

Summary
The human mitochondrial genome comprises a distinct genetic system transcribed as precursor polycistronic transcripts that are subsequently cleaved to generate individual mRNAs, tRNAs and rRNAs. Here we provide a comprehensive analysis of the human mitochondrial transcriptome across multiple cell lines and tissues. Using directional deep sequencing and parallel analysis of RNA ends, we demonstrate wide variation in mitochondrial transcript abundance and precisely resolve transcript processing and maturation events. We identify previously undescribed transcripts, including small RNAs, and observe the enrichment of several nuclear RNAs in mitochondria. Using high-throughput in vivo DNaseI footprinting, we establish the global profile of DNA-binding protein occupancy across the mitochondrial genome at single nucleotide resolution, revealing regulatory features at mitochondrial transcription initiation sites and functional insights into disease-associated variants. This integrated analysis of the mitochondrial transcriptome reveals unexpected complexity in the regulation, expression, and processing of mitochondrial RNA, and provides a resource for future studies of mitochondrial function (accessed at mitochondria.matticklab.com).
Introduction
Following its endosymbiosis from an α-proteobacterial ancestor, the mitochondrial genome has been streamlined into a small, high copy, bioenergetically specialized genetic system that allows individual mitochondria to respond, by gene expression, to changes in membrane potential and maintain oxidative phosphorylation (Lane and Martin, 2010). Given this dedicated role, it is not surprising that the mitochondrial genome is regulated and expressed in a unique manner.
Human mitochondria contain a compact circular genome that is 16,569 bp in length (Anderson et al., 1981). Replication and transcription of mitochondrial DNA (mtDNA) is initiated from a small noncoding region, the D-loop, and is regulated by nuclear-encoded proteins that are post-translationally imported into mitochondria. Mitochondrial RNAs are transcribed as long polycistronic precursor transcripts from both strands (historically termed ‘heavy’ and ‘light’) that are processed according to the ‘tRNA punctuation model’ whereby 22 interspersed tRNAs are excised to concomitantly release individual rRNAs and mRNAs (Ojala et al., 1981). The liberated RNAs then undergo maturation that involves polyadenylation of the 3′ ends of mRNAs and rRNAs, and specific nucleotide modifications and addition of CCA trinucleotides to the 3′ ends of tRNAs (Nagaike et al., 2005). Together, this comprises a unique genetic system that is able to translate the mitochondria-encoded genes into 13 protein subunits of the electron transport chain.
Little is known about the fine features of the mitochondrial transcriptome, in particular about the regulation of transcript abundance, sites of RNA processing and modification, and the possible presence of noncoding RNAs. The recent advent of deep sequencing has provided a global profile of the nuclear transcriptome, revealing unforeseen transcriptional complexity that includes prevalent post-transcriptional processing and abundant noncoding RNAs (Jacquier, 2009). We have taken a similar approach to investigate the mitochondrial transcriptome. Although the atypical features of mitochondrial gene expression require special considerations in sequencing and analysis, the small size of the genome affords a depth of coverage that provides unprecedented opportunities to sample the total RNA population and characterize even rare and transient events.
Here we provide the first comprehensive map of the human mitochondrial transcriptome by near-exhaustive deep sequencing of long and short RNA fractions from purified mitochondria. Despite their common polycistronic origin, we observe wide variation between individual tRNAs, mRNAs and rRNA amounts, attesting to the importance of post-transcriptional cleavage and processing mechanisms in the regulation of mitochondrial gene expression. By parallel analysis of RNA ends (PARE) sequencing, we provide a global profile that precisely resolves these cleavage processes, as well as indicating further unexpected non-canonical cleavage events. During this analysis we were also able to discern the contribution of nuclear RNAs to the mitochondrial transcriptome, accounting for co-purifying contaminants by a two-phase sequencing approach.
Finally, we have analyzed in vivo DNaseI protection patterns by massively parallel sequencing to generate a profile of protein-DNA interactions across the entire mitochondrial genome at single nucleotide resolution. Analysis of these sites of DNA-protein interaction, in combination with transcriptional profiling, provides novel regulatory insight into mitochondrial genome dynamics. The integration of these maps reveals unexpected complexity of the regulation, expression and processing of the mitochondrial transcriptome, and comprises an important resource for the future study of mitochondrial function and disease. This resource comprises 10 new datasets that, combined with meta-analyses of 20 publicly available sets (Figure 1; Table S1), are accessible within the mitochondria-specific genome browser hosted at mitochondria.matticklab.com.
Figure 1.

Map of the human mitochondrial genome indicating the following features (from centre with outer tracks corresponding to heavy strand and inner track corresponding to light strand); DNase sensitivity profile (central black track, opacity indicates strength according to central key), RNA secondary structures predicted by SISSIz (green track), annotated gene features (blue/red gradient indicates fraction of total mitochondrial gene expression according to central key), RNA sequencing (black histogram on grey background, scale 0-900000[log10]), 5′ RNA cleavage sites determined by PARE (black histogram on pink background, scale 0-8000), 3′ cleavage sites determined by non-template polyadenylation RNAseq reads (black histogram on purple background, scale 0-8000) and small RNA profile (black histogram on blue background, scale 0-2500).
Results
Mitochondrial gene expression
To capture the distinct features of the mitochondrial transcriptome we first performed RNA sequencing (RNAseq) on purified human 143B cell mitochondria. Mitochondrial preparations from human 143B cells were initially pre-treated with RNaseA to reduce contaminating RNA, and a clean separation of mitochondria from cytoplasmic and nuclear contaminants was confirmed by immunoblotting and qRT-PCR assessment of known compartment-specific proteins and RNAs (Figure S1A,B). We then employed a strand-specific RNA sequencing approach with random hexamer priming, no rRNA or tRNA depletion, and smaller cDNA fragmentation size to provide an expression profile that encompassed all annotated mRNAs, tRNAs and rRNAs (Figure 2). In total, we aligned 1.4 million sequenced reads uniquely to the human mitochondrial genome, finding almost the entire mitochondrial genome to be transcribed (99.9% heavy and 97.6% light strands; Figure S1C). Furthermore, less than 3.8% of the mitochondrial genome sequence was represented singly within the sequenced libraries, indicating the near-exhaustive depth of this sequencing.
Figure 2.

(A) Mitochondrial gene expression. Proportion of RNAseq reads aligning to three primary polycistronic transcripts transcribed for the heavy major (HSP; light blue), heavy minor (HSP; dark blue) and light strand promoters (LSP; grey). (B) Proportion of RNAseq reads mapping to rRNA, tRNA, mRNA and intergenic or antisense regions of the mitochondrial genome. (C) Mitochondrial gene expression (blue) determined by RNAseq in human 143B mitochondria (mitoplast) preparations (error bars indicate 95%CI). Antisense transcription (green) to each gene is also shown with notable antisense transcription to ND5 indicated (asterisk). (D) Genome browser illustration of heavy and light strand showing ND5 and ND6 mRNAs (blue) and complementary antisense regions. Black histogram indicates RNAseq read distribution across browser view, including extended region of high transcription antisense to ND5 mRNA, PARE (red) and poly-A (blue) histogram indicate transcript 5′ and 3′ ends, respectively. (E) Hierarchical clustering of mitochondrial gene expression across 16 human tissues. (Pearson's correlation [r2] indicated. Rpbm; sequenced reads per base per million) (F) Histogram indicates the proportion of total polyA+ RNA mapping uniquely and exactly to the mitochondrial genome across 16 human tissues.
Mitochondrial RNAs are derived from precursor transcripts that traverse almost the entire heavy and light mtDNA strands. These precursor transcripts are subsequently processed into individual mRNAs that exhibit considerable variation in steady-state expression levels (Figure 2B,C; Table S2A; Figure S1D,E). A similarly wide variation in the abundance of different mitochondrial tRNAs was also observed (Figure 1; Table S2A). Given their common source, the wide variation in steady-state levels of mature RNAs indicates the importance of post-transcriptional mechanisms, including those affecting transcript stability, in regulating tRNA and mRNA abundance (Piechota et al., 2006).
The strand-specific sequencing of the mitochondrial transcriptome also provided an opportunity to detect stable antisense transcripts. We distinguished several highly expressed, stable antisense RNAs against the background of unprocessed polycistronic transcripts (Figure 2C; Table S2A). Like their mRNA counterparts, these antisense RNAs exhibit considerable variation in gene expression. A notable example is the antisense transcript associated with the ND5 and ND6 genes, which are organized tail-to-tail, where strand-specific sequencing, confirmed by qRT-PCR (Figure S1E), showed a highly expressed RNA antisense to the length of the ND5 gene, mirroring the ND5 3′UTR that is antisense to the ND6 gene (Figure 2D).
We next examined mitochondrial gene expression across 16 human tissues sequenced as part of the Illumina Body Tissue Atlas (Supplementary Methods). We performed hierarchical clustering to identify tissue-specific differences (Figure 2E; Table S2B), noting some differences in mitochondrial transcript abundance in samples from human tissue and 143B cells due to the alternative use of polyadenylated or total RNA during library preparation (see below). We found that the variation in mitochondrial gene expression was consistent between tissues, with the analysis segregating two distinct gene clusters, and that mitochondrial transcript abundance is higher in tissues with high-energy demands, such as heart and muscle. Because RNA libraries are derived from whole tissues, we could also determine the proportion of the tissue mRNA content derived from mitochondria. Since both mitochondrial genome copy number and transcriptional activity contribute to this content, this analysis provides an indirect metric of the demand each tissue places upon the mitochondria (Figure 2F). In the heart, mitochondrial transcripts comprise almost 30% of total mRNA, whereas mitochondria contribute a lower bound of ∼5% to the total mRNA of tissues with lower energy demands (adrenal, ovary, thyroid, prostate, testes, lung, lymph and white blood cells).
To determine if tissue-specific differences in mitochondrial expression are coordinated with changes in nuclear gene expression, we added 1,013 nuclear-encoded genes implicated in mitochondrial function to our gene expression analysis (Pagliarini et al., 2008) (Table S2C). Hierarchical clustering of nuclear gene expression showed a significant correlation to mitochondrial gene expression (r2 = 0.78, p-value < 0.01), resolving two distinct gene sets that correspond to the pair of mitochondrial gene clusters (Figure S1F) that segregate according to tissue energy demand, reflecting a close coordination between nuclear and mitochondrial genomes in relation to the energy requirements of each tissue.
Nuclear-encoded RNAs in mitochondria
The sequencing of purified mitochondrial RNA returned a surprisingly large number of reads that uniquely aligned to the nuclear genome (Figure 3A). Although it has been shown that nuclear RNAs may be imported into human mitochondria (Entelis et al., 2001), the strong enrichment for genes associated with protein translation (p-value < 10-78) suggests that cytosolic ribosomes closely associated with the mitochondrial outer membrane co-purify with mitochondria and confound efforts to identify bona-fide imported transcripts (Sylvestre et al., 2003). Therefore, to distinguish RNA within mitochondria from co-purifying contaminants, we employed a subtractive approach that compared the RNA content of whole mitochondria to that of mitochondrial preparations stripped of their outer membrane (mitoplasts). This two-phase approach is analogous to that successfully employed to characterize the mitochondrial proteome (Pagliarini et al., 2008) and is based on the observation that RNAs within the mitochondrial matrix are enriched, whereas RNAs that co-purify with the outer membrane are depleted, during mitoplast preparation.
Figure 3.

(A,B) Relative enrichment of nuclear-encoded RNAs in mitochondria or mitoplast preparations. Proportion of sequenced reads aligning to nuclear (red) and mitochondrial (blue) genomes in libraries derived from mitochondria (A) and mitoplast (B) preparations. Comparison between libraries indicates the depletion of co-purifying contaminant transcripts in mitoplast preparations. (C) Box whisker plot indicates enrichment for mitochondrial encoded (blue) mRNAs, rRNA and tRNA in mitoplast relative to whole mitochondrial RNA preparations. While the majority of nuclear-encoded (red) mRNAs and rRNAs are depleted in mitoplast preparations, nuclear tRNAs are enriched in mitoplasts. (D,E) qRT-PCR confirmation for mitoplast depletion of mRNAs (D) and miRNAs (E) for which we observed the highest expression in whole mitochondrial preparations. (F) Scatter-plot indicating the normalised expression for nuclear (red) and mitochondrial (blue) tRNAs, showing the enrichment of several nuclear tRNAs, including tRNA-LeuUAA (p-value<0.001) and tRNA-GlnUUG (p-value=0.003) in mitoplasts. (G) qRT-PCR shows the depletion of nuclear tRNA-AlaAGC and tRNA-LysUUU in mitoplasts, and enrichment of tRNA-GluUUG (p-value=0.025) and presence of tRNA-LeuUAA within mitoplasts. 12S rRNA included as positive control (all error bars indicate SEM).
We performed matched sequencing of RNA isolated from mitoplasts and aligned a total of 4.7 million sequenced reads to the genome, representing a 5.9-fold enrichment relative to whole mitochondrial preparations (Figure 3B; Figure S2A,B). While the expression of mitochondrial genes was closely correlated between whole mitochondrial and mitoplast libraries (r2 = 0.98, p-value < 0.001; Figure S2C), there was a selective depletion of nuclear-encoded mRNAs and rRNAs, indicating that these transcripts are associated with the mitochondrial outer membrane (Figure 3C). Furthermore, there was no enrichment of cytoplasmic mRNAs encoding mitochondrial proteins in mitoplasts, consistent with their proteins being post-translationally imported into mitochondria. We confirmed by qRT-PCR the strong depletion in mitoplasts for 6 mRNAs and 3 miRNAs that showed the greatest enrichment in mitochondrial preparations (Figure 3D,E).
We next compared matched mitochondrial and mitoplast libraries to determine whether other nuclear transcripts are enriched in mitoplasts (Supplemental Experimental Procedures). Firstly, to validate the two-phase sequencing approach, we considered three noncoding RNAs (ncRNAs), 5S rRNA, MRP and RNase P, that have been previously shown to be present in the mitochondrial matrix (Wang et al., 2010), finding all transcripts, though lowly expressed, enriched in mitoplasts (Figure S2D). We also investigated whether other nuclear-encoded ncRNAs, including snoRNAs, scRNAs and novel ncRNAs predicted by Evofold (Pedersen et al., 2006) or RNAz (Washietl et al., 2005), were present within mitoplasts. Although the vast majority of such ncRNAs are either absent or depleted from mitoplasts, several novel ncRNAs were enriched in mitoplasts, although the relative amount of these ncRNAs was a fraction of that observed for mitochondria-encoded RNAs (Figure S2E-G).
Lower eukaryotes, such as trypanosomes and yeast, require the import of tRNAs into mitochondria (Entelis et al., 1998; Hancock et al., 1992) and tRNA-Gln was recently shown to be naturally imported into human mitochondria (Rubio et al., 2008). We compared the expression of nuclear tRNAs between mitochondria and mitoplasts to identify additional candidate tRNAs present within mitochondria (Supplemental Experimental Procedures). We found several nuclear-encoded tRNA species to be enriched within mitoplasts relative to mitochondrial preparations, including the previously detected tRNA-GlnUUG, an enrichment that was confirmed by qRT-PCR (Figure 3F,G). Furthermore, we noted a prevalence of sequencing mismatches at known modified nucleotide positions indicating that these tRNAs were present in a mature processed form.
Processing of primary mitochondrial polycistronic transcripts
Next we investigated the processing of polycistronic precursor transcripts into individual functional RNAs (Temperley et al., 2010b). To profile the opening stage of polycistronic processing, the RNase P-dependent cleavage of tRNA 5′ ends (Rossmanith and Holzmann, 2009), we performed parallel analysis of RNA ends (PARE) (German et al., 2008) on purified human 143B mitochondria to capture the exposed 5′-monophosphate residue of cleaved transcripts. Despite this methodology being originally developed for identification of miRNA-directed cleavage sites in mRNAs, sequenced PARE reads exhibit a distinct enrichment at the 5′ end of tRNAs, confirming the ability of this technique to detect polycistronic processing sites (Figure 4A,B). In total, using PARE, we identified 80 high-confidence cleavage sites across the mitochondrial transcriptome, of which 51.6% corresponded to 5′ tRNA cleavage events (Table S3A). To provide a complementary profile of 3′ tRNA cleavage events, we ‘rescued’ sequenced reads from our directional sequencing of purified 143B mitochondria that contain non-templated 3′-CCA nucleotides added during tRNA maturation. The majority of rescued reads (77.8%) were associated with tRNA 3′ termini (Figure 4C; Table S3B), providing a quantitative indication of tRNA 3′ cleavage frequency, which exhibited high correlation with total tRNA expression (r2 = 0.94, p-value < 0.001) and 5′ processing (r2 = 0.74, p-value < 0.001). In combination, these two techniques provided a global profile of tRNA processing from precursor transcripts (Figure 1). We did, however, note a number of cases, such as the adjacent tRNA-Ala and –Asn loci, where 5′ and 3′ processing do not correlate, suggesting incomplete or aberrant processing of tRNAs and the existence of stable conjoined tRNA transcripts (Figure S3A,B).
Figure 4.
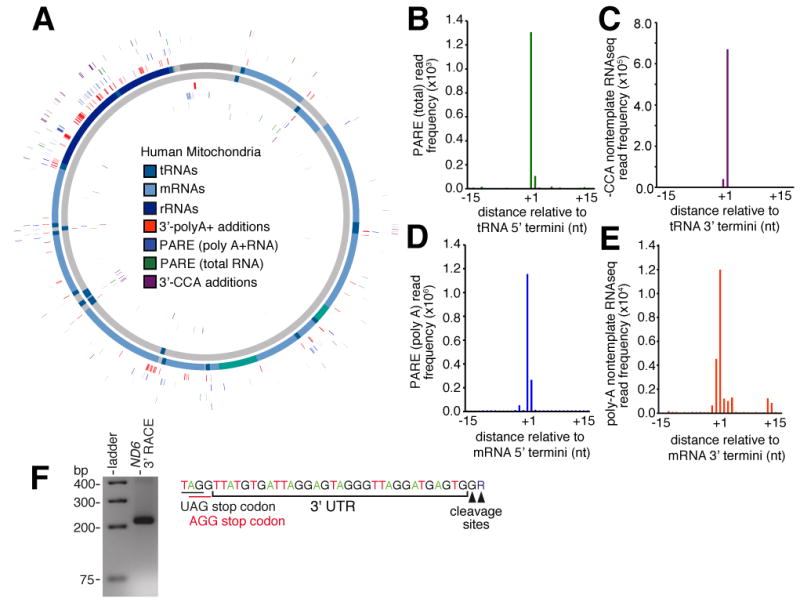
(A) Mitochondrial position of 5′ cleavage sites. Cleavage sites determined by PARE with total (green) and polyA+ (blue) human RNA and 3′ cleavage sites determined by RNAseq reads containing non-template –CCA (purple) and polyA additions (orange) are indicated. (B-E). Frequency distribution of (B) PARE reads from total RNA across window centred on annotated tRNA 5′ end, (C) non-template –CCA sites across window centred on annotated tRNA 3′ end. (D) Frequency distribution of PARE reads from poly-A+ RNA across window centred on annotated mRNA 5′ end, (E) non-template poly-A sites across window centred on annotated mRNA 3′ end. (F) Annotation of ND6 3′UTR by 3′ RACE. The 3′UTR sequence with stop codon (black) and -1 shifted codon (red) is indicated (top panel). Gel electrophoresis of product retrieved from ND6 3′RACE shown in lower panel.
Next, we examined the cleavage of mitochondrial mRNAs. We supplemented our analysis with publicly available PARE libraries generated from human Hek, HeLa cells and brain (Karginov et al., 2010; Shin et al., 2010) that, despite being generated from whole cell preparations, provided better coverage of mitochondrial mRNAs as a result of greater sequencing depth and the use of polyA+ RNA to generate these libraries. For example, in PARE libraries from Hek cells, 15.4 million (34% of total) PARE sequenced reads aligned to the mitochondrial genome. We found cleavage occurred at a single dominant 5′ nucleotide in the majority of mRNAs (Figure 4D) and, in some cases, we noted a divergence from the predicted cleavage sites in current gene annotations (Table S4A). Moreover, we did not observe a cleavage site associated with the CO3 gene, suggesting the absence of a 5′ terminal mono-phosphate residue. In addition to canonical cleavage sites, we also found evidence of intra-exonic cleavage sites within mRNAs. We focused on events that, similar to canonical sites, are characterised by a distinct single nucleotide peak that is positionally conserved in all three samples (Figure S3C,D). However, these events occur at a lower frequency (161-fold lower than canonical cleavage events associated with the 5′ ends of mRNAs) indicating that, though present, non-canonical cleavage events are relatively rare. Similar truncated mRNAs have been detected in mitochondria with aberrant RNA surveillance pathways (Szczesny et al., 2010).
A concluding stage in precursor processing involves 3′ polyadenylation of mRNAs and rRNAs. We identified mitochondrial polyadenylation events by rescuing unmapped sequenced reads from our directional sequencing of 143B mitochondria after removing non-templated 3′ adenine nucleotides and realigning the reads to the mitochondrial genome (Figure S3E). To increase the sensitivity of this approach, we also undertook a complementary analysis of directional read sequenced as part of the Illumina Body Tissue Atlas that, due to greater sequencing depth and polyA priming, allowed us to detect even rare polyadenylation sites. The majority (64.5%) of sites corresponded to the annotated 3′ ends of mRNAs, rRNAs and, surprisingly, some tRNAs (Figure 4E; Figure S3E-G; Table S4B), with the exception of ND6 mRNA, for which we observed no polyadenylation, as previously reported (Temperley et al., 2003). To identify the 3′ terminus of the ND6 transcript, we used 3′ rapid amplification of cDNA ends (RACE), and found a 33/34 nt untranslated region following the AGG stop codon (Figure 4F) that would completely encompass the stable secondary structure predicted to promote -1 frameshifting in mitochondrial ribosomes (Temperley et al., 2010a). In addition to ND6, the variation between RNA polyadenylation frequencies that did not correlate with gene expression (r2 = -0.14, p-value = 0.64) suggests other mitochondrial RNAs are also present in a non-polyadenylated form (Slomovic et al., 2005). To identify non-polyadenylated mRNAs, we compared gene expression between matched polyA+ and polyA- RNAseq libraries (Morin et al., 2008). We found the relative abundance of mRNAs varies between polyA+ and polyA- libraries suggesting the differential existence of non-polyadenylated isoforms (Figure S3H,I). For example, some mRNAs, such as CO1, are enriched in the polyA- fraction, whereas mRNAs that require polyadenylation to complete an in-frame stop codon (Ojala et al., 1981), such as ND4L/4, are diminished in the polyA- fraction (1.7-fold, p-value = 0.060). The differential polyadenylation frequencies also resolved differences between gene expression observed earlier in polyA+ and total RNA sequencing libraries (see above).
The previous identification of non-canonical transcriptional units has implied the existence of cleavage events not accounted for by the tRNA punctuation model (Guan et al., 1998; Ojala et al., 1981; Sbisa et al., 1992). At every stage of polycistronic precursor transcript cleavage, our analysis also uncovered non-canonical processing events. Given canonical processing requires the precursor transcript to locally fold into a tRNA structure, we considered whether non-canonical events identified within our analysis also associate with novel RNA secondary structures. We surveyed the mitochondrial transcriptome for evidence of evolutionary selection for RNA secondary structures using SISSIz (Gesell and Washietl, 2008), predicting 173 RNA secondary structures (Z-score > 2, P95) (Figure 1; Table S4C) of which 85 correspond to tRNAs and 82 to rRNA substructures, which include all previously annotated RNA structures in the mitochondrial genome. We identified 14 new structural clusters within the ND1, ND2, CO3 and ND5 genes, the majority of which were associated with cleavage sites, albeit at lower frequency (Figure S4A-F).
Small RNAs in mitochondria
A diversity of small RNAs (sRNAs) that regulate a range of cellular processes have been characterized in viral, bacterial and nuclear genomes (Zhang, 2009). To determine whether sRNAs are encoded within the human mitochondrial genome, we sequenced sRNAs (size range 15-30 nt) isolated from purified 143B mitochondria. We obtained 575,553 sRNA reads that mapped uniquely to the mitochondrial genome (Figure 5A). Of these, only 0.6% were represented as single reads (compared to 14.1% of sRNAs from 143B whole cell preparations being singletons) indicating that our sequencing of mitochondrial sRNAs is almost exhaustive (Mayr and Bartel, 2009). In total, this mitochondrial sRNA population comprised 3.1% of the whole cell sRNA population.
Figure 5.

(A) Mitochondrial positions of small RNAs (red). Internal pie-graph indicates majority of highly expressed small RNAs primarily associated with tRNAs (dark blue). (B) Size distribution for small RNAs derived from human 143B whole cells (green), mitochondria (blue) and highly expressed (>1,048 reads; red). (C) Small RNA frequency distribution associated with mitochondria tRNA genes indicates two distinct species immediately downstream of 5′ and 3′ processing sites.
The sRNA sequencing yielded an unexpectedly large number of sRNAs that mapped to the mitochondrial genome. To distinguish sRNAs from potential fragmented intermediates of RNA degradation, we considered only sRNAs that were expressed at greater than 1,048 reads per million, the median level of miRNA expression within 143B cells. This filtering step omitted most sRNAs between 15-18 nt in size, and revealed two distinct 21 nt and 26 nt sRNA size classes (Figure 5B). We then compared the expression of sRNA sequences between long and small RNA sequencing libraries to determine whether sRNA expression is a function of overlapping gene expression, as might be expected from transitory intermediates of RNA degradation. We found highly expressed sRNA expression was not significantly correlated (r2 = 0.2, p = 0.243) with overlapping gene expression, in contrast to the omitted sRNAs (r2 = 0.61, p = 0.003). Furthermore, we observed a large and dynamic range of expression of the highly expressed mitochondrial sRNAs within whole cell sRNA libraries derived from eight different cell types (Figure S5A-F). Together, this process annotated 31 novel sRNAs expressed from 17 distinct loci (Table S5A).
When mapped to the mitochondrial genome, we found that the majority (84%) of these abundant sRNAs were derived from tRNA genes (Figure 5A). It was recently shown that nuclear tRNAs are processed by both Dicer- and RNaseZ-dependent pathways into a range of small RNA species that are subsequently incorporated into RNA silencing pathways (Haussecker et al., 2010; Lee et al., 2009). Like nuclear tRNAs, we observed two distinct species of sRNAs associated with mitochondrial tRNA genes, one immediately downstream of the 5′ cleavage site and a second less abundant species immediately downstream of the 3′ cleavage site (Figure 5C; Figure S5A-F). However, unlike nuclear tRNAs, we did not observe a Dicer-dependent sRNA species cleaved from the 3′ stem loop of nuclear tRNAs (Pagliarini et al., 2008) (Figure S5H,I). Furthermore, we did not observe a significant correlation in the expression of the two mt-tRNA sRNA species. To investigate whether 5′ associated sRNAs are derived from mature tRNAs, we looked for sequencing mismatches as an indication of host tRNA nucleotide modifications. We found overwhelming enrichment of sequencing mismatches at known modified nucleotide positions, such as the methylated N1-A9 residue, indicating that the 5′ sRNA species was derived from the tRNA stem (Figure S5G).
Although, during this analysis, we filtered out lowly expressed sRNAs, we expect this omitted fraction to contain additional sRNAs encoded in the mitochondrial genome. For example, we observed lowly expressed sRNAs whose 5′ termini align with the origin of light strand replication (Ol) and 3′ termini map to downstream sites where RNA to DNA transition occurs, likely reflecting primers utilized by POLG during replication initiation (Fuste et al., 2010) (Figure S5J). Therefore, we anticipate the mitochondrial transcriptome includes additional sRNAs other than those annotated within this study.
Identification of DNA-binding protein occupancy by analysis of in vivo DNaseI protection patterns
Transcription initiation and termination are mediated by a suite of proteins that bind directly to sites within the mitochondrial genome (Scarpulla, 2008). Deep analysis of in vivo DNaseI cleavage sites by massively parallel sequencing enables systematic identification of DNaseI footprints and hence regulatory factor occupancy on genomic DNA (Hesselberth et al., 2009). We performed a DNaseI protection assay, whereby DNaseI-cleaved fragments are sequenced to sufficient depth to allow identification of sites protected by proteins. We sequenced DNaseI-cleaved fragments of total DNA from seven human cell lines, following which, given the abundant and exposed nature of mtDNA, we were able to align an average ∼71 million reads to the mitochondrial genome to achieve greater than 4,000-fold coverage (Figure 6A).
Figure 6.

(A) DNaseI footprinting across the mitochondrial genome provides global profile of protein-DNA interactions at single nucleotide resolution. Cleavage tracks indicate normalised percentile DNaseI cleavage frequency relative to the mtDNA (0-75%). Annotated genomic features including mRNA (light blue), tRNA (dark blue) and rRNA (green) with D-loop and MTERF1 binding site are indicated. (B) Detailed view of D-loop region. Top panel histogram showing PARE (red) and polyA+ (blue) read frequency distribution, indicating transcription initiation and termination events. Previously identified conserved elements (CS; green bars) and replication primer site (blue bars; retrieved from www.mitomap.org) and novel motif (#59; grey bar) also indicated. DNaseI cleavage profile indicates the normalised percentile DNaseI cleavage frequency (0-25%).
We observed considerable variation in DNaseI sensitivity across the genome that included numerous short stretches of protected nucleotides (‘footprints’) that marked putative sites of protein-DNA interactions. The DNaseI cleavage patterns were highly reproducible between two biological replicates (r2 = 0.86, p < 0.0001). We identified DNaseI footprints systematically by requiring 8 or more contiguous nucleotides to exhibit a depressed cleavage frequency relative to flanking regions. The contrast between internal to flanking DNaseI cleavage frequency was used to ascribe each footprint a sensitivity score (F-score). By these criteria, we identified an average of 159 putative footprints across the mitochondria genome of each cell line that collectively encompass ∼8.4% of the mitochondrial genome (Table S6A). As expected, we observed the highest density of footprints in the regulatory D-loop region where a number of footprints corresponded to sites previously identified by traditional Southern-based DNaseI footprinting assays (Fisher et al., 1992; Ghivizzani et al., 1994), confirming the validity of the footprints gleaned from the deep sequencing data.
We next performed hierarchical clustering of footprints according to DNaseI sensitivity (Figure 7A). This approach identified a core set of footprints present in all cell lines, including sites overlapping the origin of light strand replication (Figure S6A), termination sites at the D-loop, and previously described mitochondrial transcription factor A (TFAM) and mitochondrial transcription termination factor 1 (MTERF1) binding sites (Figure 7B). This approach also revealed several novel cell-specific sites (Figure 7C; Figure S6B,C).
Figure 7.

(A) Hierarchical cluster diagram indicating the sensitivity of top 100 indentified DNaseI footprints. Footprints corresponding to TFAM and MTERF1 biding sites are indicated. (B) DNaseI footprint at mTERF binding site. Cleavage events associated with transcription termination are indicated by PARE (blue histogram) and non-template polyadenylation RNAseq reads (red histogram). DNaseI cleavage frequency in HeLa cells indicated by black histogram and frequency in other cell lines indicated by density plot. Underlined green nucleotides indicate MTERF1 binding. (C) Commonly recurring novel sequence motif #59 (top panel; error bars show twice sample correction according to WebLogo; Crooks et al., 2004) and example of novel cell-specific footprint (bottom panel) (D) Association of T16189C SNP variant with distorted footprint. An extended footprint (blue bar) is present in 3 cell lines with C variant (green) but absent in cell lines with T variant (blue). (E) Histogram indicates significant increase in DNaseI cleavage frequency in cell lines containing variant 16189T allele as compared to 16189C allele (error bars indicate SEM).
To gain insight into the identity of the regulatory factors occupying mitochondrial DNaseI footprints, we performed de novo motif discovery on protected regions, and aligned the results with known transcription factor recognition sequences from public databases (Table S6B). This analysis returned motifs matching the consensus recognition sequences for CREB, STAT3 and T3R transcription factors, all of which have previously been identified within mitochondria, as well as numerous other transcription factors (Cannino et al., 2007) (Figure S6C; Table S6A,B). This analysis also identified several novel motifs that recur prevalently within mitochondrial DNaseI footprints, suggesting the existence of additional unidentified proteins that regulate the mitochondrial genome and its expression (Figure 7C).
Protein-DNA interactions at single-nucleotide resolution
In addition to providing a global map of DNaseI footprints, DNaseI sensitivity mapping has the potential to reveal fine scale DNA-protein interactions within DNaseI footprints at single-nucleotide resolution (Hesselberth et al., 2009). To verify the reproducibility of the nucleotide-resolution DNaseI cleavage patterns within identified footprints, we compared two HeLa cell biological replicates, finding 86% of internal footprint patterns to be significantly correlated (mean r2=0.88, p < 0.05). To illustrate the single nucleotide resolution of DNaseI cleavage patterns relative to protein structural features, we considered the binding of MTERF1 to the tRNA-LeuUUR termination DNA sequence, the structure of which was recently solved by X-ray crystallography (Yakubovskaya et al., 2010). Aligning the nucleotide-level DNaseI cleavage profile with this resolved structure showed sharply delineated DNaseI protection corresponding to the MTERF1 DNA recognition region and additional internal low frequency cleavage events corresponding to phosphodiester bonds selectively occluded by the MTERF1 DNA-protein interface (Figure 7B; Figure S6D).
Transcription termination within the D-loop
The D-loop, where mitochondrial replication and transcription initiates, is the major site of transcriptional regulation (Scarpulla, 2008), reflected in the complex DNaseI footprint profiles observed within this region (Figure 6B). These complex profiles include footprints associated with TFAM binding sites (Fisher et al., 1992), light strand transcription initiation, transcription termination and sites of RNA to DNA transitions. In HeLa cells, we integrated the DNaseI cleavage data with transcriptomic maps to provide insight into regulation within the D-loop. Firstly, using PARE sequencing, we identified reads from HeLa cells that aligned to the light (4.0 × 105 reads) and minor heavy strand (1.4 × 105 reads) promoters. The similarly high number of PARE reads aligning to both the light and heavy strand promoters supports the bidirectional nature of mitochondrial transcription initiation (Fisher et al., 1987) but contrasts with the dominant expression of the heavy strand relative to the light strand as indicated by RNAseq. Given that transcription termination is the key mechanism that determines the ratio of mitochondrial rRNA to mRNA expression (Scarpulla, 2008), we next considered whether termination also modulates light strand expression stoichiometry. We found the largest component of light strand polyadenylation events (35.5% of total) occurs just ∼160-185 nt downstream of the light-strand promoter, coincident with the abortion of transcription observed in RNAseq (Figure S6E) (Suissa et al., 2009). The strong DNaseI footprints at this site, encompassing several recurrent novel motifs, suggest the involvement of DNA-binding proteins in this termination event that, like MTERF1, contribute to the ratio of asymmetric polycistronic precursor expression (Figure 6B).
Identification of functional sequence variants within DNaseI footprints
The accurate alignment of sequence reads from DNaseI mapping experiments required us to firstly genotype the mitochondrial genome of each cell line and identify sequence variation. This allowed us to consider whether the identified sequence variation occurred within footprints and could potentially interfere with protein-DNA interactions. For this analysis, we extended the sample set to include DNaseI hypersensitivity mapping data from an additional 8 less deeply sampled ENCODE cell types, thereby increasing our sample size to 15 cell types derived from different individuals (Consortium, 2011). In total we identified 215 mtDNA variants relative to the hg18 reference human sequence (Table S6C), 50 of which occurred within DNaseI footprints. Of these, 4 have previously been associated with disease (Crimi et al., 2003; Hauswirth and Clayton, 1985; Khogali et al., 2001; Lu et al., 2010). Of note, an additional 49 disease-associated mitochondrial variants that were not present in the cell lines examined also localize within DNaseI footprint regions.
We next analyzed the consequence of sequence variation on protein-DNA interactions by quantifying DNaseI protection at the variant position in affected cell types and comparing this with cell types containing the reference sequence. By this approach we identified a T16189C sequence variation that lies adjacent to a previously defined control element (Suissa et al., 2009) and associates with significant differences in DNaseI footprint occupancy (Figure 7E). We found the DNaseI cleavage profile over this variant position to be significantly distorted (p < 0.001) in the 3 cell lines containing the C allele compared with all 12 cell lines harboring the T allele (Figure 7D,E). Previously, the T16189C variant has been associated with an increased risk of dilated cardiomyopathy and type 2 diabetes (Khogali et al., 2001; Bhat et al., 2007). These results suggest a mechanistic basis for the contribution of the T16189C variant to human disease, and demonstrate the potential for mitochondrial DNaseI footprinting to simultaneously identify sequence variation and its functional consequences.
Higher-order mitochondrial genome organisation
Little is known about the higher order structure of the mitochondrial genome in vivo. Periodic trends in DNaseI cleavage patterns sites provide information on higher order DNA structure, such as the 10bp periodicity of minor groove DNaseI cleavages characteristic of DNA apposed to nucleosomes or other surfaces (Noll, 1974). We therefore considered whether periodic trends within DNaseI cleavage profiles might similarly provide insight into the higher order organization of the mitochondrial genome. We firstly identified periodicity of DNaseI cleavage by WOSA (Welch's Overlapped Segment Averaging) estimators. This revealed reproducible peaks at ∼10 nt phasing that deviate from random expectations (Figure S7A-G) (Percival D.B., 1993), suggesting that mitochondrial DNA is tightly apposed to a surface in vivo (Noll, 1974). The existence of additional higher order organization of mtDNA is also suggested by an enrichment for DNase cleavage at ∼80 nt periodicity, although the significance of this pattern within the structural model of the mitochondrial nucleoid (Holt et al., 2007) is unknown.
Discussion
The application of DNA and RNA sequencing technologies has transformed our understanding of the nuclear genome and revealed many novel features to a highly complex human transcriptome. In comparison to the nuclear genome, the mitochondrial genome has evolved its own unique solutions to regulate gene expression. The streamlined mitochondrial genome is initially transcribed in its entirety before a variety of post-transcriptional mechanisms act to liberate individual gene products and regulate their expression. We reasoned that these same approaches would be similarly well suited to profile the mitochondrial transcriptome at single-nucleotide resolution and at genome-wide scale.
Sequencing of the mitochondrial transcriptome revealed wide variation in gene expression which, given their common source, indicates the existence of complex post-transcriptional regulatory mechanisms. Analysis of the data also identified novel mitochondrial small RNAs, including small RNAs derived from tRNAs, as well as previously unknown antisense RNAs. The existence of these novel RNAs is demonstrated by PARE sequencing, which, in combination with other signatures of RNA processing, resolves the precise sites and frequencies by which individual gene products are fashioned from a common substrate to assemble a complete genetic system. This process is fundamental to the coordination of mitochondrial individual gene expression, and underscores the complexity of the mitochondrial transcriptome. Our analysis also revealed numerous non-canonical processing sites, including transient or rare un- or mis-processed intermediate events. This technique, in combination with the reference maps provided within this study, provides a powerful tool to investigate the hierarchy of cleaved intermediates and the proteins responsible for these processes (Temperley et al., 2010b).
Our analysis also adds to our growing understanding of the regulatory protein factors that bind to the mitochondrial genome. Recent chIP-seq studies have revealed that regulatory factors bind at many sites across the nuclear genome, often remotely to gene promoters. The small size of the mitochondrial genome permitted us to conduct a DNaseI protection analysis at unprecedented depth and resolution, and thereby identify regions of the genome protected by proteins. This revealed many novel binding motifs, for which the majority of interacting proteins remain to be discovered. Strong protein footprints are also associated with a major site of transcript termination and cleavage (as shown by RNaseA and PARE data) immediately downstream of the light strand promoter, which likely controls the asymmetric strand expression of the mitochondrial genome.
A broad spectrum of degenerative diseases has been associated with mtDNA mutations. Mitochondrial mutations can result in defective oxidative phosphorylation and energy metabolism, and may affect numerous downstream pathological effects, such as changes in apoptosis and DNA repair (Taylor and Turnbull, 2005). The maps collated within this resource, particularly the protein-binding profiles, provide a ready context in which to interpret the functional consequence of such genetic variation. Whether mediated at the level of protein-DNA interactions, post-transcriptional processing or gene expression, our data will provide a richer framework to understand the mechanisms by which these mutations mediate their phenotypic effects.
During our survey of the mitochondrial transcriptome, we were greeted at all levels with unanticipated novelty and complexity. We anticipate the continued interrogation of the datasets presented herein to reveal many previously unknown features of mitochondrial gene expression, function and regulation. As previously for the nuclear transcriptome, we expect that the application of sequencing approaches will transform our understanding of the distinct genetic system contained within human mitochondria.
Supplementary Material
Acknowledgments
This work was supported by grants and fellowships from the Australian Research Council, the National Health and Medical Research Council of Australia, the Queensland Government Department of Employment, Economic Development and Innovation, the Australian Stem Cell Centre, and NIH Grants U54HG004592 and U54HG004592-S1. We thank Rob King from GeneWorks (Adelaide, SA) for advice with RNA sequencing; Andreas Gruber from the Institute for Theoretical Chemistry in Vienna for providing his software for merging and fragmenting alignment files; Robert Thurman, Alex Reynolds and Dave Shechner for help with DNaseI hypersensitivity analysis; and Illumina (Hayward, CA) for providing early-access to the human ‘Body Map’ RNA sequencing atlas.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Bhat A, Koul A, Sharma S, Rai E, Bukhari SI, Dhar MK, Bamezai RN. The possible role of 10398A and 16189C mtDNA variants in providing susceptibility to T2DM in two North Indian populations: a replicative study. Hum Genet. 2007;120:821–826. doi: 10.1007/s00439-006-0272-4. [DOI] [PubMed] [Google Scholar]
- Cannino G, Di Liegro CM, Rinaldi AM. Nuclear-mitochondrial interaction. Mitochondrion. 2007;7:359–366. doi: 10.1016/j.mito.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Consortium TEP. A User's Guide to the Encyclopedia of DNA Elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimi M, Del Bo R, Galbiati S, Sciacco M, Bordoni A, Bresolin N, Comi GP. Mitochondrial A12308G polymorphism affects clinical features in patients with single mtDNA macrodeletion. Eur J Hum Genet. 2003;11:896–898. doi: 10.1038/sj.ejhg.5201056. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entelis NS, Kieffer S, Kolesnikova OA, Martin RP, Tarassov IA. Structural requirements of tRNALys for its import into yeast mitochondria. Proc Natl Acad Sci U S A. 1998;95:2838–2843. doi: 10.1073/pnas.95.6.2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entelis NS, Kolesnikova OA, Martin RP, Tarassov IA. RNA delivery into mitochondria. Adv Drug Deliv Rev. 2001;49:199–215. doi: 10.1016/s0169-409x(01)00135-1. [DOI] [PubMed] [Google Scholar]
- Fisher RP, Lisowsky T, Parisi MA, Clayton DA. DNA wrapping and bending by a mitochondrial high mobility group-like transcriptional activator protein. J Biol Chem. 1992;267:3358–3367. [PubMed] [Google Scholar]
- Fisher RP, Topper JN, Clayton DA. Promoter selection in human mitochondria involves binding of a transcription factor to orientation-independent upstream regulatory elements. Cell. 1987;50:247–258. doi: 10.1016/0092-8674(87)90220-0. [DOI] [PubMed] [Google Scholar]
- Fuste JM, Wanrooij S, Jemt E, Granycome CE, Cluett TJ, Shi Y, Atanassova N, Holt IJ, Gustafsson CM, Falkenberg M. Mitochondrial RNA polymerase is needed for activation of the origin of light-strand DNA replication. Mol Cell. 2010;37:67–78. doi: 10.1016/j.molcel.2009.12.021. [DOI] [PubMed] [Google Scholar]
- German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol. 2008;26:941–946. doi: 10.1038/nbt1417. [DOI] [PubMed] [Google Scholar]
- Gesell T, Washietl S. Dinucleotide controlled null models for comparative RNA gene prediction. BMC Bioinformatics. 2008;9:248. doi: 10.1186/1471-2105-9-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghivizzani SC, Madsen CS, Nelen MR, Ammini CV, Hauswirth WW. In organello footprint analysis of human mitochondrial DNA: human mitochondrial transcription factor A interactions at the origin of replication. Mol Cell Biol. 1994;14:7717–7730. doi: 10.1128/mcb.14.12.7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan MX, Enriquez JA, Fischel-Ghodsian N, Puranam RS, Lin CP, Maw MA, Attardi G. The deafness-associated mitochondrial DNA mutation at position 7445, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase subunit ND6 gene expression. Mol Cell Biol. 1998;18:5868–5879. doi: 10.1128/mcb.18.10.5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock K, LeBlanc AJ, Donze D, Hajduk SL. Identification of nuclear encoded precursor tRNAs within the mitochondrion of Trypanosoma brucei. J Biol Chem. 1992;267:23963–23971. [PubMed] [Google Scholar]
- Haussecker D, Huang Y, Lau A, Parameswaran P, Fire AZ, Kay MA. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA. 2010;16:673–695. doi: 10.1261/rna.2000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth WW, Clayton DA. Length heterogeneity of a conserved displacement-loop sequence in human mitochondrial DNA. Nucleic Acids Res. 1985;13:8093–8104. doi: 10.1093/nar/13.22.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ, He J, Mao CC, Boyd-Kirkup JD, Martinsson P, Sembongi H, Reyes A, Spelbrink JN. Mammalian mitochondrial nucleoids: organizing an independently minded genome. Mitochondrion. 2007;7:311–321. doi: 10.1016/j.mito.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Jacquier A. The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat Rev Genet. 2009;10:833–844. doi: 10.1038/nrg2683. [DOI] [PubMed] [Google Scholar]
- Karginov FV, Cheloufi S, Chong MM, Stark A, Smith AD, Hannon GJ. Diverse endonucleolytic cleavage sites in the mammalian transcriptome depend upon microRNAs, Drosha, and additional nucleases. Mol Cell. 2010;38:781–788. doi: 10.1016/j.molcel.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khogali SS, Mayosi BM, Beattie JM, McKenna WJ, Watkins H, Poulton J. A common mitochondrial DNA variant associated with susceptibility to dilated cardiomyopathy in two different populations. Lancet. 2001;357:1265–1267. doi: 10.1016/S0140-6736(00)04422-6. [DOI] [PubMed] [Google Scholar]
- Lane N, Martin W. The energetics of genome complexity. Nature. 2010;467:929–934. doi: 10.1038/nature09486. [DOI] [PubMed] [Google Scholar]
- Lee YS, Shibata Y, Malhotra A, Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs) Genes Dev. 2009;23:2639–2649. doi: 10.1101/gad.1837609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Qian Y, Li Z, Yang A, Zhu Y, Li R, Yang L, Tang X, Chen B, Ding Y, et al. Mitochondrial haplotypes may modulate the phenotypic manifestation of the deafness-associated 12S rRNA 1555A>G mutation. Mitochondrion. 2010;10:69–81. doi: 10.1016/j.mito.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin R, Bainbridge M, Fejes A, Hirst M, Krzywinski M, Pugh T, McDonald H, Varhol R, Jones S, Marra M. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. Biotechniques. 2008;45:81–94. doi: 10.2144/000112900. [DOI] [PubMed] [Google Scholar]
- Nagaike T, Suzuki T, Katoh T, Ueda T. Human mitochondrial mRNAs are stabilized with polyadenylation regulated by mitochondria-specific poly(A) polymerase and polynucleotide phosphorylase. J Biol Chem. 2005;280:19721–19727. doi: 10.1074/jbc.M500804200. [DOI] [PubMed] [Google Scholar]
- Noll M. Internal structure of the chromatin subunit. Nucleic Acids Res. 1974;1:1573–1578. doi: 10.1093/nar/1.11.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470–474. doi: 10.1038/290470a0. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput Biol. 2006;2:e33. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival DB, W AT. Spectral Analysis for Physical Applications: Multitaper and Conventional Univariate Techniques. Cambridge: Cambridge University Press; 1993. [Google Scholar]
- Piechota J, Tomecki R, Gewartowski K, Szczesny R, Dmochowska A, Kudla M, Dybczynska L, Stepien PP, Bartnik E. Differential stability of mitochondrial mRNA in HeLa cells. Acta Biochim Pol. 2006;53:157–168. [PubMed] [Google Scholar]
- Rossmanith W, Holzmann J. Processing mitochondrial (t)RNAs: new enzyme, old job. Cell Cycle. 2009;8:1650–1653. doi: 10.4161/cc.8.11.8502. [DOI] [PubMed] [Google Scholar]
- Rubio MA, Rinehart JJ, Krett B, Duvezin-Caubet S, Reichert AS, Soll D, Alfonzo JD. Mammalian mitochondria have the innate ability to import tRNAs by a mechanism distinct from protein import. Proc Natl Acad Sci U S A. 2008;105:9186–9191. doi: 10.1073/pnas.0804283105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbisa E, Tullo A, Nardelli M, Tanzariello F, Saccone C. Transcription mapping of the Ori L region reveals novel precursors of mature RNA species and antisense RNAs in rat mitochondrial genome. FEBS Lett. 1992;296:311–316. doi: 10.1016/0014-5793(92)80311-4. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, Bartel DP. Expanding the microRNA targeting code: functional sites with centered pairing. Mol Cell. 2010;38:789–802. doi: 10.1016/j.molcel.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomovic S, Laufer D, Geiger D, Schuster G. Polyadenylation and degradation of human mitochondrial RNA: the prokaryotic past leaves its mark. Mol Cell Biol. 2005;25:6427–6435. doi: 10.1128/MCB.25.15.6427-6435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suissa S, Wang Z, Poole J, Wittkopp S, Feder J, Shutt TE, Wallace DC, Shadel GS, Mishmar D. Ancient mtDNA genetic variants modulate mtDNA transcription and replication. PLoS Genet. 2009;5:e1000474. doi: 10.1371/journal.pgen.1000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvestre J, Vialette S, Corral Debrinski M, Jacq C. Long mRNAs coding for yeast mitochondrial proteins of prokaryotic origin preferentially localize to the vicinity of mitochondria. Genome Biol. 2003;4:R44. doi: 10.1186/gb-2003-4-7-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny RJ, Borowski LS, Brzezniak LK, Dmochowska A, Gewartowski K, Bartnik E, Stepien PP. Human mitochondrial RNA turnover caught in flagranti: involvement of hSuv3p helicase in RNA surveillance. Nucleic Acids Res. 2010;38:279–298. doi: 10.1093/nar/gkp903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temperley R, Richter R, Dennerlein S, Lightowlers RN, Chrzanowska-Lightowlers ZM. Hungry codons promote frameshifting in human mitochondrial ribosomes. Science. 2010a;327:301. doi: 10.1126/science.1180674. [DOI] [PubMed] [Google Scholar]
- Temperley RJ, Seneca SH, Tonska K, Bartnik E, Bindoff LA, Lightowlers RN, Chrzanowska-Lightowlers ZM. Investigation of a pathogenic mtDNA microdeletion reveals a translation-dependent deadenylation decay pathway in human mitochondria. Hum Mol Genet. 2003;12:2341–2348. doi: 10.1093/hmg/ddg238. [DOI] [PubMed] [Google Scholar]
- Temperley RJ, Wydro M, Lightowlers RN, Chrzanowska-Lightowlers ZM. Human mitochondrial mRNAs--like members of all families, similar but different. Biochim Biophys Acta. 2010b;1797:1081–1085. doi: 10.1016/j.bbabio.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Chen HW, Oktay Y, Zhang J, Allen EL, Smith GM, Fan KC, Hong JS, French SW, McCaffery JM, et al. PNPASE regulates RNA import into mitochondria. Cell. 2010;142:456–467. doi: 10.1016/j.cell.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Hofacker IL, Lukasser M, Huttenhofer A, Stadler PF. Mapping of conserved RNA secondary structures predicts thousands of functional noncoding RNAs in the human genome. Nat Biotechnol. 2005;23:1383–1390. doi: 10.1038/nbt1144. [DOI] [PubMed] [Google Scholar]
- Yakubovskaya E, Mejia E, Byrnes J, Hambardjieva E, Garcia-Diaz M. Helix unwinding and base flipping enable human MTERF1 to terminate mitochondrial transcription. Cell. 2010;141:982–993. doi: 10.1016/j.cell.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. Novel functions for small RNA molecules. Curr Opin Mol Ther. 2009;11:641–651. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.