


Abstract
Genomic sequences obtained through high-throughput sequencing are not uniformly distributed across the genome. For example, sequencing data of total genomic DNA show significant, yet unexpected enrichments on promoters and exons. This systematic bias is a particular problem for techniques such as chromatin immunoprecipitation, where the signal for a target factor is plotted across genomic features. We have focused on data obtained from Illumina’s Genome Analyser platform, where at least three factors contribute to sequence bias: GC content, mappability of sequencing reads, and regional biases that might be generated by local structure. We show that relying on input control as a normalizer is not generally appropriate due to sample to sample variation in bias. To correct sequence bias, we present BEADS (bias elimination algorithm for deep sequencing), a simple three-step normalization scheme that successfully unmasks real binding patterns in ChIP-seq data. We suggest that this procedure be done routinely prior to data interpretation and downstream analyses.
INTRODUCTION
High-throughput sequencing provides a rapid and inexpensive platform for large-scale genome-wide studies, including transcriptome profiling, genomic variation identification and chromatin structure and organization analyses (1–8). While this technique has unprecedented advantages in large-scale biological studies, such as higher resolution and sensitivity, it also poses challenges in data analysis. The recovery of sequenced DNA fragments is not uniform along the genome. GC-rich sequences are often over-represented (9,10). The read mapping procedure generates regional bias (1,11). Because sequence reads that map to multiple sites in the genome are usually discarded, genomic regions with high sequence degeneracy show lower mapped read coverage than unique regions, creating systematic bias. Furthermore, the local structure of DNA or chromatin can lead to coverage inhomogeneity. For instance, the resistance of heterochromatic regions to shearing can result in their under-representation in chromatin samples (1,12). Sequence coverage variability has been observed with different sequencing platforms (13). Because genomic features such as protein-coding exons are often higher in GC content, systematic biases in sequencing data could lead to a false inflation of read counts in such regions (14). These biases are a particular problem in analyses of chromatin immunoprepcipitation followed by high-throughput sequencing (ChIP-seq) data, where regions of factor enrichment are sought. Therefore, removal of bias is necessary to determine real enrichment patterns.
An obvious approach to correct for biases would be to divide experimental signals by those obtained from input control DNA, assuming that ChIP and control samples would have the same biases. Many peak calling algorithms use input control for significance testing of enriched regions (11,15–21), but none actually corrects for biases in the data. USeq and SPP subtract input control from ChIP sample to try to normalize the data prior to peak calling (20,21). PeakSeq accounts for mappability differences when selecting candidate enrichment regions (11). However, none of the peak calling methods generates bias-free versions of the original sequencing data. Having the biases corrected globally in the entire data set is essential for studies that are not based upon peak calling results, e.g. to determine patterns of factor binding across genomic features such as promoters (3,22).
Here, we show that due to sample to sample variation in the amount of systematic biases in different sequencing data sets, relying on input control, a normalizer is not generally appropriate. We present BEADS (bias elimination algorithm for deep sequencing), a simple three-step normalization scheme that estimates and corrects for data set-specific biases. After normalization, ChIP-seq data sets retained enrichments on genomic features previously determined by other methods, whereas unexpected patterns were removed. Our results demonstrate that BEADS successfully unmasks real binding patterns in ChIP-seq data.
MATERIALS AND METHODS
Caenorhabditis elegans input sequence and ChIP library preparation
Samples of synchronized mid-L3 larvae were prepared by growing starved L1s in liquid culture at 20°C. Larvae were cleaned by sucrose flotation and flash frozen in liquid nitrogen. The ChIP protocol was as described previously (22). Briefly, frozen worm powder was fixed in 1% formaldehyde. The cross-linked chromatin was sonicated using a Bioruptor (Diagenode) to an average range of 100–300 bp. Prior to adding antibody, 10% of the volume used for ChIP was taken for the input sequence sample. Remaining extract aliquots were incubated with each specific antibody overnight at 4°C and immunoprecipitated with protein A Dynabeads. After decross-linking and purification, DNA was amplified using the ChIP-Seq DNA Sample Prep Kit (Illumina, IP-102-1001). Fragments in the 250- to 350-bp range were size selected by gel extraction and sequenced. The antibodies used were: anti-H3K4me3 Active Motif AR0169, H3K9me3 Upstate 07-442, H3K36me3 Abcam ab9050, and anti-DPY-27 (23). Sequence data sets are available from GEO: Input controls 1-5 (GSM706164, GSM706166, GSM727910, GSM727911, GSM706165, respectively); H3K4me3 (GSM727906); H3K9me3 (GSM727907); H3K36me3 (GSM727908); DPY-27 (GSM727909).
Human input sequence and genomic DNA sequencing data
We obtained sequence reads of human ChIP input control DNA from a published study (1), where the data set from replicate 1 was used as the experimental input sequence and data sets from the other two replicates were used to generate the master control set. The human genomic DNA sequencing data shown in Supplementary Figure S2 was obtained from the publicly available 1000 Genomes Project (Pilot data NA12878, SRR014603). These human data shown in the paper were confined to chromosome 1 as a proof of principle. For the human ChIP control DNA sequencing data sets, each replicate had ∼10% of all reads mapped to chromosome 1 (i.e. 623 546 chromosome 1 reads for replicate 1, 658 449 reads for replicate 2 and 676 664 reads for replicate 3 were used). For the human genomic DNA sequencing data shown in Supplementary Figure S2, 430 579 chromosome 1 reads (∼8% of all reads) were used in the analysis.
Data shown in Supplementary Figure S3b were derived from entire sets of short reads generated by multiple sequencing runs of the same library, taken from 1000 Genomes Project (Library Solexa-3623; Experiment ID ‘SRX000259′).
Read mapping and signal profiling
Sequence reads of 35 bp were obtained on Solexa/Illumina sequencing platforms and aligned to the C. elegans genome WS190 [25-bp sequence reads from (1) were aligned to the human genome GRCh37] using MAQ (24) version 0.7.1 with default settings. Aligned reads with MAQ mapping quality less than 10 were removed from subsequent analyses to ensure the remaining reads are mapped unambiguously to unique positions.
Mapped sequence reads were extended to 200 bp, which was the estimated mean insert size targeted in the size selection step when preparing the libraries. A genomic profile of signal levels was then generated by counting the number of extended sequence reads (i.e. read count) overlapping sampling points spaced at regular 50-bp intervals across the genome.
Division by input control data without BEADS
Read counts of a set of sequencing data (either a ChIP sample or an input control) were divided by corresponding read counts of an input sequence at the sampled 50-bp intervals, after being linearly scaled (using a genome wide read count coefficient between the two libraries) to account for sequence depth disparity between dividend and divisor.
Weighting reads by GC content
Reads that could be mapped confidently onto the reference genome were extended to 200 bp. Extended reads of experimental sequencing data were divided amongst 201 bins (i.e. from GC = 0 to GC = 200) according to the total number of guanines and cytosines residing in the 200-bp fragments. Similarly, nucleotide compositions of 200-bp fragments from the reference genome were calculated (a 200-bp fragment was selected every 50 bp). Frequency plots were generated for the GC distributions of the sequenced library and of the genome. For ChIP-seq data sets, only fragments that did not overlap with identified enrichment regions were used to construct the two GC frequency distributions whereas for input sequence data sets the whole set of mapped sequences was used.
A sequence read with 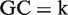 is given a weight, wk:
is given a weight, wk:
where  and
and 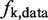 are the frequencies of the GC distribution bins from the reference genome and the sequencing data, respectively. This normalization step adjusts the experimental GC distribution of the sequencing data to match the genomic GC distribution. At the end of this step, each sequence read was associated with a GC weight; GC-normalized read counts were collected at 50-bp regular intervals across the genome.
are the frequencies of the GC distribution bins from the reference genome and the sequencing data, respectively. This normalization step adjusts the experimental GC distribution of the sequencing data to match the genomic GC distribution. At the end of this step, each sequence read was associated with a GC weight; GC-normalized read counts were collected at 50-bp regular intervals across the genome.
Regions of ChIP enrichment in H3K4me3, H3K9me3 and H3K36me3 ChIP-seq data were identified using USeq (20), applying the ‘sum’ method without a control library. Window size was set to 500 bp. For Supplementary Figure S7, score thresholds of 300, 150, 80, 50 and 40 were used on the H3K4me3 data to identify enrichment regions with most stringent, stringent, moderate, lax and most lax criteria, respectively, and thresholds of 200, 50 and 20 were used on the H3K36me3 data to identify enrichment regions with stringent, moderate and lax criteria, respectively. Enriched regions identified using moderate criteria were used for all other analyses. For Supplementary Figure S5, score threshold of 30 was used on the H3K9me3 data to identify enrichment regions. Any peak-calling software can be used for enrichment identification (with or without input control), but we recommend users to visually check the results to ensure that the identified regions reasonably capture most enriched regions before applying the normalization procedure.
Quantifying mappability and adjusting for its variation
To determine the level of sequence read mappability across the genome, we generated a simulated set of 35-bp sequence reads (25-bp for human data) covering the whole genome at 1-bp resolution. These sequences were mapped onto the genome using MAQ (24) in the same way that we treated experimental sequence data (i.e. using default settings and only sequence reads that were aligned with a mapping quality 10 or above were retained). This procedure exhaustively maps all 35-bp (or 25-bp) sequences in the genome that can theoretically be mapped. Each mappable read was extended to 200-bp (the expected average size of the sequenced fragments), and mappability at a given position was quantified as the number of overlapping extended reads. Because a read can be either positive or negative stranded, for 200 bp fragments, the maximum number of mapped reads at a genomic location is 400 (i.e. 100% mappability) and the minimum is zero (0%). We sampled mappability at 50-bp intervals (at the same positions as used at the end of GC correction step) and generated a mappability track.
To correct for mappability variations for a given genomic location i, we applied a weighting function to the signal, Si, which is the sum of GC-corrected weights of the overlapping reads:
where mi is the mappability of location i and mmax is a constant of the mappability upper bound value (400 in our case). A cut-off value was applied to the data such that genomic locations with mappability lower than 100 (25% of maximum) were removed from subsequent analyses, since low sampling could result in unreliable corrections in these regions (Supplementary Figure S11).
Local correction and fold-change estimation using input control data
Three C. elegans input sequence data sets [or sequence reads from two replicates of human data (1)] were individually GC and mappability corrected, then pooled according to total read counts so that they contributed equally. At the sampled 50-bp intervals, experimental GC- and mappability-normalized read counts were divided by the pooled GC and mappability-normalized input read counts after linear scaling to match total read counts in experimental and pooled input data sets. Fold-change values were assigned to every genomic location by taking the value of the nearest sample point of data.
Simulated sequence reads for fragment size analysis
We simulated two sets of theoretical input control libraries with no genuine enrichment by randomly sampling 30 million DNA fragments from the C. elegans genome (WS190). Each of these two independent sets of DNA fragments follows a realistic size distribution with a mean value of 150 bp (Supplementary Figure S9a). We then transformed these two sets of DNA fragments into sequence reads by introducing a realistic GC bias (i.e. throwing away fragments in each set until the remaining fragments represent the GC distribution of a sequenced input library; the resulting sets contained 10 million reads) and keeping only 35 bp of one end of each fragment (selected randomly). At this point, we had two sets of sequence reads from simulated input control libraries whose actual average fragment size are 150 bp. These reads were mapped onto the C. elegans genome using MAQ (24) in the same way that we treated experimental sequence data.
Genomic features
We extracted C. elegans genomic features from Ensembl database of Wormbase release WS190 (http://www.wormbase.org) and human genomic features from Ensembl database assembly GRCh37. We selected protein coding exons that were at least 200-bp long, flanked by at least 500 bp of introns and excluded the first or last exons in the transcript, resulting in 2105 internal exons for C. elegans genome and 1021 internal exons for human chromosome 1 meeting these criteria. We aligned these internal exons at the intron/exon and exon/intron splice junctions and scaled the exons to a pseudosize of 250 bp.
We defined isolated transcript start site (TSS) as the first base of an annotated transcript that was at least 100-bp long and with no other transcripts within 1 kb, resulting in 4624 TSSs selected for C. elegans genome and 516 TSSs for human chromosome 1. H3K4me3 and H3K36me3 are histone marks for active promoters and gene bodies respectively and their patterns are most clearly seen when only active genes are considered (22). Therefore, in addition to using a complete set of genes, we also assembled a subset of C. elegans TSSs for highly expressed genes by selecting only isolated TSSs from genes that are in the top 10% of expression level (22) (a set of 332 TSSs).
In Supplementary Figure S5a, H3K9me3 ChIP-seq signals were plotted across internal exons of a set of previously identified C. elegans ubiquitous genes (25) that were at least 200-bp long, flanked by at least 500-bp of introns (resulting in 931 exons). In Supplementary Figure S5b, H3K9me3 ChIP-seq signals were plotted across the gene bodies of 2591 ubiquitous genes (25) and 415 silent genes (22) with 250-bp flanking the start and end of genes.
In Supplementary Figure S10b, DPY-27 ChIP-seq signals were plotted across a set of chromosome X foci defined by Ercan et al. (23). Because the published X foci locations were based on an older C. elegans assembly (WS120), the genomic coordinates were lifted over to assembly WS190 using the UCSC LiftOver tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver). Signals were plotted across X foci and 1-kb upstream and downstream. In Supplementary Figure S10d, BLMP-1 ChIP-seq signals were plotted across a set of previously identified binding sites (26).
Data visualization
In Figure 1a and Supplementary Figure S7a, signals of sequencing data across chromosomal regions were displayed using the Affymetrix Integrated Genome Browser (27).
Figure 1.

DNA fragments in high-throughput sequencing data are not uniformly distributed over the genome. (a) The patterns of raw sequencing signals of independent C. elegans input sequence extracts and genomic DNA samples (black) are similar to underlying GC content (red) and mappability (blue). Positions 11 075 000–11 098 000 of chromosome I of the C. elegans genome are shown. (b) GC frequency distributions of the C. elegans genome (solid line) and a set of input sequence reads (dashed line). (c) GC frequency ratio between input sequence data and the C. elegans genome.
To visualize the signals of sequencing data across internal exons and around TSSs, we took samples at 10-bp intervals across the resulting landscape model, then collected all sequencing data mapping to each sampling point around the relevant features, and calculated 95% confidence intervals on likely values of the mean signal by bootstrapping. In Supplementary Figure S7b and c, only the profiles of mean signals but not confidence intervals were shown for the sake of clarity.
Software and data availability
BEADS is not limited to the specific choices of parameters specified above. For example, sequence reads can be of any length other than 35 bp and extended to fragments of any expected sizes other than 200 bp; and users can use any aligner of their choice to map sequence reads onto the reference genome and customize the criteria to filter mapped reads, etc. as long as the parameters chosen are consistent throughout the whole process.
BEADS is publicly available at http://beads.sourceforge.net/.
RESULTS AND DISCUSSION
We focus here on the application of high-throughput sequencing using the Illumina Genome Analyser to chromatin immunoprepcipitation (ChIP-seq) for genome-wide identification of DNA–protein interaction sites, although the technical issues are also relevant to other sequencing applications and platforms. In a typical ChIP-seq protocol, extracts are prepared by sonication of formaldehyde cross-linked chromatin. DNA fragments associated with a target protein are co-immunoprecipitated from this extract using an antibody; in parallel, a portion of the total DNA in the extract is used as an input control sample. The sequences of the purified immunoprecipitated DNA and input control DNA are determined by sequencing. Usually, ∼35 bp of sequence is read from millions of DNA fragments in the ∼150- to 300-bp range (28), then these sequence reads are mapped onto a reference genome.
Because a ChIP input control library should, in theory, equally represent all regions of the genome, we expected that its sequence reads would be distributed relatively uniformly over the genome. However, we observed that C. elegans ChIP input control DNA sequence data (hereafter referred to as input sequence) display strong reproducible patterns (Figure 1a). These patterns do not appear to be due to cross-linking and incomplete solubilization of DNA fragments in the input extract because similar patterns are evident in published genomic DNA sequence data (7), where the DNA was deproteinated prior to fragmentation and not cross-linked (Figure 1a). The reproducibility of the patterns indicates that they are not due to random sampling variations. Human input sequence has also been shown to have biased genomic coverage (11).
We found that the input sequence read counts correlate with the GC contents of the underlying genomic sequences (Figure 1a and Supplementary Table S1). To examine this potential GC bias in more detail, we compared the GC frequency distribution of the C. elegans input sequence reads with the expected distribution of the genome. This demonstrated an over-representation of GC-rich and an under-representation of AT-rich sequences in the input sequence data (Figures 1b and c). A similar bias was seen with C. elegans and human genomic DNA sequence (Supplementary Figures S1a and S2).
Because genomic features can deviate substantially from the mean genomic GC content, a GC bias in sequencing data could lead to an artefactual inflation of read counts in such regions. For example exons are generally more GC-rich than introns (14), as visualized by plotting GC content across a set of aligned internal exons and their flanking introns (Figure 2a and g). As expected from the GC bias in input sequence data, we found a strong enrichment of sequence reads across C. elegans and human exons (Figure 2c and i). Similarly, raw input sequence data showed enrichment near aligned transcript start sites (TSSs) which resembles the GC content plots of the same regions (Figure 2d, f, j and l). We also observed exonic and TSS peaks in raw C. elegans genomic DNA sequence (Supplementary Figure S1b and c).
Figure 2.

Bias in GC content (a, d, g, j), mappability (b, e, h, k) and raw input sequence signals (c, f, i, l) across internal exons and around transcript start sites in C. elegans and human. The error bars represent the 95% confidence intervals of the estimated mean GC, mappability or sequence signal values.
A second source of potential bias lies in the read mapping procedure. Short sequence reads obtained in ChIP-seq experiments can only be confidently mapped on the reference genome if their sequences are unique. After mapping, reads are usually either extended to the expected fragment size or shifted towards the centre of the distributions made up of forward- and reverse-strand reads (28). Signals are then sampled at regular intervals to construct a genomic profile. Therefore, non-mappable regions and adjacent areas will have lower than expected read counts. We quantified mappability across the entire genome (see ‘Materials and Methods’ section) and found that it is higher in exons than in introns in both C. elegans and human genome (Figure 2b and h). A region of higher mappability was also noticed near TSSs (Figure 2e and k), as observed previously in human genome (11). Correction of the biases in GC content and mappability of high-throughput sequencing data is clearly necessary for determining true enrichment patterns in ChIP experiments.
We first explored whether a simple division by input control would correct biases in sequence data. We sequenced several different libraries of input control DNA (biological replicates) and found that each had a different extent of GC bias (Supplementary Figure S3a). Moreover, we found that data from multiple sequencing runs of a library (technical replicates) can have different GC compositions (Supplementary Figure S3b; data from human 1000 genomes project). These observations suggest that division by input would not correctly remove bias. Indeed, we found that dividing one input control by input controls with different GC biases resulted in apparent exonic enrichment or depletion (Supplementary Figure S4). Similarly, we show below that dividing ChIP-seq data sets by different input controls leads to different resulting enrichment patterns. Therefore, data set-specific biases in both input and ChIP-seq data need to be corrected independently.
To estimate GC bias in a data set and determine correction factors individually for each library, we compared GC histograms of the sequence reads to the expected GC distribution of the genome. For each GC bin a correction factor was calculated by dividing the frequency expected to that observed. This factor was applied to each sequence read in a given bin, with the effect of shifting the experimental distribution to that of the genome. After applying this GC correction to C. elegans and human input sequence data, the enrichments on exons and near TSSs were greatly reduced in magnitude (green plots in Figure 3a, b, g and h), indicating that GC bias plays a major role in creating systematic biases.
Figure 3.

BEADS normalization of high-throughput sequence reads of C. elegans input sequence (a, b), H3K4me3 ChIP (c, d), H3K36me3 ChIP (e, f) and human input sequence (g, h) libraries. Shown are the results following each step of correction: Raw (uncorrected), GC corrected, GC + mappability corrected, and GC + mappability + local corrected signals. Plots show signals across internal exons and around transcript start sites of all genes; except in d and f, only transcript start sites of highly expressed genes were used (see ‘Materials and Methods’ section). The normalized signals are plotted as relative normalized read counts (left-hand y-axis). The fully normalized signal is also plotted as fold-change relative to the genomic average (right-hand y-axis). Solid lines show average signal and shaded regions show 95% confidence intervals. Genomic read-count averages for the C. elegans input-control, H3K4me3, H3K36me3 and human input-control libraries are 33.0, 29.8, 14.6 and 0.4, respectively.
To calculate a mappability correction, we simulated sequence reads where each possible sequence in the genome was represented once, and mapped these onto the reference genome using the same criteria for processing experimental data (see ‘Materials and Methods’ section). We then extended each read to the estimated fragment size and quantified mappability of a given genomic position by counting the number of possible overlapping extended mapped reads. At each genomic location, we scaled the GC-weighted input sequence signal according to mappability, with higher weights given to locations of low mappability. We introduced an adjustable cutoff filter to remove regions with very low mappability prior to subsequent analyses because the low read coverage of these regions makes it difficult to reliably correct the signals (see ‘Materials and Methods’ section). We found that the mappability adjustment step further flattened the exonic peak of input sequence data (blue plots in Figure 3a and g). This step also further levelled the signals around the aligned TSSs (Figure 3b and h), but the effect was smaller presumably because mappability varies less around TSSs than across exons (Figure 2b, e, h and k). The GC and mappability corrections removed most of the bias in the input sequence data, but a small amount of bias still remained after these steps.
As part of sequencing library generation, DNA molecules are physically broken down into smaller fragments (e.g. by sonication or other methods) and this process could generate bias due to differences in underlying DNA structure. For example, heterochromatin is found to be more refractory to shearing, resulting in an under-representation of these DNA fragments compared to euchromatin (12). Smaller-scale structural differences due to nucleotide composition might also cause DNA to be differentially susceptible to breakage. Such chromatin or DNA structural effects might contribute to the coverage inhomogeneity in high-throughput sequencing data.
In order to correct for such local biases, after GC and mappability correction, we pooled several independently generated input sequence data sets to create a ‘master’ control data set to reflect reproducible local biases. We then applied a local correction by dividing the signals of the experimental data set by the master control data set at each position in the genome. The input sequence data being studied was not included in the master control data set. We found that the local correction step further removed the unexpected enrichments of C. elegans input sequence data seen on exons and near TSSs (Figures 3a and b). The signals from fully normalized input sequence data also no longer followed the patterns of GC content or mappability of underlying sequences (Figure 1a). For human input sequence, the division step did not have a large correctional effect on the overall signal profile. The GC and mappability corrections appeared sufficient to remove biases across TSSs and exons (Figure 3g and h). It is possible that the DNA structural differences were too subtle to be captured by the sequenced data due to its poor read coverage (∼0.04×).
We next addressed normalization of C. elegans ChIP-seq data. For this, we focused on three well-studied histone modifications, trimethylation of lysine 4, lysine 9 and lysine 36 of histone H3 (H3K4me3, H3K9me3 and H3K36me3). We previously showed using chromatin immunoprecipitation followed by microarray hybridization (ChIP-chip) in C. elegans, that similar to other organisms, H3K4me3 shows discrete peaks of enrichment near TSSs of transcribed genes, H3K9me3 shows higher enrichment across silent genes than active genes, and H3K36me3 is broadly enriched on the actively transcribed regions of genes (22). Further, there is a significant enrichment of H3K36me3 on exonic compared to intronic chromatin. In contrast, H3K4me3 and H3K9me3 do not show general exon enrichment.
Chromatin immunoprecipitation enriches for DNA bound to factors of interest, but 60–99% of ChIP reads are estimated to be background noise (29). Given the observed biases in input sequence data, this background is likely to cause artefactual patterns. Indeed, in addition to the expected signals, we found that H3K4me3 and H3K9me3 ChIP-seq data also showed enrichment on exons that was not seen in ChIP-chip mapping experiments (22) (Figure 3 and Supplementary Figure S5). An expected exonic enrichment was observed in raw H3K36me3 ChIP-seq data (Figure 3e), but it is possible that background bias contributes to this signal, making analysis of amount of enrichment difficult.
We tested the effects of normalization through division by input control sequence data. We carried out ChIP of H3K4me3 and H3K36me3 each from a different wild-type extract, and then we prepared libraries and sequenced ChIP and input DNA samples. When the input sequence of matched extract was used as a divisor for H3K4me3, we observed an unusual pattern: loss of the expected enrichment just downstream of the TSS and lower signal in exons relative to introns. Using the input sequence of non-matched extract as a divisor resulted in an expected promoter peak and removed most of the exonic enrichment (Supplementary Figure S6a and b). For H3K36me3, dividing by the extract matched input control produced an exonic enrichment as expected but using the other input resulted in an exon depletion (Supplementary Figure S6c and d). These different and unexpected enrichment patterns are likely to be due to the input and ChIP samples having different amounts of technical bias. We conclude that dividing ChIP signals by input signals is not a generally appropriate method for correcting systematic biases in sequencing data.
We next applied BEADS to remove bias from H3K4me3, H3K9me3, and H3K36me3 ChIP-seq data. Because a factor of interest might have a true binding preference for AT- or GC-rich sequences (e.g. H3K36me3 is enriched on exons, which are also GC-rich), using the entire set of ChIP-seq reads as we did for input control data could cause either over- or under-correction. Therefore, we attempted to separate the reads into two components, potential enriched regions and background, and used only background reads for estimating GC bias. We defined sequence reads as background if they did not overlap regions of potential enrichment identified using a peak calling program [(20); see ‘Materials and Methods’ section]. After deriving GC correction factors from background reads, we applied the correction to the entire set of ChIP sequence reads. Therefore, an AT or GC preference for factor enrichment would not affect estimation of GC bias in the sequence data set. After BEADS normalization of H3K4me3 ChIP-seq data, the TSS peak was still present, as expected, but the exon peak disappeared (Figure 3c and d). Similarly, H3K9me3 enrichment on exons was removed by BEADS normalization, but enrichment on silent genes was maintained (Supplementary Figure S5). In contrast, exonic enrichment of H3K36me3 was still observed after BEADS normalization (Figure 3e and f).
We found that generation of the background data set was relatively insensitive to over- or under-identification of peak regions (Supplementary Figure S7). In addition, deriving GC correction from only a subset of sequence reads did not introduce apparent artefacts. Using only reads in the H3K4me3 or H3K36me3 background regions to calculate GC correction factors for input sequence data yielded similar results to using the entire set of input sequence reads (Supplementary Figure S8). However, we found that use of the correct average library fragment size is necessary for effective bias removal. Under- or over- extending read lengths led to the generation of artefactual patterns (Supplementary Figure S9).
We also tested the effect of BEADS normalization on the distributions of two proteins with punctate binding patterns: DPY-27, a component of the C. elegans dosage compensation complex that has been shown to bind specifically to foci on chromosome X (23), and BLMP-1, a C. elegans transcription factor (26). Similar to raw H3K4me3 ChIP-seq data, raw DPY-27 and BLMP-1 ChIP-seq data showed an unexpected enrichment on exons in addition to the sharp peak detected on known binding sites (Supplementary Figure S10). After applying BEADS normalization to the data set, the expected peaks on chromosome X foci for DPY-27 and on binding sites of BLMP-1 were still sharp and high in magnitude, whereas the unexpected exon peaks were removed (Supplementary Figure S10).
We have demonstrated that BEADS removes systematic biases present in high-throughput sequencing data. Before normalization, raw input sequence and ChIP data sets showed artefactual enrichments not reflective of true biology. After BEADS normalization, these enrichments were removed, but previously documented enrichments determined by other methods (such as ChIP-chip or qPCR) remained intact. Bias correction was thus essential for the analysis of these ChIP-seq data. Furthermore, when factor enrichments are low, false signals from systematic biases are likely to dominate. However, we note that since BEADS uses ‘background’ reads to derive GC correction factors, it is difficult to apply to data sets where background reads cannot be easily identified (e.g. nucleosome or RNA-seq data).
We expect BEADS to be useful in other applications of high-throughput sequencing besides ChIP-seq. For example, BEADS normalization could aid the detection of genome copy number variations, where bias in the distribution of mapped sequence reads could mask or enlarge differences in copy number. Bias correction using BEADS might also improve the performance of peak-calling algorithms.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Human Genome Research Institute modENCODE grant 1-U01-HG004270-01; Wellcome Trust Senior Research Fellowship (054523) and Cambridge Newton Trust funding to J.A.; Wellcome Trust Research Career Development Fellowship to T.A.D.; core funding from the Wellcome Trust and Cancer Research UK to J.A. and T.D. Funding for open access charge: Wellcome Trust.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Raymond Auerbach for the human input sequence data and Oliver Hobert for the C. elegans genomic DNA sequence data. The authors are also grateful to Ron Chen and Paulina Kolasinska-Zwierz for helpful comments on the manuscript, and Marco Canini for technical discussion on the software package. MSC developed the software, performed the analyses and drafted the manuscript. TAD provided insights throughout the project and drafted the manuscript. IL performed the ChIP experiments and contributed to data analyses. JA conceived the study, gave direct supervision and drafted the manuscript. All authors read and approved the final manuscript.
REFERENCES
- 1.Auerbach RK, Euskirchen G, Rozowsky J, Lamarre-Vincent N, Moqtaderi Z, Lefrançois P, Struhl K, Gerstein M, Snyder M. Mapping accessible chromatin regions using Sono-Seq. Proc. Natl Acad. Sci. USA. 2009;106:14926–14931. doi: 10.1073/pnas.0905443106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, Soldatov A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Platts AE, Land SJ, Chen L, Page GP, Rasouli P, Wang L, Lu X, Ruden DM. Massively parallel resequencing of the isogenic Drosophila melanogaster strain w(1118); iso-2; iso-3 identifies hotspots for mutations in sensory perception genes. Fly. 2009;3:192–203. doi: 10.4161/fly.3.3.9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarin S, Prabhu S, O’Meara MM, Pe’er I, Hobert O. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat. Methods. 2008;5:865–867. doi: 10.1038/nmeth.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz S, Meshorer E, Ast G. Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 2009;16:990–995. doi: 10.1038/nsmb.1659. [DOI] [PubMed] [Google Scholar]
- 9.Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, Swerdlow H, Turner DJ. A large genome center's improvements to the Illumina sequencing system. Nat. Methods. 2008;5:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rozowsky J, Euskirchen G, Auerbach RK, Zhang ZD, Gibson T, Bjornson R, Carriero N, Snyder M, Gerstein MB. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat. Biotechnol. 2009;27:66–75. doi: 10.1038/nbt.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teytelman L, Ozaydin B, Zill O, Lefrançois P, Snyder M, Rine J, Eisen MB. Impact of chromatin structures on DNA processing for genomic analyses. PLoS One. 2009;4:e6700. doi: 10.1371/journal.pone.0006700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harismendy O, Ng PC, Strausberg RL, Wang X, Stockwell TB, Beeson KY, Schork NJ, Murray SS, Topol EJ, Levy S, et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu L, Zhang Y, Zhang W, Yang S, Chen JQ, Tian D. Patterns of exon-intron architecture variation of genes in eukaryotic genomes. BMC Genomics. 2009;10:47. doi: 10.1186/1471-2164-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat. Biotechnol. 2008;26:1293–1300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tuteja G, White P, Schug J, Kaestner KH. Extracting transcription factor targets from ChIP-Seq data. Nucleic Acids Res. 2009;37:e113. doi: 10.1093/nar/gkp536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods. 2008;5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;25:1952–1958. doi: 10.1093/bioinformatics/btp340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nix DA, Courdy SJ, Boucher KM. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics. 2008;9:523. doi: 10.1186/1471-2105-9-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol. 2008;26:1351–1359. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ercan S, Giresi PG, Whittle CM, Zhang X, Green RD, Lieb JD. X chromosome repression by localization of the C. elegans dosage compensation machinery to sites of transcription initiation. Nat. Genet. 2007;39:403–408. doi: 10.1038/ng1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rechtsteiner A, Ercan S, Takasaki T, Phippen TM, Egelhofer TA, Wang W, Kimura H, Lieb JD, Strome S. The histone H3K36 methyltransferase MES-4 acts epigenetically to transmit the memory of germline gene expression to progeny. PLoS Genet. 2010;6:e1001091. doi: 10.1371/journal.pgen.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicol JW, Helt GA, Blanchard SG, Jr, Raja A, Loraine AE. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009;25:2730–2731. doi: 10.1093/bioinformatics/btp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat. Methods. 2009;6:S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.