

Abstract
High throughput gene expression screens provide a quantitative picture of the average expression signature of biological samples. However, the analysis of spatial gene expression patterns with single cell resolution requires quantitative in-situ measurement techniques. Here we describe recent technological advances in RNA fluorescent in-situ hybridization (FISH) techniques that facilitate detection of individual fluorescently labeled mRNA molecules of practically any endogenous gene. These methods, which are based on advances in probe design, imaging technology, and image processing, enable the absolute measurement of transcript abundance in individual cells with single-molecule resolution.
High-throughput measurements of gene expression on a genomic scale using microarray technology or high throughput sequencing contributed tremendously to our understanding of how genetic networks coordinately function in normal cells and tissues and how they malfunction in disease. Such measurements allow one to infer the function of genes based on their co-expression patterns1, to detect which genes have altered expression in disease2, and to identify expression signatures that are predictive of cancer progression3,4. However, the variability in single cell gene expression in most biological systems and especially in tissues and tumors suggests that bulk transcriptome measurements should be complemented by techniques aimed at characterizing gene expression programs in individual cells5. This review will describe advances in single-molecule transcript imaging, which yield integer counts of transcripts in single cells in suspension and within intact tissues.
Biological samples are inherently heterogeneous
Bulk transcriptome measurements inform on the average gene expression in a sample. Thus in a heterogeneous sample, containing several cell types with different gene expression signatures, only the most abundant signature will be captured. Such heterogeneity is present in practically any biological sample. Even bacterial and yeast cells that are derived from isogenic monoclonal populations have been shown to exhibit pronounced cell to cell variation in the expression of many genes, stemming from stochastic events such as bursting transcriptional dynamics and cell cycle dependence6. Expression heterogeneity is even more pronounced in tissues, which are usually composed of several types of cells with profoundly different gene expression programs. In many epithelial tissues, such as the skin and the intestine, there is a clear hierarchical partition into stem cells and diverse differentiated epithelial progenies, each of which displays distinct phenotypic and morphological features. The precise location of cells within the tissue translates to constant changes in the levels of niche-secreted morphogens, which give rise to position-modulated gene expression programs. Thus two adjacent cells could harbor dramatically different expression programs.
Solid tumors represent a particular case of cell heterogeneity. Most solid tumors consist of a mixture of cancer and stromal cells. Additionally, cancer cells often show profound diversity not only in their transcript content but also in their genotype. This diversity stems from increased mutation rates, rapid cell proliferation and spatially varying selection forces. Single cells from a wide range of colorectal cancer cell lines change their chromosomal copy number on average once every five cell divisions in vitro7, and there is a dramatic heterogeneity in copy numbers observed in tissue cross-sections8,9. A bulk measurement of gene expression in tumors only captures the most dominant tumor clone and is masked by stromal signals, therefore providing only partial information on the different expression signatures co-existing in the tumor. Moreover, some tumors have been shown to contain a minority of cells with unlimited proliferative capacity and increased oncogenic ability termed cancer stem cells10. Bulk measurements of tumors cannot capture the expression signature of such minor populations.
Attempts to address this underlying heterogeneity are based on enriching for certain subpopulations from tissues or tumors. An example of such enrichment is the use of laser capture microdissection, enabling extracting regions within a tissue based on morphological features or gene expression markers11. This technique is frequently applied to selectively collect tumor cells rather than a mixture of tumor and stroma cells for down-stream expression analysis. Alternatively a fluorescence activated cell sorter can be used to specifically isolate cells expressing a small number of defined gene expression markers. These cells can then be used for either bulk measurements or for single cell measurements that require cell lysis, such as quantitative RT-PCR12 or digital RT-PCR13. Similarly, sorting GFP positive cells from transgenic mice expressing GFP under the control of a cell-type specific promoter of interest enables enriching for the cells expressing this gene and allows characterization of the expression program of these particular cells, as has been done with tissue stem cells14. The main drawback of these methods is the fact that they involve dissociating the tissue and thus result in the complete loss of spatial information. Moreover, specific expression of a single gene in only one cell type is uncommon, and thus the enriched cell population would generally still consist of a mixture of different cell populations. In addition, the variability resulting from the reverse transcription and the exponential PCR amplification steps often limits the sensitivity and reproducibility of these approaches12. Thus quantitatively unraveling the gene expression programs of single cells within heterogeneous samples while preserving spatial context requires transcript measurement methods in intact tissues.
Traditional approaches for measuring in-situ gene expression
The traditional method for measuring gene expression in individual cells in intact tissues is RNA in-situ hybridization (ISH) and its variant RNA fluorescent in-situ hybridization (FISH)15. FISH methods have originally been developed for DNA analysis16, and are based on the specific binding of long fluorescently labeled oligonucleotide probes to their complementary sequences in fixed and permeabilized samples. While there are many nuances to FISH techniques most entail the following steps (Fig. 1). Tissues or cells are fixed, permeabilized, and then hybridized with fluorescently labeled oligonucleotide probes. Probes are either directly coupled to fluorescent molecules, or alternatively coupled to haptens, typically biotin or digoxigenin (DIG), to which either avidin or anti-DIG antibody bind respectively. In turn these are either directly conjugated to fluorophores, or to enzymes such as alkaline phosphatase or horseradish peroxidase, which generate either fluorescent (in FISH) or chromogenic (in ISH) products from specific added substrates. Indirect labeling offers the advantage that signal amplification can be achieved via the use of primary and secondary antibodies, of which only the secondary antibody elicits a light emitting reaction17.
Figure 1.

Common steps in ISH. Dashed boxes are method-specific steps. Hapten coupling requires a secondary light-emitting reaction. Single-molecule FISH requires image enhancement in the form of deconvolutionor high-pass filtering of the image stacks, followed by automatic spot localization.
Coupling fluorophores to the oligonucleotide probes has traditionally been achieved by enzymatic means such as nick translation or in-vitro transcription, methods in which dye-modified nucleotides are stochastically incorporated along the probe (Fig. 2a). While detection of single molecules using traditional FISH has been reported for a specific probe in Drosophila18, in most cases traditional ISH and FISH techniques can only give qualitative information on gene expression. One drawback is the random distribution of dyes along the linear sequence, which has been shown to impede the hybridization quality by destabilizing the probe-target duplex, often leading to mutual quenching of adjacent dyes19. Also, long probes show poor permeability, high background and low sensitivity. Since it has been estimated that more than 80% of yeast genes are expressed at lower than two mRNA copies per cell20, it would be impossible to study the majority of the eukaryotic transcriptome with traditional FISH.
Figure 2.
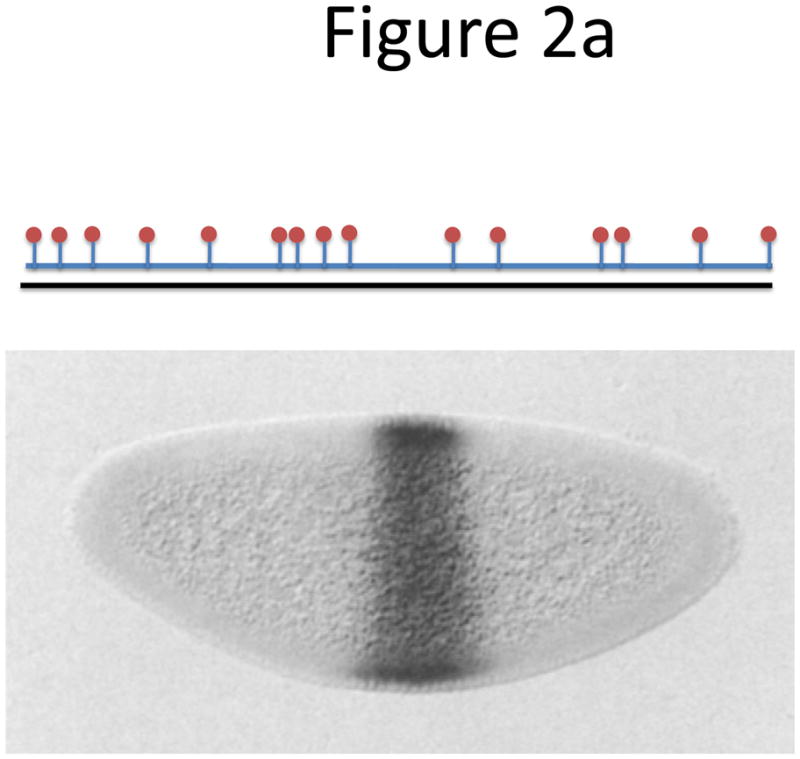

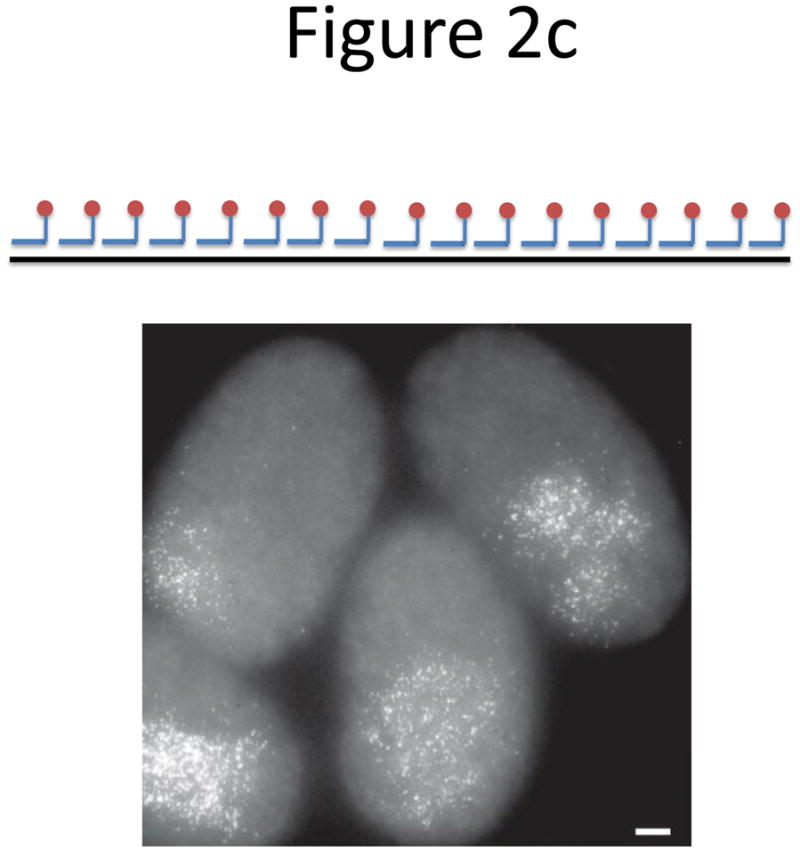
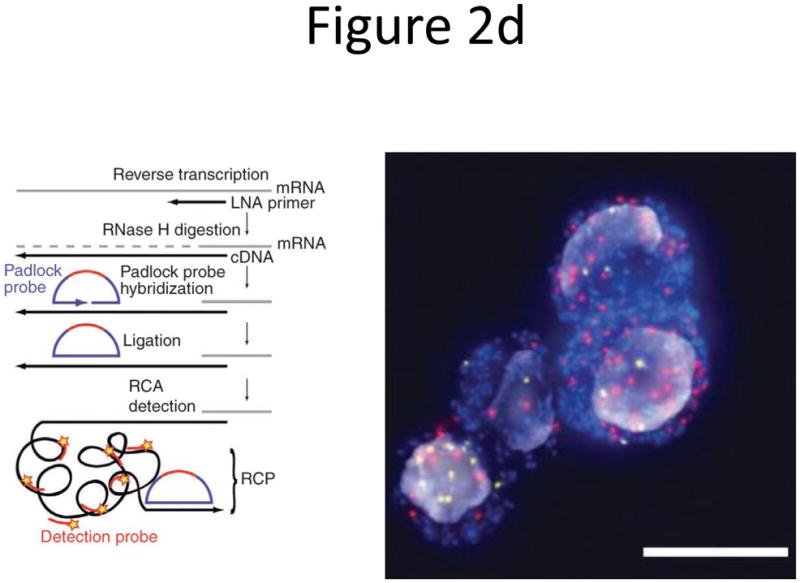
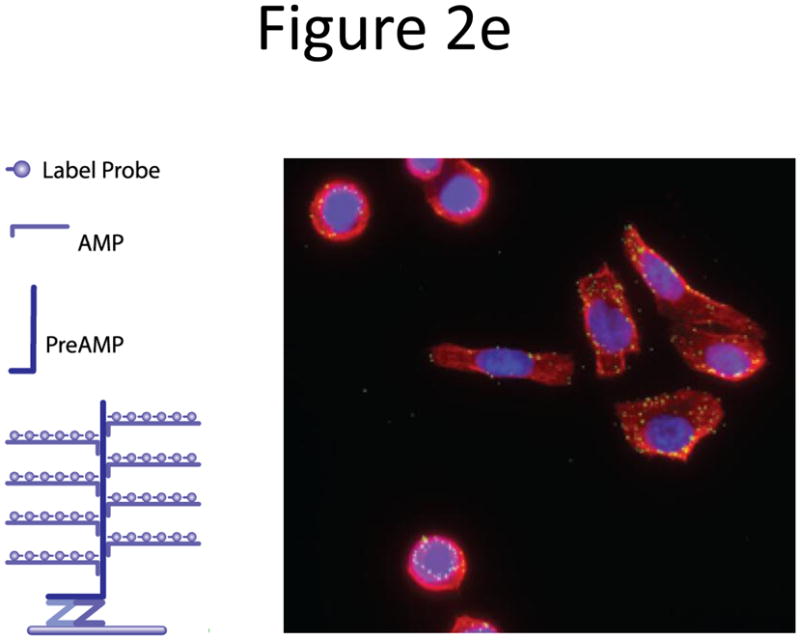
Probe designs for ISH. (a) Schematic of long probes with randomly distributed labeled nucleotides (top) and Drosophila embryo hybridized with probes complementary to the hunchback gene (bottom; reprinted from ref. 77). (b) Schematic of shorter probes, each coupled to 3–5 labeled nucleotides (top) and fluorescence image, showing single mRNA transcripts in mammalian cells (bottom; reprinted from ref. 26). Scale bar, 5 μm. (c) Schematic of multiple singly labeledshorter probes (top) and C. elegans embryos hybridized with probes complementary to the end-1 gene (bottom; reprinted from ref. 31). Scale bar, 5 μm. (d) Illustration of rolling-circle amplification (RCA) of padlock probes34; fluorescent probes bind their complementary sequences in the rolling circle product (RCP) (left). Fluorescence image (right) illustrates detection of ERBB2 (also known as HER2; red), MYC (yellow) and ACTB (blue) transcripts in primary human fibroblast cultures (reprinted from ref. 34). Scale bar, 20 μm. (e) Schematic of a branched DNA detection method using four probe sets. Multiple label probes bind multiple amplifier molecules that are in turn bound to a pre-amplifier molecule that binds the transcript-specific probe pairs. Image of ERBB2 (green) and 18S rRNA (red) in SKBR3 cells (right; reprinted from http://www.panomics.com/).
Single-molecule transcript imaging
Recent improvements in probe designs, imaging technology and image processing software have emerged that enable highly specific and robust hybridization of fluorescently labeled probes to an arbitrary transcript of interest with high spatial localization, yielding diffraction limited spots under a fluorescence microscope (Table 1). Thus an integer number of transcripts in individual cells can be determined by simply counting fluorescent dots. Moreover, the precise sub-cellular localization of the individual transcripts can be detected.
Table 1.
Technologies for single-molecule transcript imaging
| Method | Single - molecule resolution | Live-cell imaging | Advantages | Disadvantages | References |
|---|---|---|---|---|---|
| Traditional FISH | no | no | Probes can be designed for a wide range of genes | Limited sensitivity and specificity; impossible to detect very short transcripts | 15 |
| Probes labeled with multiple fluorophores | yes | no | Generic approach suited for most transcripts; established protocols exist | High variability in the number of probes bound to target | 21–26,39 |
| Multiple probes labeled with single fluorophores | yes | no | Generic approach suited for most transcripts, low variability in the number of probes bound to target; commercially available labeled probes | Limited in the detection of very short transcripts | 28,30,31 http://info.biosearchtech.com/singlemoleculefish |
| Rolling circle amplification of Padlock probes | yes | no | Can detect very short probes and discern between single nucleotide differences | Protocol requires reverse transcription and amplification steps | 34 |
| Branched DNA probes | yes | no | Commercially available labeled probes | Protocol requires amplification | 38 www.panomics.com |
| Quantum dot-labeled probes | yes | yes | Bright and photostable; huge spectral range | Large physical size detrimental to target binding and can cause cell penetration problems | 70–75 |
| Sub-diffraction microscopy | yes | yes | Achieves a spatial resolution of 20 nanometers | Slow, expensive instrumentation | 64–68 |
| MS2-GFP | yes | yes | No need for external interventions (for example microinjection); yields spatial information | Requires generating transgenes, mRNA tend to form clumps | 43,44,78 |
| Molecular beacons | yes | yes | High specificity with no clumping; labeled probes are commercially available | Require micro-injecting probes | 52–54 |
Short probes labeled with multiple fluorophores
Robert Singer and colleagues pioneered single mRNA molecule imaging techniques21. The key improvement was the replacement of long probes with 50 bp probes, that are complementary to sequential parts of the target mRNA and are each coupled to typically 3–5 fluorescent dyes at predefined positions (Fig. 2b). A minimal spacing between incorporated dyes along the linear oligonucleotide sequence was set to prevent quenching of adjacent dyes22. Coupled probes were separated from non-coupled probes and free dyes using chromatography. Singer and colleagues optimized the GC content of all probes to be as close as possible to 50%, and imaged the hybridized samples with a fluorescence microscope that captured stacks of images every fraction of microns, a high numerical aperture objective and a low-noise CCD camera. These improvements yielded three dimensional digital images in which the specific accumulation of fluorescent molecules in the small volume occupied by a single mRNA molecule appeared as diffraction limited spots.
Several research groups have since applied the use of multiply labeled probes for single mRNA counting to provide a detailed characterization of transcripts distribution in yeast23–25 and in mammalian cells26. In mammalian tissue, this method did not yet achieve single transcript resolution but enabled the detection of transcription sites in paraffin embedded human tumors27. But the approach also has some drawbacks, mainly a high variability in the number of probes bound to the target21. While ideally each spot would be composed of the same number of probes, in fact most fluorescent spots were estimated to originate from only one or two probes. This made it difficult to differentiate the true specific binding to the legitimate target from non-specific binding. Since an inefficient binding of one or two probes can lead to a high variability in spot intensity when only a handful of probes are used, it is important to carefully select the probes using this method. An additional source of variability using probes designed to be coupled to several fluorescent molecules is in the number of fluorescent molecules actually coupled to each probe -it is difficult to separate fully coupled probes from partially coupled ones.
Short probes with single fluorophores
Raj et al.28 modified the Singer method to create probe libraries consisting of many (typically between 48 and 96) short, 17–22 bp oligos labeled with only a single fluorophore at their 3′ termini28 (Fig. 2c). This allowed more efficient purification and the variability in spot intensity caused by inefficient binding of any single probe was lower when multiple probes were used. By optimizing probe design to have a uniform GC content of around 45% and a minimum gap of three nucleotides between successive probes, robust specific hybridization was achieved with a set of 48 probes or less. Raj et al.28 demonstrated simultaneous transcript counting of three transcripts coupled to distinct fluorophores. The use of a specialized mounting medium containing an oxygen scavenging system inhibited oxygen-dependent, light initiated pathways that destroy fluorophores, thus increasing the photostability of the dyes. This is particularly important in the optical setup in which the same field of view is illuminated multiple times as the optical sections are gathered22.
Several research groups have successfully applied singly labeled probes to a wide range of samples, ranging from yeast29 and mammalian cells28,30, to Drosophila28 and C. elegans embryos31. The approach is appealing mainly for its simplicity and generality, aided by a simple web interface to design optimal probes for arbitrary sequences (www.singlemoleculefish.com). Probe libraries are typically prepared with a 96-position DNA synthesizer, pooled and then simultaneously coupled to a fluorophore of choice. This format facilitates additional flexibility by enabling coupling of the same library to several different fluorescent dyes for simultaneous hybridization to other genes of interest.
A current limitation is the difficulty in detecting short transcripts. Xie and coworkers recently demonstrated single mRNA detection with only one 20bp fluorescently labeled probe in E. coli32. The ability to detect single transcripts in mammalian cells using one probe is much more challenging due to the larger volumes and increased off-target sequences involved. In-situ detection of short transcripts using oligonucleotide probes can be achieved by using modified nucleic acids to increase specificity and by applying signal amplification to increase sensitivity. Next we will discuss these two approaches.
Short probes with modified backbones
When the target sequences are too short, one runs into specificity problems, as any single probe has a non-negligible probability to bind a different target. Increasing the sensitivity and specificity of individual probes could enable the detection of very short transcripts or of single microRNA molecules, which typically span 19–24 bps33, as well as allow distinguishing between transcript variants that differ by single nucleotides34. This can be achieved with probes containing modified nucleic acids. The most commonly used modifications are peptide nucleic acids (PNA) and locked nucleic acids (LNA). PNA have an uncharged peptide-like backbone35 and therefore hybridizes more stably to RNA compared to DNA probes. LNA is a 2′-O, 4′-C-methylene-linked ribonucleotide derivative of RNA, enabling more specific hybridization with RNA and DNA, compared to DNA or RNA probes. PNA probes have been used to detect telomeres - repetitive hexamer sequences at chromosome ends36, whereas LNA probes facilitated the detection of microRNAs33,37, but with the use of signal amplification.
Signal amplification of single-molecule probes
Detection of short transcripts is still hampered by insufficient fluorescence from a single bound probe. Amplifying the fluorescent signal can solve this sensitivity limitation. Single microRNA molecules have been detected in situ using a single LNA probe and an alkaline phosphatase–based signal amplification33. Larsson et al.34 introduced the use of padlock probes that can distinguish transcripts that differ by only a single base pair. The authors first reverse-transcribed the mRNA into cDNA using LNA primers, then hybridized linear padlock probes to juxtaposed segments of the target sequence, enzymatically ligated them and used them as templates for rolling circle amplification by Phi29 DNA polymerase (Fig. 2d)34. This created a single strand of DNA containing tandem repeats of the padlock probe sequence that, after hybridization to fluorescently labeled probes, yielded bright diffraction-limited spots. Larsson et al.34 demonstrated multiplex detection of up to three genes in cells and frozen mouse embryonic tissue.
In the branched DNA approach38 hybridization is performed with four distinct probe sets: a gene-specific probe set composed of ten or more oligonucleotide probe pairs that are complementary to the target sequence, a pre-amplifier probe that hybridizes to gene-specific probes, multiple amplifier probes that hybridize to the pre-amplifier probe and labeled probes that attach to the amplifier probes. The resulting construct yields bright concentrated fluorescence (Fig. 2e). This technology is commercially named QuantiGene ViewRNA38.
Spectral barcoding
The number of spectrally resolvable fluorophores, typically three, limits the number of simultaneously measured genes in transcript imaging techniques. ‘Spectral barcoding’ is an approach developed by Singer and colleagues to increase the number of simultaneously detected transcripts21,39. The technology, which was based on a similar method used for DNA FISH40,41, entailed the division of the probe set that is complementary to a given transcript of interest into groups, each of which is coupled to a different fluorophore. By precisely registering the images from the different fluorescence channels one can determine not only whether a spot is present or not, but also how many colors compose it. With n spectrally resolvable fluorophores one can in principle achieve 2n-1 different probe color combinations. This limit could be increased if the detection method is sensitive enough to detect not only whether a given fluorophore is present or absent at a diffraction limited spot, but to also estimate the number of oligonucleotides coupled to the specific fluorophore of interest39.
Single-molecule transcript imaging in live cells
A limitation of transcript imaging using FISH approaches is that these require the samples to be fixed. The ability to temporally measure gene expression in living cells offers a rich source of additional information, including a detailed description of the life of an mRNA molecule, from transcription through intra-cellular trafficking, translation and degradation. Transcript imaging in live cells is considerably more challenging than in fixed cells, both because of the gentler chemical conditions that must be applied to preserve cell integrity, and the more intricate image processing steps, requiring tracking the trajectory of a transcript with time, a task that is often confounded by the high diffusion rates of molecules and the proximity to other molecules42. Two main technologies enable real-time measurements of transcript levels in single cells – the MS2 method and molecular beacons.
The coat protein of the bacteriophage MS2 binds a specific RNA hairpin loop. In the MS2 system, a fluorescent reporter protein gene such as GFP is fused to the MS2 coat protein gene, and several successive target RNA hairpin–encoding sequences are inserted into the 3′ untranslated region of a target gene of interest. Simultaneous binding of several MS2-GFP proteins to the target transcript yields a concentrated fluorescence signal. This method has been used to follow the dynamics of gene expression in bacteria43, yeast44 and mammalian cells45,46, achieving single-transcript resolution. Moreover, a general method to tag and image any mRNA in yeast has recently become available47. A limitation of the MS2-GFP approach is the high background fluorescence associated with the MS2-GFP molecules that are not bound to the target RNA. Split-GFP approaches48–51 alleviate this limitation, as each fragment is attached to a different RNA-binding protein that targets a distinct RNA motif. By constructing these motifs in tandem at the3′untranslated region of the target, an intact fluorescent GFP is assembled only when bound to the target transcript. The main limitation of the MS2 and split-GFP methods is the requirement to generate transgenes for the GFP construct. Yoshio Umezawa and colleagues recently demonstrated the ability to target endogenous mRNA by fusing the GFP fragments to the RNA-binding domain of the Pumilio protein, which can be designed to facilitate specific binding to an arbitrary RNA sequence51. A remaining drawback is that the binding of multiple proteins to the target mRNA could modify its intracellular dynamics, thus providing a nonrepresentative picture of the endogenous conditions.
Another technique for following individual transcripts in live cells uses molecular beacons52,53—nucleic acid probes that form hairpins coupled to a fluorophore and a quencher on opposite ends. Specific hybridization to the target sequence causes a conformational change that physically separates the quencher and fluorophore, resulting in light emission. Transcript detection in live cells is more challenging owing to their decreased stability caused by intracellular RNases and their non-homogenous intracellular distribution49. Probes with 2′-O-methylribonucleotide, which is not a target of RNase H, have been shown to alleviate this problem54–56. Molecular beacons require microinjection into cells, a procedure with high yield but one that may affect cell viability and can often result in a rapid drift of the microinjected probes into the nucleus. As an alternative, Santangelo et al.56 used reversible permeabilization of the plasma membrane with pore-forming toxins, such as streptolysin-O, to deliver multiply labeled tetravalent RNA probes with minimal cytotoxicity. These probes, which are complementary to the target RNA and labeled with three fluorescent molecules each, tetramerize through their additional binding to streptavidin, thus yielding increased accumulation of fluorophores at the target transcript and allowing visualization of RNA dynamics56.
Imaging technologyfor single-molecule FISH
Detection of single mRNA molecules imposes specific requirements on the imaging platform in terms of spatial resolution and sensitivity. For lens-based systems the maximum resolution can be estimated by the Rayleigh criteria, which says the resolution is equal to (0.61)λ/NA, in which λ is the illumination wavelength, and NA is the numerical aperture of the lens. In practice, this limits the lateral resolution to 200–400 nm. Optimizing spatial resolution typically requires the use of a high-numerical-aperture oil-immersion lens and immersion oil that exactly matches the refractive index of the lens and the cover glass. Achromat or apochromat objectives minimize chromatic aberrations and are thus necessary when multiplexing different fluorophores42. Resolving diffraction-limited volumes also requires using a camera with sufficient spatial resolution that does not compromise the optimal optical resolution. CCD cameras have pixel sizes of 2–40 μm, which translates to a size of 20–400 nm at the image plane with a 100× objective depending on the optical setup. To achieve optimal resolution, it is crucial to determine the size of the image being projected onto the camera and verify that there are 2.5–3 pixels per unit resolution. If pixel binning is used to increase the signal at the camera, the number of required pixels must be multiplied accordingly. Exposure times should be optimized: long exposure times lead to higher precision in transcript localization but could accelerate sample bleaching57.
Image signal is often masked by noise stemming from both instrument-related factors such as dark current, pixelation noise and CCD readout noise but mostly from out-of-focus light from cellular autofluorescence57,58. Maximizing signal-to-noise ratio requires increasing photon output of the fluorescent labels and limiting background noise. Wide-field epi-illuminated microscopes use a broadband light source with a spectrum from 250 nm to 1,100 nm. Excitation filters can narrow this band to the fluorophore excitation wavelength, but autofluorescence always remains. Collecting light from a wide field also results in an increase in background. Confocal microscopes have the advantage of using a pinhole or slit to block detection of light from outside the focal region, thus potentially increasing relevant fluorescence and decreasing autofluorescence. Disadvantages of confocal microscopes over epi-fluorescence microscopes include their higher cost, the lower throughput owing to the necessity to scan the image (partly alleviated by using spinning disk confocal microscopes), more stringent safety requirements and the additional descanning optics necessary to guide the detected light to the detector, which can limit transmission efficiency. Multiplex detection of several fluorophores can be achieved by using filter wheels for epifluorescence microscopes and multimode lasers for confocal microscopes. Instrument-related noise, such as dark current can be substantially minimized by cooling the CCD chip to −80 °C58.
Data analysis
Extracting gene expression from single-molecule FISH experiments requires automatic detection of spots representing single mRNA molecules in digital three dimensional image stacks. The image processing steps usually include image enhancement - either deconvolution or high pass filtering, followed by thresholding and localization of connected components21,22,28,31,34. A point source in the illuminated image plane is expanded and distorted by the microscope and the detector optics resulting in a blurred three dimensional version, called the point spread function (PSF). Because a fluorescence emission pattern can be treated as a sum of point sources, a recorded image is a convolution of the real image and the PSF. When an accurate description of the PSF is available one can use deconvolution algorithms to reconstruct the original image59. Such algorithms deblur images and reassign photons emanating from out-of-focus z-dimension planes back to their original plane. Successful deconvolution requires precise measurements of the PSF, which is typically achieved by imaging sub-diffraction fluorescent beads21,22. An alternative to deconvolution is to filter the image stack with a three dimensional high-pass filter (such as a 3D Laplacian of Gaussian filter) to enhance features of the relevant spot dimension28,31. The next step after image enhancement usually consists of applying a threshold to the enhanced image yielding a binary image, in which connected components can be localized. Since a connected component spans more than a single pixel its centroid achieves sub-pixel resolution for the localization of the source. The value of the image thresholds can affect the number of spots detected and a reasonable choice for this value is one at which the number of detected spots is least sensitive to threshold selection28,31. Alternatively least-square Gaussian fitting algorithms can be used to localize the point source in a gray scale image57,60.
Outlook
One appealing application of single transcript imaging is the validation of regulatory interactions. A traditional approach entails engineering mice in which a regulatory gene of interest is deleted, and searching for putative target genes with modified expression levels61. Single-molecule FISH can enable detecting such targets in wild-type tissue by hybridizing to a sample probes for the regulatory gene and for a putative target gene. A measured positive correlation in the transcript abundance could imply either a direct regulation or alternatively regulation by a common upstream component. Single-molecule transcript imaging can also provide valuable information on the behavior of network motifs, modular circuit components such as feedback and feedforward loops which are highly abundant in transcriptional networks and often comprise only a few genes62. The simultaneous quantitative in-situ measurements of a handful of different transcripts can shed light on the behavior of these motifs within their tissue context. In a tumor single-molecule transcript imaging can highlight the role of transcriptional heterogeneity in tumor progression and the relation between spatial context and phenotypic states of cells, represented by their expression signatures.
Single-molecule transcript imaging techniques can be combined with high-throughput expression analysis in two complementary ways. One would be to start out with large gene expression screens that would suggest putative genes of interest, the detailed in-situ expression of which would then be described using single-molecule FISH. An alternative approach would be to start with an unbiased mapping of a tissue using a panel of single-molecule FISH probes to detect an interesting expression pattern in terms of spatial distribution within a tissue or an unusual co-expression pattern of a few genes in isolated cells. One could then enrich for such cells using FACS or laser capture and extend this core gene expression signature with high-throughput genome-wide expression measurements. This approach could provide a detailed description of rare cell populations residing within a tumor, such as putative cancer stem cells
A technical limitation of single-molecule transcript imaging is the inability to spatially resolve single molecules when they are closer than the diffraction limit, typically 200 nm. This could be a significant problem in highly expressed genes such as ribosomal components especially in smaller organisms such as yeast25, and when mRNA molecules are physically localized in transport particles63. Techniques that can address this limitation are sub-diffraction-limit microscopy methods such as STED64, STORM65 and PALM66, which enable resolving fluorescent molecules with nanometer resolution67. While sub-diffraction microscopy outperforms other technologies in spatial resolution, enabling probing molecular structures in fixed and even live cells68,69, scaling the technology to comprehensively measure gene expression in-situ in many cells and in tissues is still a challenge due to long recording times, high intensity illumination, the prolonged use of which could potentially be harmful to the sample and expensive instrumentation.
Another exciting recent development is the use of probes coupled to quantum dots70–73. The brightness of quantum dots makes them especially attractive for studying tissues, where cellular autofluorescense often masks the signal. Oligonucleotide probes labeled with quantum dots have been used to detect transcripts in cells and tissues70–72, in paraffin embedded tissue74 and even in live cell imaging75. Some limitations of using quantum dots, such as reduced permeability and steric hindrance difficulties when binding the targets, mainly caused by their relatively large size compared to conventional probes, as well as their tendency to turn on and off (‘blinking’)76 still limit the use of quantum-dot labeled probes for transcript imagingbut their attractive photophysical properties suggest a huge potential for single-molecule detection.
Finally, a challenge for the future is to combine transcript imaging approaches with quantitative measurements of other cellular constituents, namely proteins and DNA. While protocols tailored to simultaneously perform RNA FISH and immunofluorescense have been shown to be successful in some cases56,69, their generic use is still limited by the variabilities inherent in immunofluorescense. Combination of RNA FISH with immunofluorescense or with GFP protein measurements will facilitate decoupling the relative contributions of transcriptional and translational regulation in cells and tissues, whereas combination with DNA FISH can address the expression variability of different clones within a tumor tissue. Such analysis can provide important insights into the combined regulation of protein expression in complex tissues.
Acknowledgments
We thank Stefan Semrau, Jan Philipp Junker, Shankar Mukherji and Anat Lavi-Itzkovitz for valuable comments. This work was supported by the NIH/NCI Physical Sciences Oncology Center at MIT (U54CA143874) and a NIH Pioneer award (1DP1OD003936) to A.v.O. S.I. acknowledges support from EMBO, the Human Frontiers Science Program and the Machiah Foundation.
References
- 1.Eisen MB, Spellman PT, Brown PO, et al. Proc Natl Acad Sci U S A. 1998;95(25):14863. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heller RA, Schena M, Chai A, et al. Proc Natl Acad Sci U S A. 1997;94(6):2150. doi: 10.1073/pnas.94.6.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sorlie T, Perou CM, Tibshirani R, et al. Proc Natl Acad Sci U S A. 2001;98(19):10869. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van ‘t Veer LJ, Dai H, van de Vijver MJ, et al. Nature. 2002;415(6871):530. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 5.Levsky JM, Singer RH. Trends Cell Biol. 2003;13(1):4. doi: 10.1016/s0962-8924(02)00002-8. [DOI] [PubMed] [Google Scholar]
- 6.Raj A, van Oudenaarden A. Cell. 2008;135(2):216. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lengauer C, Kinzler KW, Vogelstein B. Nature. 1997;386(6625):623. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 8.Park SY, Gonen M, Kim HJ, et al. J Clin Invest. 2010;120(2):636. doi: 10.1172/JCI40724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navin N, Krasnitz A, Rodgers L, et al. Genome Res. 2010;20(1):68. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reya T, Morrison SJ, Clarke MF, et al. Nature. 2001;414(6859):105. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 11.Emmert-Buck MR, Bonner RF, Smith PD, et al. Science. 1996;274(5289):998. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- 12.Freeman WM, Walker SJ, Vrana KE. Biotechniques. 1999;26(1):112. doi: 10.2144/99261rv01. [DOI] [PubMed] [Google Scholar]
- 13.Warren L, Bryder D, Weissman IL, et al. Proc Natl Acad Sci U S A. 2006;103(47):17807. doi: 10.1073/pnas.0608512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Flier LG, van Gijn ME, Hatzis P, et al. Cell. 2009;136(5):903. doi: 10.1016/j.cell.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 15.Levsky JM, Singer RH. J Cell Sci. 2003;116(Pt 14):2833. doi: 10.1242/jcs.00633. [DOI] [PubMed] [Google Scholar]
- 16.van der Ploeg M. Eur J Histochem. 2000;44(1):7. [PubMed] [Google Scholar]
- 17.Gregorieff A, Clevers H. Curr Protoc Stem Cell Biol. Chapter 2(Unit 2F):1. doi: 10.1002/9780470151808.sc02f01s12. [DOI] [PubMed] [Google Scholar]
- 18.Pare A, Lemons D, Kosman D, et al. Curr Biol. 2009;19(23):2037. doi: 10.1016/j.cub.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Randolph JB, Waggoner AS. Nucleic Acids Res. 1997;25(14):2923. doi: 10.1093/nar/25.14.2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larson DR, Singer RH, Zenklusen D. Trends Cell Biol. 2009;19(11):630. doi: 10.1016/j.tcb.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Femino AM, Fay FS, Fogarty K, et al. Science. 1998;280(5363):585. doi: 10.1126/science.280.5363.585. This pioneering work demonstrated single-molecule transcript imaging in fixed cells using multiply labeled fluorescent probes. [DOI] [PubMed] [Google Scholar]
- 22.Femino AM, Fogarty K, Lifshitz LM, et al. Methods Enzymol. 2003;361:245. doi: 10.1016/s0076-6879(03)61015-3. [DOI] [PubMed] [Google Scholar]
- 23.Maamar H, Raj A, Dubnau D. Science. 2007;317(5837):526. doi: 10.1126/science.1140818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zenklusen D, Larson DR, Singer RH. Nat Struct Mol Biol. 2008;15(12):1263. doi: 10.1038/nsmb.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan RZ, van Oudenaarden A. Mol Syst Biol. 2010;6:358. doi: 10.1038/msb.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raj A, Peskin CS, Tranchina D, et al. PLoS Biol. 2006;4(10):e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capodieci P, Donovan M, Buchinsky H, et al. Nat Methods. 2005;2(9):663. doi: 10.1038/NMETH786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raj A, van den Bogaard P, Rifkin SA, et al. Nat Methods. 2008;5(10):877. doi: 10.1038/nmeth.1253. Single-molecule transcript imaging in cells and in fixed tissues was achieved using multiple singly labeled probes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.To TL, Maheshri N. Science. 2010;327(5969):1142. doi: 10.1126/science.1178962. [DOI] [PubMed] [Google Scholar]
- 30.Khalil AM, Guttman M, Huarte M, et al. Proc Natl Acad Sci U S A. 2009;106(28):11667. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raj A, Rifkin SA, Andersen E, et al. Nature. 2010;463(7283):913. doi: 10.1038/nature08781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taniguchi Y, Choi PJ, Li GW, et al. Science. 2010;329(5991):533. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu J, Tsourkas A. Nucleic Acids Res. 2009;37(14):e100. doi: 10.1093/nar/gkp482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larsson C, Grundberg I, Soderberg O, et al. Nat Methods. 2010;7(5):395. doi: 10.1038/nmeth.1448. This work introduced a sensitive single-molecule transcript imaging technique that utilizes padlock probes and in situ target-primed rolling-circle amplification in cells and tissues to specifically detect targets with a single nucleotide difference. [DOI] [PubMed] [Google Scholar]
- 35.Wittung P, Nielsen PE, Buchardt O, et al. Nature. 1994;368(6471):561. doi: 10.1038/368561a0. [DOI] [PubMed] [Google Scholar]
- 36.Lansdorp PM, Verwoerd NP, van de Rijke FM, et al. Hum Mol Genet. 1996;5(5):685. doi: 10.1093/hmg/5.5.685. [DOI] [PubMed] [Google Scholar]
- 37.Kloosterman WP, Wienholds E, de Bruijn E, et al. Nat Methods. 2006;3(1):27. doi: 10.1038/nmeth843. [DOI] [PubMed] [Google Scholar]
- 38.Player AN, Shen LP, Kenny D, et al. J Histochem Cytochem. 2001;49(5):603. doi: 10.1177/002215540104900507. [DOI] [PubMed] [Google Scholar]
- 39.Levsky JM, Shenoy SM, Pezo RC, et al. Science. 2002;297(5582):836. doi: 10.1126/science.1072241. This work substantially increased the number of targets that can be simultaneously imaged by using combination of probes labeled with spectrally distinct fluorophores. [DOI] [PubMed] [Google Scholar]
- 40.Nederlof PM, van der Flier S, Vrolijk J, et al. Cytometry. 1992;13(8):839. doi: 10.1002/cyto.990130806. [DOI] [PubMed] [Google Scholar]
- 41.Nederlof PM, van der Flier S, Wiegant J, et al. Cytometry. 1990;11(1):126. doi: 10.1002/cyto.990110115. [DOI] [PubMed] [Google Scholar]
- 42.Park HY, Buxbaum AR, Singer RH. Methods Enzymol. 472:387. doi: 10.1016/S0076-6879(10)72003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Golding I, Paulsson J, Zawilski SM, et al. Cell. 2005;123(6):1025. doi: 10.1016/j.cell.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 44.Bertrand E, Chartrand P, Schaefer M, et al. Mol Cell. 1998;2(4):437. doi: 10.1016/s1097-2765(00)80143-4. This work introduced the MS2 method for single mRNA detection in live cells. [DOI] [PubMed] [Google Scholar]
- 45.Fusco D, Accornero N, Lavoie B, et al. Curr Biol. 2003;13(2):161. doi: 10.1016/s0960-9822(02)01436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shav-Tal Y, Darzacq X, Shenoy SM, et al. Science. 2004;304(5678):1797. doi: 10.1126/science.1099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haim L, Zipor G, Aronov S, et al. Nat Methods. 2007;4(5):409. doi: 10.1038/nmeth1040. [DOI] [PubMed] [Google Scholar]
- 48.Rackham O, Brown CM. Embo J. 2004;23(16):3346. doi: 10.1038/sj.emboj.7600341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tyagi S. Nat Methods. 2009;6(5):331. doi: 10.1038/nmeth.1321. [DOI] [PubMed] [Google Scholar]
- 50.Valencia-Burton M, McCullough RM, Cantor CR, et al. Nat Methods. 2007;4(5):421. doi: 10.1038/nmeth1023. [DOI] [PubMed] [Google Scholar]
- 51.Ozawa T, Natori Y, Sato M, et al. Nat Methods. 2007;4(5):413. doi: 10.1038/nmeth1030. [DOI] [PubMed] [Google Scholar]
- 52.Tyagi S, Kramer FR. Nat Biotechnol. 1996;14(3):303. doi: 10.1038/nbt0396-303. This work introduced molecular beacons for the detection of single mRNA in live cells. [DOI] [PubMed] [Google Scholar]
- 53.Vargas DY, Raj A, Marras SA, et al. Proc Natl Acad Sci U S A. 2005;102(47):17008. doi: 10.1073/pnas.0505580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bratu DP, Cha BJ, Mhlanga MM, et al. Proc Natl Acad Sci U S A. 2003;100(23):13308. doi: 10.1073/pnas.2233244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lifland AW, Zurla C, Santangelo PJ. Bioconjug Chem. 2010 doi: 10.1021/bc9003876. [DOI] [PubMed] [Google Scholar]
- 56.Santangelo PJ, Lifland AW, Curt P, et al. Nat Methods. 2009;6(5):347. doi: 10.1038/nmeth.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thompson RE, Larson DR, Webb WW. Biophys J. 2002;82(5):2775. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grunwald D, Singer RH, Czaplinski K. Methods Enzymol. 2008;448:553. doi: 10.1016/S0076-6879(08)02627-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McNally JG, Karpova T, Cooper J, et al. Methods. 1999;19(3):373. doi: 10.1006/meth.1999.0873. [DOI] [PubMed] [Google Scholar]
- 60.Smith CS, Joseph N, Rieger B, et al. Nat Methods. 2010;7(5):373. doi: 10.1038/nmeth.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van der Flier LG, Clevers H. Annu Rev Physiol. 2009;71:241. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 62.Alon U. Nat Rev Genet. 2007;8(6):450. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 63.Bassell G, Singer RH. Curr Opin Cell Biol. 1997;9(1):109. doi: 10.1016/s0955-0674(97)80159-7. [DOI] [PubMed] [Google Scholar]
- 64.Klar TA, Jakobs S, Dyba M, et al. Proc Natl Acad Sci U S A. 2000;97(15):8206. doi: 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rust MJ, Bates M, Zhuang X. Nat Methods. 2006;3(10):793. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Betzig E, Patterson GH, Sougrat R, et al. Science. 2006;313(5793):1642. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 67.Hell SW. Science. 2007;316(5828):1153. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 68.Shroff H, Galbraith CG, Galbraith JA, et al. Nat Methods. 2008;5(5):417. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bassell GJ, Zhang H, Byrd AL, et al. J Neurosci. 1998;18(1):251. doi: 10.1523/JNEUROSCI.18-01-00251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choi Y, Kim HP, Hong SM, et al. Small. 2009;5(18):2085. doi: 10.1002/smll.200900116. [DOI] [PubMed] [Google Scholar]
- 71.Chan P, Yuen T, Ruf F, et al. Nucleic Acids Res. 2005;33(18):e161. doi: 10.1093/nar/gni162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gonzalez-Maeso J, Ang RL, Yuen T, et al. Nature. 2008;452(7183):93. doi: 10.1038/nature06612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith AM, Duan H, Mohs AM, et al. Adv Drug Deliv Rev. 2008;60(11):1226. doi: 10.1016/j.addr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tholouli E, Hoyland JA, Di Vizio D, et al. Biochem Biophys Res Commun. 2006;348(2):628. doi: 10.1016/j.bbrc.2006.07.122. [DOI] [PubMed] [Google Scholar]
- 75.Ishihama Y, Funatsu T. Biochem Biophys Res Commun. 2009;381(1):33. doi: 10.1016/j.bbrc.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 76.Weil TT, Parton RM, Davis I. Trends Cell Biol. 20(7):380. doi: 10.1016/j.tcb.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tautz D, Pfeifle C. Chromosoma. 1989;98(2):81. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- 78.Bloom KS, Beach DL, Maddox P, et al. Methods Cell Biol. 1999;61:369. doi: 10.1016/s0091-679x(08)61990-1. [DOI] [PubMed] [Google Scholar]