN respectively; (-): no enrichment at the P<1.0E-03 level.
: gene process ontology;
: gene function ontology;
: gene component ontology.
Discussion
Using a full pair-wise genome-wide association scan we generated a profile of epistasis in SUA levels in the Italian MICROS population. No epistatic SNP pairs reached the Bonferroni adjusted threshold for the pair-wise genome scan, which was not a surprise because of the relatively small population size. Nonetheless, we found that the SLC2A9 gene may be an important epistatic locus interacting with multiple loci across the genome more frequently than expected by chance (Tables 2 and S1). Two SLC2A9 epistatic pairs (NFIA – SLC2A9 and SLC2A9 – ESRRAP2) were significant in SNP-genome scans and jointly explained 8.0% of the phenotypic variance in SUA levels where 3.4% was explained by their interaction components. However, caution should be taken in light of the billions of tests performed, the potential overestimation of the variance explained and the limited replication of the two pairs. The NFIA (nuclear factor I/A) gene is known as a cellular transcription DNA replication factor and its haploinsufficiency is associated with a central nervous system malformation syndrome and ureteral and renal defects [32]. Recent GWA studies showed NFIA was responsible for celiac disease [33] and ventricular depolarization and conduction [34]. The ESRRAP2 (estrogen-related receptor alpha pseudogene 2) is not yet known to have any related functions.
The SLC2A9 gene is known to interact with sex, age and dietary patterns [35], [36], [37] and GWA studies have identified several common variants mapping to various introns and exons of the locus [11], [38], [39], [40]. The protein GLUT9 encoded by SLC2A9 is a class II glucose/fructose transporter as well as high-capacity/low-affinity urate transporter with two isoforms expressed in basolateral and apical membranes of proximal renal tubular cells respectively [41]. The complexity in the SLC2A9 polymorphism was demonstrated further in two recent studies linking SUA levels with human cognitive aging [42] and Parkinson's disease [7]. Here we showed that several GWA significant SNPs of SLC2A9 were involved in epistatic interactions, which adds another dimension of studying the SLC2A9 polymorphism and provides new clues of the genetic mechanism underlying SUA levels.
The impact of SUA levels on human cognitive dysfunction has been investigated in recent years [7], [42], [43], [44]. It is still unclear though whether high SUA levels causes disease associated aging or vice versa. Considering most SUA associated genes identified from GWA studies encode transporters, it is natural to speculate these transporter genes interact with others that have moderate or low marginal effects (thus not yet discovered in GWA studies) but with certain regulatory roles. Our epistasis results and GO enrichment analyses supported the speculation and showed that numerous SLC2A9 interacting genes had clear neuronal influence (e.g. synapse, glutamate receptors) and/or membrane functions (Tables 2, S2, S3). It is noteworthy that the GO enrichment results were almost identical after removing SLC2A9 and genes that interacted with it (8 out of 910 unique genes in MICROS). The epistatic genes encoding glutamate receptors (common in both populations, Table S2) are particularly interesting because glutamate receptors are implicated in the pathologies of a number of neurodegenerative diseases due to their central role in excitotoxicity and their prevalence throughout the central nervous system [45], [46]. A rat study showed that uric acid could protect the neurons from glutamate-induced toxicity [47]. Furthermore, a number of shared epistatic genes in the two populations (Table S2) suggested links between SUA levels and various diseases such as autism (CNTNAP2, POU6F2, MYO1D), schizophrenia and bipolar disorder (ERC2, ROBO1, ROBO2, CNTNAP2, GRM7, SYNE1, CNTN5, ANKS1B), Alzheimer's disease (EPHA4, CNTN4, ADCY8, PCSK5, CUBN, SORCS1, RORA, GRIN2A), Parkinson's disease (DLG2), sclerosis (RGS7, CNTN4, IGF2R, SH3GL2, GPC5, GPC6, GRIN2A, MYH9), and diabetes (CD69, PTPRD, SORCS1, PREX1).
Statistically detecting and replicating an epistatic pair of SNPs is far more challenging than for a single SNP signal for a number of reasons. Whereas a single SNP association is dependent on strong LD between the marker SNP and causative variant, detection of epistasis requires strong LD for both loci. Small allele frequency changes (e.g. 10%) between detection and replication populations can lead to a dramatic loss of power in replication. Such small allele frequency changes are not uncommon across the small and/or isolated populations. The issue of missing joint genotype class in replication populations (an extreme case of allele frequency change) increases the difficulty of replication, especially when the epistatic pair for test has a rare genotype class (e.g. less than 4 individuals in Table 2). These challenges support the case for exploring epistatic effects and evidence for their replication at the gene and/or pathway levels [48].
The lack of statistical replication at the SNP level may be not too surprising considering that the epistatic signals detected in one isolated sample were to replicate in another isolated sample (CROATIAN) or a less isolated but smaller sample (i.e. SOCCS). Even when all the retained SNP pairs were considered regardless, we just found two SNP pairs (out of 212933) were replicated at the SNP level. When testing for replication at the GO level, we found 13 GO enriched terms (>50%) were replicated. Clearly replication improved dramatically as the level changes from SNP to GO annotation. A high level replication (i.e. gene or GO/pathway) does not guarantee a SNP level replication but is valuable to understand the biology of interest. Therefore, we recommend testing for replication of epistasis signals at all three levels. For example, rs737267 (SLC2A9) – rs4085921 (GPC6) was replicated at all three levels, rs737267 (SLC2A9) – rs2302558 (CLEC16A) at the SNP and gene levels but not the GO level as CLEC16A is not associated with an enriched GO term. Both GPC6 (glypican 6) and CLEC16A (C-type lectin domain family 16, member A) are associated with multiple sclerosis [49], [50], [51], [52].
In addition to epistasis signal replication, this study also raises a number of issues in GWA epistasis studies. First, it is difficult to detect and replicate genome-wide epistasis signals in small samples because of low power [53] and the problem of missing joint genotype classes (i.e. 26.3% of the total SNP pairs skipped in MICROS). It is suboptimal to ignore SNP pairs with missing genotype classes as some might be true epistatic signals. Methods such as the allelic model used in PLINK [17] and Pseudohaplotype [54] may be helpful to this situation but require further investigation. A large sample size and use of relatively common SNPs (e.g. minor allele frequency >5%) are a partial solution. Second, proper genome-wide thresholds remain to be defined. Bonferroni adjusted thresholds may be appropriate when the total number of tests is close to the effective number of independent tests [55] but may become over stringent when more SNPs are genotyped in GWA studies. Permutation can be a good option but is not yet feasible for GWA epistasis studies due to the excessive computing demand. A set of consensus thresholds [26] will be very useful to guide future GWA epistasis studies. Third, trait normality is critical to the detection of epistasis so as to avoid inflated test statistics simply because more parameters are fitted in an epistatic model than an additive model. Increasing the sample size is a good way to recover the power reduced by the use of quantile normalisation. When it is not possible to do so, a good alternative is to use GO enrichment and pathway analyses [56] to rescue some true signals that are not genome-wide significant but jointly important biologically. It is arguable what threshold should be used to select a proper set of less significant epistatic pairs that includes most true signals and excludes noise as much as possible. A different threshold (other than −log10Pint of 6.5 used here) could be chosen as long as the big cluster of SLC2A9 pairs was mostly excluded (Figure 2) and sufficient epistatic genes were available for the GO enrichment analysis. If we considered a threshold of 7.62, there would be only 143 epistatic genes (from 111 SNP pairs) in MICROS and 97 genes (from 96 SNP pairs) in CROATIAN available for the enrichment analyses. The SNP-gene annotation method could affect the GO analysis as well. Here we took a commonly used method based on map distances and allowing only one gene per SNP. This method is generally effective but has a problem when genes overlap or the distances to two genes are equal. In that case, either gene could be quoted depending on which appears first in the annotation results. Such cases were rare so our GO enrichment results were little affected. Nonetheless, new methods are needed to make even more effective use of epistasis signals than our demonstration where epistatic genes were treated independently and more than 40% epistatic SNPs were not gene annotated and hence not considered.
In summary, this study characterized epistasis signals in SUA levels in the MICROS population and shows that SLC2A9 may be an important epistatic locus interacting with multiple loci across the genome, including those with neuronal influence and/or associating with neurological disorders. Two SLC2A9 epistatic pairs were genome-wide significant and explained additional phenotypic variance in SUA levels via interactions. We conclude that GWA epistasis study is useful and can provide new insights into the trait of interest in conjunction with GO/pathway enrichment analysis utilising less significant epistatic signals.
Supporting Information
The QQ plots for single-SNP based genome-wide association scans in the MICROS and CROATIAN populations.
(TIF)
The joint genotype – phenotype maps for two genome-wide significant epistatic pairs.
(TIF)
Gene component ontology terms enriched by epistatic genes in the MICROS population.
(TIF)
Gene component ontology terms enriched by epistatic genes in the CROATIAN populations.
(TIF)
SLC2A9 involved epistatic pairs (−log10Ppair>6.5 and −log10Pint>6.5) in MICROS.
(PDF)
Epistatic genes in the replicated GO terms shared by the MICROS and CROATIAN populations.
(PDF)
Epistatic pairs with at least one shared GO gene and replicated in MICROS and CROATIAN.
(PDF)
Acknowledgments
We are grateful to Dr. Bill Hill (MRC Human Genetics Unit) for the excellent computing support and Dr. Attila Gyenesei and Asta Laiho for suggestions on GO enrichment analysis. We like to thank all of the staff and participants in the MICROS, VIS, KORCULA and SOCCS studies, in particular those who contributed to data and sample collection, genotyping, trait measurement and data analysis. We acknowledge the Wellcome Trust Clinical facility (Edinburgh) for genotyping the VIS study and Peter Lichner and the Helmholtz Zentrum Munchen genotyping staff (Munich, Germany) for genotyping the KORCULA and MICROS cohorts.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: The authors acknowledge the financial support provided by the MRC Core Fund. BBSRC funded WHW and CSH's time in part (grant reference: BB/H024484/1). The MICROS study was supported by the Ministry of Health and the Department of Educational Assistance, University and Research of the Autonomous Province of Bolzano, and the South Tyrolean Sparkasse Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Heinig M, Johnson RJ. Role of uric acid in hypertension, renal disease, and metabolic syndrome. Cleve Clin J Med. 2006;73:1059–1064. doi: 10.3949/ccjm.73.12.1059. [DOI] [PubMed] [Google Scholar]
- 2.Hooper DC, Spitsin S, Kean RB, Champion JM, Dickson GM, et al. Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A. 1998;95:675–680. doi: 10.1073/pnas.95.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spitsin S, Koprowski H. Role of uric acid in multiple sclerosis. Curr Top Microbiol Immunol. 2008;318:325–342. doi: 10.1007/978-3-540-73677-6_13. [DOI] [PubMed] [Google Scholar]
- 4.Doherty M. New insights into the epidemiology of gout. Rheumatology. 2009;48:ii2–ii8. doi: 10.1093/rheumatology/kep086. [DOI] [PubMed] [Google Scholar]
- 5.Dehghan A, van Hoek M, Sijbrands EJ, Hofman A, Witteman JC. High serum uric acid as a novel risk factor for type 2 diabetes. Diabetes Care. 2008;31:361–362. doi: 10.2337/dc07-1276. [DOI] [PubMed] [Google Scholar]
- 6.Annanmaki T, Pessala-Driver A, Hokkanen L, Murros K. Uric acid associates with cognition in Parkinson's disease. Parkinsonism & related disorders. 2008;14:576–578. doi: 10.1016/j.parkreldis.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Facheris MF, Hicks AA, Minelli C, Hagenah JM, Kostic V, et al. Variation in the Uric Acid Transporter Gene SLC2A9 and Its Association with AAO of Parkinson's Disease. J Mol Neurosci. 2011;43:246–250. doi: 10.1007/s12031-010-9409-y. [DOI] [PubMed] [Google Scholar]
- 8.Roddy E, Zhang W, Doherty M. The changing epidemiology of gout. Nat Clin Pract Rheumatol. 2007;3:443–449. doi: 10.1038/ncprheum0556. [DOI] [PubMed] [Google Scholar]
- 9.Whitfield JB, Martin NG. Inheritance and alcohol as factors influencing plasma uric acid levels. Acta Genet Med Gemellol (Roma) 1983;32:117–126. doi: 10.1017/s0001566000006401. [DOI] [PubMed] [Google Scholar]
- 10.Doring A, Gieger C, Mehta D, Gohlke H, Prokisch H, et al. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat Genet. 2008;40:430–436. doi: 10.1038/ng.107. [DOI] [PubMed] [Google Scholar]
- 11.Kolz M, Johnson T, Sanna S, Teumer A, Vitart V, et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genetics. 2009;5:e1000504. doi: 10.1371/journal.pgen.1000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nature Genetics. 2008;40:437–442. doi: 10.1038/ng.106. [DOI] [PubMed] [Google Scholar]
- 13.Eichler EE, Flint J, Gibson G, Kong A, Leal SM, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibson G. Hints of hidden heritability in GWAS. Nat Genet. 2010;42:558–560. doi: 10.1038/ng0710-558. [DOI] [PubMed] [Google Scholar]
- 15.Wei WH, Knott S, Haley CS, de Koning DJ. Controlling false positives in the mapping of epistatic QTL. Heredity. 2010;104:401–409. doi: 10.1038/hdy.2009.129. [DOI] [PubMed] [Google Scholar]
- 16.Ma L, Yang J, Runesha HB, Tanaka T, Ferrucci L, et al. Genome-wide association analysis of total cholesterol and high-density lipoprotein cholesterol levels using the Framingham heart study data. BMC Med Genet. 2010;11:55. doi: 10.1186/1471-2350-11-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schupbach T, Xenarios I, Bergmann S, Kapur K. FastEpistasis: a high performance computing solution for quantitative trait epistasis. Bioinformatics. 2010;26:1468–1469. doi: 10.1093/bioinformatics/btq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pattaro C, Marroni F, Riegler A, Mascalzoni D, Pichler I, et al. The genetic study of three population microisolates in South Tyrol (MICROS): study design and epidemiological perspectives. BMC Med Genet. 2007;8:29. doi: 10.1186/1471-2350-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam AC, Powell J, Wei WH, de Koning DJ, Haley CS. A combined strategy for quantitative trait loci detection by genome-wide association. BMC Proc. 2009;3(Suppl 1):S6. doi: 10.1186/1753-6561-3-s1-s6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vitart V, Biloglav Z, Hayward C, Janicijevic B, Smolej-Narancic N, et al. 3000 years of solitude: Extreme differentiation in the island isolates of Dalmatia, Croatia. European Journal of Human Genetics. 2006;14:478–487. doi: 10.1038/sj.ejhg.5201589. [DOI] [PubMed] [Google Scholar]
- 21.Polasek O, Marusic A, Rotim K, Hayward C, Vitart V, et al. Genome-wide association study of anthropometric traits in Korcula Island, Croatia. Croat Med J. 2009;50:7–16. doi: 10.3325/cmj.2009.50.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tenesa A, Farrington SM, Prendergast JGD, Porteous ME, Walker M, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nature Genetics. 2008;40:631–637. doi: 10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 25.Aulchenko YS, de Koning DJ, Haley C. Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics. 2007;177:577–585. doi: 10.1534/genetics.107.075614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 27.VanderWeele TJ. Epistatic interactions. Stat Appl Genet Mol Biol. 2010;9:Article 1. doi: 10.2202/1544-6115.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jannink JL, Jansen R. Mapping epistatic quantitative trait loci with one-dimensional genome searches. Genetics. 2001;157:445–454. doi: 10.1093/genetics/157.1.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kooperberg C, Leblanc M. Increasing the power of identifying gene x gene interactions in genome-wide association studies. Genet Epidemiol. 2008;32:255–263. doi: 10.1002/gepi.20300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marsglia G, Tsang WW, Wang J. Evaluating Kolmogorov's distribution. Journal of Statistical Software. 2003;8:1–4. [Google Scholar]
- 31.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu W, Quintero-Rivera F, Fan Y, Alkuraya FS, Donovan DJ, et al. NFIA haploinsufficiency is associated with a CNS malformation syndrome and urinary tract defects. PLoS Genet. 2007;3:e80. doi: 10.1371/journal.pgen.0030080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sotoodehnia N, Isaacs A, de Bakker PI, Dorr M, Newton-Cheh C, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–1076. doi: 10.1038/ng.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brandstatter A, Kiechl S, Kollerits B, Hunt SC, Heid IM, et al. Sex-specific association of the putative fructose transporter SLC2A9 variants with uric acid levels is modified by BMI. Diabetes Care. 2008;31:1662–1667. doi: 10.2337/dc08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brandstatter A, Lamina C, Kiechl S, Hunt SC, Coassin S, et al. Sex and age interaction with genetic association of atherogenic uric acid concentrations. Atherosclerosis. 2010;210:474–478. doi: 10.1016/j.atherosclerosis.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeroncic I, Mulic R, Klismanic Z, Rudan D, Boban M, et al. Interactions between genetic variants in glucose transporter type 9 (SLC2A9) and dietary habits in serum uric acid regulation. Croat Med J. 2010;51:40–47. doi: 10.3325/cmj.2010.51.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guan M, Zhou D, Ma W, Chen Y, Zhang J, et al. Association of an intronic SNP of SLC2A9 gene with serum uric acid levels in the Chinese male Han population by high-resolution melting method. Clin Rheumatol. 2011;30:29–35. doi: 10.1007/s10067-010-1597-x. [DOI] [PubMed] [Google Scholar]
- 39.Hollis-Moffatt JE, Xu X, Dalbeth N, Merriman ME, Topless R, et al. Role of the urate transporter SLC2A9 gene in susceptibility to gout in New Zealand Maori, Pacific Island, and Caucasian case-control sample sets. Arthritis Rheum. 2009;60:3485–3492. doi: 10.1002/art.24938. [DOI] [PubMed] [Google Scholar]
- 40.Rule AD, de Andrade M, Matsumoto M, Mosley TH, Kardia S, et al. Association between SLC2A9 transporter gene variants and uric acid phenotypes in African American and white families. Rheumatology (Oxford) 2011;50:871–878. doi: 10.1093/rheumatology/keq425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wright AF, Rudan I, Hastie ND, Campbell H. A ‘complexity’ of urate transporters. Kidney Int. 2010;78:446–452. doi: 10.1038/ki.2010.206. [DOI] [PubMed] [Google Scholar]
- 42.Houlihan LM, Wyatt ND, Harris SE, Hayward C, Gow AJ, et al. Variation in the uric acid transporter gene (SLC2A9) and memory performance. Human Molecular Genetics. 2010;19:2321–2330. doi: 10.1093/hmg/ddq097. [DOI] [PubMed] [Google Scholar]
- 43.Schretlen DJ, Inscore AB, Vannorsdall TD, Kraut M, Pearlson GD, et al. Serum uric acid and brain ischemia in normal elderly adults. Neurology. 2007;69:1418–1423. doi: 10.1212/01.wnl.0000277468.10236.f1. [DOI] [PubMed] [Google Scholar]
- 44.Vannorsdall TD, Jinnah HA, Gordon B, Kraut M, Schretlen DJ. Cerebral Ischemia Mediates the Effect of Serum Uric Acid on Cognitive Function. Stroke. 2008;39:3418–3420. doi: 10.1161/STROKEAHA.108.521591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madden DR. The structure and function of glutamate receptor ion channels. Nat Rev Neurosci. 2002;3:91–101. doi: 10.1038/nrn725. [DOI] [PubMed] [Google Scholar]
- 46.Markowitz AJ, White MG, Kolson DL, Jordan-Sciutto KL. Cellular interplay between neurons and glia: toward a comprehensive mechanism for excitotoxic neuronal loss in neurodegeneration. Cellscience. 2007;4:111–146. [PMC free article] [PubMed] [Google Scholar]
- 47.Du Y, Chen CP, Tseng C-Y, Eisenberg Y, Firestein BL. Astroglia-mediated effects of uric acid to protect spinal cord neurons from glutamate toxicity. Glia. 2007;55:463–472. doi: 10.1002/glia.20472. [DOI] [PubMed] [Google Scholar]
- 48.Moore JH, Williams SM. Epistasis and its implications for personal genetics. Am J Hum Genet. 2009;85:309–320. doi: 10.1016/j.ajhg.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoffjan S, Akkad DA. The genetics of multiple sclerosis: an update 2010. Mol Cell Probes. 2010;24:237–243. doi: 10.1016/j.mcp.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 50.Lorentzen AR, Melum E, Ellinghaus E, Smestad C, Mero IL, et al. Association to the Glypican-5 gene in multiple sclerosis. J Neuroimmunol. 2010;226:194–197. doi: 10.1016/j.jneuroim.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Nischwitz S, Cepok S, Kroner A, Wolf C, Knop M, et al. More CLEC16A gene variants associated with multiple sclerosis. Acta Neurol Scand. 2011;123:400–406. doi: 10.1111/j.1600-0404.2010.01421.x. [DOI] [PubMed] [Google Scholar]
- 52.Mero IL, Ban M, Lorentzen AR, Smestad C, Celius EG, et al. Exploring the CLEC16A gene reveals a MS-associated variant with correlation to the relative expression of CLEC16A isoforms in thymus. Genes Immun. 2011;12:191–198. doi: 10.1038/gene.2010.59. [DOI] [PubMed] [Google Scholar]
- 53.Gauderman WJ. Sample size requirements for association studies of gene-gene interaction. Am J Epidemiol. 2002;155:478–484. doi: 10.1093/aje/155.5.478. [DOI] [PubMed] [Google Scholar]
- 54.Wu X, Dong H, Luo L, Zhu Y, Peng G, et al. A Novel Statistic for Genome-Wide Interaction Analysis. PLoS Genet. 2010;6:e1001131. doi: 10.1371/journal.pgen.1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet Epidemiol. 2008;32:227–234. doi: 10.1002/gepi.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang K, Li M, Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat Rev Genet. 2010;11:843–854. doi: 10.1038/nrg2884. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The QQ plots for single-SNP based genome-wide association scans in the MICROS and CROATIAN populations.
(TIF)
The joint genotype – phenotype maps for two genome-wide significant epistatic pairs.
(TIF)
Gene component ontology terms enriched by epistatic genes in the MICROS population.
(TIF)
Gene component ontology terms enriched by epistatic genes in the CROATIAN populations.
(TIF)
SLC2A9 involved epistatic pairs (−log10Ppair>6.5 and −log10Pint>6.5) in MICROS.
(PDF)
Epistatic genes in the replicated GO terms shared by the MICROS and CROATIAN populations.
(PDF)
Epistatic pairs with at least one shared GO gene and replicated in MICROS and CROATIAN.
(PDF)