

Abstract
Bacteria commonly grow in densely populated surface-bound communities, termed biofilms, where they gain benefits including superior access to nutrients and resistance to environmental insults. The secretion of extracellular polymeric substances (EPS), which bind bacterial collectives together, is ubiquitously associated with biofilm formation. It is generally assumed that EPS secretion is a cooperative phenotype that benefits all neighboring cells, but in fact little is known about the competitive and evolutionary dynamics of EPS production. By studying Vibrio cholerae biofilms in microfluidic devices, we show that EPS-producing cells selectively benefit their clonemates and gain a dramatic advantage in competition against an isogenic EPS-deficient strain. However, this advantage carries an ecological cost beyond the energetic requirement for EPS production: EPS-producing cells are impaired for dispersal to new locations. Our study establishes that a fundamental tradeoff between local competition and dispersal exists among bacteria. Furthermore, this tradeoff can be governed by a single phenotype.
Keywords: social evolution, cooperation, quorum sensing
Bacteria lead highly interactive lives within densely populated and often heterogeneous communities, termed biofilms (1–5). Biofilms typically grow on solid–liquid or liquid–air interfaces, and they are critical for processes ranging from biogeochemical cycling (6) and bacterial pathogenesis (7) to industrial biofouling (8). Biofilm-dwelling bacteria often secrete extracellular polymeric substances (EPS), which form a structural matrix in which cells become embedded (9). A predominant message—both implicit and explicit—in the biofilm literature is that EPS secretion is a cooperative trait that benefits neighboring bacteria and enhances a multicellular developmental program within bacterial communities (10–13). Studies examining biofilms of Pseudomonas fluorescens on the surface of liquids have suggested that EPS can benefit the bacterial community as a whole, including cells that do not contribute to production of the polymer matrix (14–16). Because they do not pay the cost of EPS production, however, such nonproducing strains invade wild-type biofilms on liquid surfaces and compromise their structural integrity (14–16).
Although the results described above certainly apply in their experimental context, it is puzzling that biofilm-dwelling bacteria often secrete EPS in natural settings if they are so readily invaded by nonproducing mutants. For example, hypermucoid strains of Pseudomonas aeruginosa and Burkholderia cepacia that secrete copious EPS are frequently isolated from the chronically infected lungs of cystic fibrosis patients (17), and numerous wild and laboratory isolates of Vibrio cholerae, which causes pandemic cholera in humans, constitutively produce EPS (18). Using spatially explicit simulations of biofilm growth, Xavier, Foster, and colleagues (19, 20) have suggested that bacteria that produce EPS can occupy spatial locations with superior access to nutrients relative to nonproducing cells. These theoretical models indicate that EPS secretion may be a competitively advantageous phenotype in natural biofilms, rather than one that easily succumbs to exploitation. In the present work we test this possibility via a combination of molecular genetics, microfluidics, and evolutionary theory.
Results and Discussion
We use the model organism V. cholerae, which generates biofilms on a range of surfaces, including the exoskeletons of marine arthropods and the intestinal tracts of animal and human hosts (21, 22). V. cholerae initiates biofilm growth after adhering to surfaces and shedding its flagella. Additionally, V. cholerae uses quorum sensing to activate EPS production at low cell density and to repress EPS production at high cell density (18). To isolate the EPS secretion phenotype and to control for the many hundreds of other genes regulated by flagellar activity and by quorum sensing, we deleted flaA, which encodes the flagellin core protein, and hapR, which encodes the quorum-sensing master regulator. The resulting ΔflaAΔhapR double mutant constitutively produces EPS and is hereafter designated EPS+. We also engineered an isogenic strain lacking vpsL, a gene required for EPS biosynthesis. The ΔflaAΔhapRΔvpsL triple mutant never produces EPS, and we call it EPS–.
Our rationale for introducing the ΔhapR and ΔflaA mutations into the EPS+ and EPS– strains is as follows: deletion of hapR ensures constitutive production of EPS in a strain of V. cholerae containing vpsL (18). We deleted flaA, rendering both strains immotile, for two reasons. First, mutation of flaA further increases EPS production in a strain containing vpsL (23). Second, a ΔhapR single mutant is relatively immotile due to constitutive production of EPS, which results in cell aggregation. A ΔhapRΔvpsL double mutant that cannot produce EPS, on the other hand, is highly motile. As a result, a ΔhapR mutant and a ΔhapRΔvpsL mutant exhibit two major differences, namely EPS production and motility. Introducing the ΔflaA mutation into both strain backgrounds ensured that EPS production is the only difference between the EPS+ and EPS– strains, providing greater clarity in the interpretation of our results.
Two versions each of the EPS+ and EPS– strains were next derived: one that constitutively expresses the teal fluorescent protein mTFP1 (24) and one that constitutively expresses the red fluorescent protein mKate (25). These fluorescent protein constructs allowed us to distinguish between EPS+ and EPS– cells in subsequent experiments, all of which were performed with each fluorescent variant to ensure that our results did not depend on which strain expressed which fluorescent protein.
We first addressed whether V. cholerae pays a cost in exchange for EPS production by growing the EPS+ and EPS– strains and their fluorescent counterparts in shaking minimal broth. The EPS+ strain exhibited a maximum growth rate of 0.139 h−1, 25% lower than that of the EPS– strain [0.183 h−1; two-tailed t test; t(4) = 9.39, P < 0.01]. These data demonstrate that the EPS+ strain pays a substantial cost for diverting resources away from biomass production and into the synthesis of EPS. Importantly, our growth curve results also show that the fluorescent protein constructs have no effect on growth rate (Figs. S1 and S2).
We next tested whether EPS production could provide a competitive advantage despite the large investment it entails, and if so, whether such an advantage is specific to the biofilm environment. We inoculated the EPS+ and EPS– strains in monoculture and in coculture at a range of initial frequencies on the glass substratum of simple straight-chamber microfluidic devices. In a companion experiment, we inoculated the two strains at a range of initial frequencies into shaken liquid culture tubes. At the end of each biofilm and mixed-liquid competition trial, we measured the frequency of each cell type using confocal scanning laser microscopy (biofilm) or conventional epifluorescence microscopy (liquid).
When grown in biofilm monocultures, the EPS+ strain accumulates more bioviolume per unit area of substratum than does the EPS– strain (Fig. 1A). When coinoculated at a 1:1 ratio in biofilms, the EPS+ strain's growth is unaffected by the presence of EPS– cells; however, the EPS– strain's biovolume accumulation decreases by more than 80%, suggesting that EPS+ cells interfere with the growth of EPS– cells (Fig. 1A). Regardless of the initial ratio of the two strains, EPS+ cells always increased in frequency at the expense of EPS– cells within biofilms (Fig. 1B). Biofilm structures rendered from confocal micrograph stacks are remarkably similar to those predicted by simulation studies (19, 20): EPS+ cells grow and divide into 3D clusters that physically displace EPS– cells, which are largely confined to the substratum (Fig. 2). The time-series shown in Fig. 2 illustrates the EPS+ strain increasing in frequency from <5% to >90% in 40 h, corresponding to a selection coefficient of 0.144 h−1 (Fig. S3). In mixed liquid culture, by contrast, EPS+ cells always decreased in frequency relative to the EPS– strain, in accordance with a simple model (Materials and Methods) that considers the lower growth rate of EPS+ cells compared with that of EPS– cells (Fig. 1B). Here, the selection coefficient is −0.044 h−1 with respect to the EPS+ strain.
Fig. 1.

Competition between EPS-producing (EPS+) and nonproducing (EPS–) V. cholerae strains in biofilms and in well-mixed liquid environments. (A) Left: When growing in biofilm monocultures, the EPS+ strain (white bar) accumulates more biovolume per unit area of substratum than does the EPS– strain (black bar). Right: The EPS+ strain is unaffected by coinoculation with the EPS– strain (white bar), whereas the EPS– strain's accumulated biovolume decreases by 80% when grown in coculture with EPS+ cells (black bar). Error bars denote SEM (n = 5). (B) Final frequency of the EPS+ strain is plotted against its initial frequency. In biofilms (closed circles), the EPS+ strain increases in frequency. In shaken liquid environments (open circles), the EPS+ strain decreases in frequency in accordance with model predictions (black line) based on its lower maximum growth rate relative to the EPS– strain. Error bars denote SD (biofilm experiments, n = 10–16; liquid experiments, n = 3).
Fig. 2.

A time-series of EPS+ cells (red) growing in a biofilm with EPS– cells (blue). The initial frequency of EPS+ cells is <5%, and its final frequency is >90%. At 36 h the upper surfaces of some EPS+ cell clusters appear flat because they have reached the ceiling of the chamber in which they are growing. Grid boxes are 11 μm on a side.
Our results indicate that EPS production is disadvantageous in mixed liquid environments, where the primary fitness currency is simply growth rate. In biofilm environments, on the other hand, a strain that produces EPS readily outcompetes an isogenic strain that does not. The EPS+ strain's advantage derives from its capacity to build structures that adhere to the substratum and resist shear stress. Biofilm simulations suggest that in building these structures, EPS+ cells could preferentially benefit themselves and their daughter cells (19, 20, 26). If so, each tower should predominantly contain cells of one lineage. To test this possibility, we initiated biofilms with only EPS+ cells, half expressing mTFP1 and half expressing mKate, and monitored biofilm growth. Although roughly the same number of cells of each color was present in the biofilms, each individual biofilm cluster largely contained cells of only one color, indicating that each cluster had indeed arisen from a single cell lineage (Fig. S4). These findings begin to explain why EPS secretion occurs in natural biofilms, many of which are submerged and subjected to flow: EPS-producing cells and their offspring gain a competitive advantage when growing on solid surfaces, and they inherently avoid exploitation by nonproducers.
Local competition is not the only factor contributing to long-term evolutionary dynamics: organisms must disperse to new resource patches as old ones are depleted or destroyed. Consistent with this broad ecological principle, biofilms of several bacterial species have been reported to break up and disperse (27). We therefore considered whether constitutive EPS production could affect dispersal ability. To explore this possibility, we inoculated the EPS+ and EPS– strains in microfluidic chambers and, at regular intervals, measured the frequency of EPS+ cells in both the biofilm and the liquid effluent. As shown above, the EPS+ strain rapidly dominates the biofilm. However, it remains a minority (although slowly increasing in frequency) in the liquid effluent over the same time period (Fig. 3). To determine whether the EPS+ strain's apparent dispersal disadvantage translates into poor colonization of new environments, we repeated the above experiment, but at 20 h and 46 h we connected the biofilm chamber's effluent tube to the inlet of fresh microfluidic devices and measured the composition of cell monolayers deposited in the new chambers. EPS+ cells were sparsely represented in the colonizing population at both time points (Fig. 4B). To simulate severe disturbance, we repeated the colonization experiment, and at 20 h and 46 h (in separate replicates) we increased flow velocity through the biofilm chambers 1,000-fold, the maximum permitted by our apparatus. The results were identical to those obtained when disturbance was mild (Fig. 4C).
Fig. 3.

A time-series showing the frequency of EPS+ cells in the biofilm and effluent phases of microfluidic flow chambers, when growing in competition with EPS– cells. The EPS+ strain rapidly increases in frequency relative to the EPS– strain within the biofilm (closed circles). In the effluent phase of the culture, EPS+ cells do increase in frequency over time (open circles), but more slowly than they do within the biofilm. Error bars denote SD (n = 4).
Fig. 4.

Competition between EPS+ and EPS– cells within biofilms and for access to new resource patches. (A) The frequency of the EPS+ strain within biofilms is plotted as a function of time. Error bars denote SD (n = 3). (B) Weak disturbance: effluent from chambers containing growing biofilms was diverted to new chambers at 20 h and 46 h. The biovolumes of EPS+ cells (white bars) and EPS– cells (black bars) in the newly formed monolayers are shown for both time points. (C) Severe disturbance: flow velocity through biofilm chambers was increased 1,000-fold at 20 and 46 h in separate replicates, and the effluent was used to colonize new chambers as in B.
Collectively, our findings suggest that the long-term dynamics of competition between EPS-producing and nonproducing cells fundamentally depend on how often resource patches are created and destroyed. Cells that produce EPS dominate local competition but are rarely dislodged from the structures they generate within biofilms. If resource patches are long-lived and new patches are rarely generated, EPS production will be favored. When resource patches are short-lived and new patches are generated often, however, cells that do not produce EPS will be favored. This system represents a canonical paradigm in ecology, the competition–colonization tradeoff. Notably, our study suggests that this tradeoff may be governed by a single phenotype among bacteria, namely EPS secretion. A clear implication is that bacteria, like metazoans, have undergone selection to strike a balance between local competition and dispersal ability, and as a result they have evolved finely tuned regulatory mechanisms, such as quorum sensing, with which they modulate the timing and strength of EPS production.
Materials and Methods
Strain Construction.
EPS+ and EPS– V. cholerae strains and fluorescent derivatives were engineered using standard protocols (28). Fluorescent protein constructs were inserted in single copy on the V. cholerae chromosome under the control of the strong constitutive promoter Ptac.
Herculase Enhanced DNA Polymerase (Stratagene) was used for all PCR cloning reactions. Restriction endonucleases, dNTPs, and T4 DNA ligase were acquired from New England Biolabs, and DNA extraction and purification kits were obtained from Qiagen and IBI Scientific. A full list of strains and plasmids used in this study is provided in Table S1.
Liquid Culture Growth Curve Experiments.
Strains were inoculated in shaking LB broth at 37 °C and grown overnight. Subsequently, cultures were diluted ≈100-fold into 2 mL of LB broth. These cultures were shaken at 37 °C and monitored until their optical density at 600 nm (OD600) was ≈0.6, corresponding to midexponential phase. The cultures were back-diluted 10,000-fold into 50-mL conical tubes (Corning) containing 40 mL of M9 minimal medium broth and 0.5% glucose. These cultures were incubated with shaking at 37 °C, and 1-mL samples were collected from each tube and their OD600 measured every hour until optical density readings saturated, corresponding to stationary phase. This experiment was replicated three times on 3 separate days. The curve-fitting software package available in MatLab (MathWorks) was used to calculate the maximum slope of each growth curve, and these data were compiled to determine the maximum growth rates of the EPS+ and EPS– strains and their derivatives (Fig. S1). To confirm that the difference in maximum growth rate between EPS+ and EPS– cells also occurred at room temperature, the temperature at which biofilm competition experiments were performed, we repeated the liquid growth curve experiments at room temperature. The maximum growth rates of both strains were lower at room temperature than at 37 °C; nonetheless, the maximum growth rate of the EPS+ strain remained at least 25% lower than that of the EPS– strain (Fig. S2).
Model of Competition Between EPS+ and EPS– Cells in Mixed Liquid Culture.
The maximum growth rate data obtained from the growth curve experiments were used to generate quantitative predictions for the outcome of competition between the EPS+ and EPS– strains in mixed liquid environments. Our model is based on the standard description of exponential population growth:
Nt is the population size at time t, N0 is the initial population size, and r is the maximum growth rate. The frequency of the EPS+ strain at time t, denoted fEPS+,t, is then equal to:
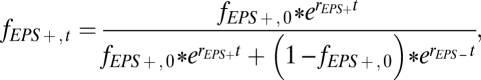 |
where fEPS+,0 is the initial frequency of the EPS+ strain, rEPS+ is the maximum growth rate of the EPS+ strain, and rEPS– is the maximum growth rate of the EPS– strain. Note that this model assumes that both strain populations are always increasing at their maximum rates. To satisfy this assumption in our liquid competition experiments, cultures were regularly diluted to prevent them from reaching stationary phase. Experimental data provided a good fit to this model, showing that the difference in maximum growth rate between the EPS+ and EPS– strains when grown alone is sufficient to explain the evolutionary dynamics of these two strains in liquid coculture. Because these cultures were maintained in exponential phase and growth rates were held constant at approximately their maximum values, we may also infer that the selection coefficient is s = rEPS+ − rEPS– = 0.139 h−1 – 0.183 h−1 = –0.044 h−1 with respect to the EPS+ strain in mixed liquid environments (29).
Competition in Mixed Liquid Cultures.
The fluorescent EPS+ and EPS– strains were inoculated in 2 mL of LB broth and grown overnight with shaking at 37 °C. EPS+mTFP1 was always paired with EPS–mKate (and vice versa), so that the two strains could be distinguished via microscopy. Overnight cultures were diluted ≈100-fold into test tubes containing 2 mL of LB broth. These cultures were shaken at 37 °C and monitored until their OD600 was ≈0.6, corresponding to midexponential phase. One-milliliter samples from these cultures were placed into Eppendorf tubes containing 0.1 mL autoclaved, acid-washed, 400- to 700-μm diameter glass beads (Sigma). Tubes were mixed by vortex for 30 s to disperse cell clusters, which primarily accumulated in EPS+ culture tubes. The bead-dispersed EPS+ and EPS– cultures were then diluted such that their OD600 measurements were equal, and from these diluted cultures a 1:1 mixture of the two strains was prepared. This sample was inspected via epifluorescence microscopy (see below) to confirm that the two strains were equally represented. If any discrepancy was noted, the pure cultures were diluted accordingly to ensure that the two strains could be combined at controlled ratios.
Using the cell density-equalized cultures, the EPS+ and EPS– strains were mixed at a range of initial frequencies and subsequently back-diluted 1,000-fold into Eppendorf tubes containing 0.1 mL autoclaved beads and 1.2 mL M9 minimal medium broth with 0.5% glucose. The competition cultures were rotated at 37 °C. Every 10 h, the tubes were mixed by vortex for 30 s to disperse cell clusters, and 1-μL samples were collected and placed into new tubes containing 0.1 mL autoclaved beads and 1.5 mL fresh M9 minimal medium with 0.05% glucose. After 40 h, the competition cultures were again mixed by vortex for 30 s, and 1-μL samples were placed onto glass slides. Agarose pads (5 × 5 mm) were placed on top of the 1-μL culture aliquots to ensure that all cells were retained in the same focal plane for inspection by epifluorescence microscopy. Three to four images were acquired per 1-μL sample, each of which captured approximately 1,000 cells. The liquid competition experiment was repeated three times on 3 separate days, and the data from all replicates were compiled and visualized using MatLab.
Competition in Biofilms.
Microfluidic devices consisted of poly(dimethylsiloxane) bonded to 22 mm × 60 mm microscope slides and were fabricated using standard methods (30). The biofilm growth chambers contained a single fluid inlet channel leading to a 2,000 μm × 200 μm × 40 μm (length × width × height) rectangular tunnel, followed by a single fluid outlet channel. Lengths of polyethylene tubing (Intramedic) with an internal diameter of 0.38 mm and an outer diameter of 1.09 mm were connected to the inlet and outlet channels.
Competition cultures containing EPS+ and EPS– cells at varying initial frequencies were prepared as described above for the liquid competition experiments. A fresh biofilm chamber was mounted onto a confocal scanning laser microscope (CSLM) before initiation of each experiment so that it need not be moved or manipulated before biofilm imaging. The premixed culture was pumped into the fresh chamber only until the culture was observed to enter the outlet tube. This initial culture was maintained in the biofilm chamber for ≈20 min, allowing cells to sink and adhere to the glass substratum. A new tube connected to a syringe containing fresh M9 minimal medium with 0.5% glucose was inserted into the chamber's inlet channel. The syringe was mounted on a syringe pump (Harvard Apparatus), which was used to supply fresh medium to the system and to clear the inoculating mixture from the biofilm chamber. The syringe pump operated at a flow velocity of 75–100 μm/s through the biofilm chamber. To measure the initial frequency of EPS+ cells in the biofilm monolayer, three to four images were immediately collected using the CSLM apparatus. After 40 h of growth at room temperature, biofilms were imaged using the CSLM apparatus (see below). Between five and eight samples were acquired per chamber, ensuring no overlap between the areas imaged, and these images were analyzed as independent replicates of biofilm growth. Data were obtained from two chambers for each initial EPS+ strain frequency, one using the EPS+mTFP1/EPS–mKate pair and one using the EPS+mKate/EPS–mTFP1 pair.
The procedure described above was performed with several modifications for the biofilm and liquid effluent time-series experiment. First, an inoculum containing 5% EPS+ cells was used to seed a biofilm monolayer, and a 0.5-cm length of tubing was connected to the outlet channel. At every sampling time point, images were acquired from nonoverlapping locations within each biofilm chamber, and the effluent that had gathered into a droplet (1–5 μL) at the top of the short outlet tube was collected. These effluent samples were immediately placed on microscope slides, covered with agarose pads, and imaged using an epifluorescence microscope. Data were quantified as described above for the liquid competition experiments.
To measure the colonization ability of the EPS+ and EPS– strains, the procedure described above was performed, and images were acquired from the chambers at regular intervals to assess biofilm composition. Twenty hours after the experiment began, the effluent tube was connected to the inlet channel of a fresh chamber. Twenty minutes later, five images of the colonizing layer in the new chamber were acquired using the CSLM apparatus to determine how well each strain had colonized the new substratum. Meanwhile, fresh medium continued to flow through the first biofilm chamber, and after 46 h, the first chamber's effluent tube was again connected to the inlet channel of a fresh chamber to allow colonization by the EPS+ and EPS– strains. Our experiment simulating severe disturbance was identical to that described above, except that we increased flow velocity through the channels 1,000-fold for 2 min at 20 h and 46 h (in separate replicates). The effluent was then loaded into a syringe and injected into a new chamber, allowed to sit for 20 min, and then imaged as described above.
Measurement of the Selection Coefficient in Biofilm Environments.
To quantify the strength of selection for the EPS production phenotype in biofilms, the selection coefficient was measured based on the experimentally observed evolutionary dynamics of the EPS+ and EPS– strains growing together in microfluidic devices. For a continuously reproducing haploid population consisting of two competing strains, the selection coefficient is defined as follows (29):
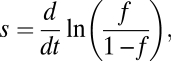 |
where f is the frequency of the focal strain; here, we take f to be the frequency of EPS+ cells in a population, such that the frequency of EPS– cells is equal to 1 − f. We determined the best fit line of 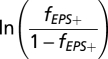 as a function of time using MatLab (Fig. S3). The average selection coefficient is equal to the slope of this line, or 0.144 h−1. This coefficient quantifies the strength of selection at the vps operon, because our two strains were otherwise isogenic, and there are no known pleiotropies associated with vps operon expression.
as a function of time using MatLab (Fig. S3). The average selection coefficient is equal to the slope of this line, or 0.144 h−1. This coefficient quantifies the strength of selection at the vps operon, because our two strains were otherwise isogenic, and there are no known pleiotropies associated with vps operon expression.
Microscopy Techniques.
Conventional epifluorescence microscopy imaging for liquid competition experiments and the effluent phase of biofilm competition experiments was performed using an inverted Nikon TE-2000U microscope fitted with GFP and RFP filter sets. Images were acquired with 40× and 100× oil objectives and an Andor iXon CCD camera cooled to −65 °C. Cells were counted using the cell counter add-on to the standard ImageJ software package (freely available from the National Institutes of Health: http://rsbweb.nih.gov/ij/). Cell counts, in turn, were used to calculate strain frequencies, and these data were analyzed and visualized in MatLab.
To visualize bacteria in biofilm competition experiments, we used an inverted Nikon Eclipse TE-2000U microscope fitted with a PerkinElmer UltraView RS spinning disk confocal unit and a Hamamatsu C9100-13 EMCCD camera cooled to −65 °C. A 488-nm laser line was used to excite the mTFP1 fluorescent protein, and a 568-nm laser line was used to excite the mKate fluorescent protein. Biofilms were imaged from bottom to top using a Z-interval of 0.4 μm. Because cells grow into dense clusters within biofilms, they could not be counted individually. Instead, the Volocity 3D imaging software package (PerkinElmer) was used to measure the total volume of red and teal fluorescent cells within biofilms. In the case of EPS+ cells, cell biovolume could be distinguished from EPS by tuning fluorescence thresholds in the Volocity software package, because EPS exhibits low levels of autofluorescence. The volumetric measurements thus obtained were taken as a proxy for the number of cells of each strain, and from these data we calculated the frequencies of the EPS+ and EPS– strains present in the biofilms. Strain frequencies were analyzed and visualized in MatLab.
Supplementary Material
Acknowledgments
We thank James Adelman, Sam Brown, Adrian de Froment, Knut Drescher, Kevin Foster, Nathan Gregory, Ned Wingreen, and members of the Bassler group and Levin group for advice; Brian Hammer and Christopher Waters for plasmids and help with genetics protocols; Jiandi Wan and Howard Stone for supporting the experiments using microfluidics; and Joseph Goodhouse for helping with confocal microscopy. C.D.N. is supported by a National Science Foundation Graduate Research Fellowship, a Princeton Centennial Fellowship, and Defense Advanced Research Planning Agency Grant HR0011-05-1-0057 via Simon A. Levin. B.L.B. is supported by the Howard Hughes Medical Institute, National Institutes of Health Grant 5R01GM065859, and National Science Foundation Grant MCB-0343821.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1111147108/-/DCSupplemental.
References
- 1.Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: From the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 2.West SA, Diggle SP, Buckling A, Gardner A, Griffins AS. The social lives of microbes. Annu Rev Ecol Evol Syst. 2007;38:53–77. [Google Scholar]
- 3.Nadell CD, Bassler BL, Levin SA. Observing bacteria through the lens of social evolution. J Biol. 2008;7:27. doi: 10.1186/jbiol87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nadell CD, Xavier JB, Foster KR. The sociobiology of biofilms. FEMS Microbiol Rev. 2009;33:206–224. doi: 10.1111/j.1574-6976.2008.00150.x. [DOI] [PubMed] [Google Scholar]
- 5.Xavier JB. Social interaction in synthetic and natural microbial communities. Mol Syst Biol. 2011;7:483. doi: 10.1038/msb.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paerl HW, Pinckney JL. A mini-review of microbial consortia: Their roles in aquatic production and biogeochemical cycling. Microb Ecol. 1996;31:225–247. doi: 10.1007/BF00171569. [DOI] [PubMed] [Google Scholar]
- 7.Fux CA, Costerton JW, Stewart PS, Stoodley P. Survival strategies of infectious biofilms. Trends Microbiol. 2005;13:34–40. doi: 10.1016/j.tim.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Jass J, Walker JT. Biofilms and biofouling. In: Walker JT, Surman S, Jass J, editors. Industrial Biofouling:Detection, Prevention and Control. New York: John Wiley & Sons; 2000. pp. 1–12. [Google Scholar]
- 9.Wingender J, Neu TR, Flemming H-C. Microbial Extracellular Polymeric Substances: Characterization, Structure. Berlin: Function; 1999. [Google Scholar]
- 10.O'Toole G, Kaplan HB, Kolter R. Biofilm formation as microbial development. Annu Rev Microbiol. 2000;54:49–79. doi: 10.1146/annurev.micro.54.1.49. [DOI] [PubMed] [Google Scholar]
- 11.Danese PN, Pratt LA, Kolter R. Biofilm formation as a developmental process. Methods Enzymol. 2001;336:19–26. doi: 10.1016/s0076-6879(01)36574-6. [DOI] [PubMed] [Google Scholar]
- 12.Hall-Stoodley L, Stoodley P. Developmental regulation of microbial biofilms. Curr Opin Biotechnol. 2002;13:228–233. doi: 10.1016/s0958-1669(02)00318-x. [DOI] [PubMed] [Google Scholar]
- 13.Webb JS, Givskov M, Kjelleberg S. Bacterial biofilms: Prokaryotic adventures in multicellularity. Curr Opin Microbiol. 2003;6:578–585. doi: 10.1016/j.mib.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 14.Rainey PB, Rainey K. Evolution of cooperation and conflict in experimental bacterial populations. Nature. 2003;425:72–74. doi: 10.1038/nature01906. [DOI] [PubMed] [Google Scholar]
- 15.Brockhurst M, Buckling A, Racey D, Gardner A. Resource supply and the evolution of public-goods cooperation in bacteria. BMC Biol. 2008;6:20. doi: 10.1186/1741-7007-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brockhurst MA, Buckling A, Gardner A. Cooperation peaks at intermediate disturbance. Curr Biol. 2007;17:761–765. doi: 10.1016/j.cub.2007.02.057. [DOI] [PubMed] [Google Scholar]
- 17.Govan JR, Deretic V. Microbial pathogenesis in cystic fibrosis: Mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev. 1996;60:539–574. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammer BK, Bassler BL. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol Microbiol. 2003;50:101–104. doi: 10.1046/j.1365-2958.2003.03688.x. [DOI] [PubMed] [Google Scholar]
- 19.Xavier JB, Foster KR. Cooperation and conflict in microbial biofilms. Proc Natl Acad Sci USA. 2007;104:876–881. doi: 10.1073/pnas.0607651104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nadell CD, Xavier JB, Levin SA, Foster KR. The evolution of quorum sensing in bacterial biofilms. PLoS Biol. 2008;6:e14. doi: 10.1371/journal.pbio.0060014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu J, Mekalanos JJ. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev Cell. 2003;5:647–656. doi: 10.1016/s1534-5807(03)00295-8. [DOI] [PubMed] [Google Scholar]
- 22.Tamayo R, Patimalla B, Camilli A. Growth in a biofilm induces a hyperinfectious phenotype in Vibrio cholerae. Infect Immun. 2010;78:3560–3569. doi: 10.1128/IAI.00048-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watnick PI, Lauriano CM, Klose KE, Croal L, Kolter R. The absence of a flagellum leads to altered colony morphology, biofilm development and virulence in Vibrio cholerae O139. Mol Microbiol. 2001;39:223–235. doi: 10.1046/j.1365-2958.2001.02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ai HW, Henderson JN, Remington SJ, Campbell RE. Directed evolution of a monomeric, bright and photostable version of Clavularia cyan fluorescent protein: Structural characterization and applications in fluorescence imaging. Biochem J. 2006;400:531–540. doi: 10.1042/BJ20060874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shcherbo D, et al. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 2007;4:741–746. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- 26.Nadell CD, Foster KR, Xavier JB. Emergence of spatial structure in cell groups and the evolution of cooperation. PLOS Comput Biol. 2010;6:e1000716. doi: 10.1371/journal.pcbi.1000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaplan JB. Biofilm dispersal: Mechanisms, clinical implications, and potential therapeutic uses. J Dent Res. 2010;89:205–218. doi: 10.1177/0022034509359403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 29.Chevin LM. On measuring selection in experimental evolution. Biol Lett. 2010;7:210–213. doi: 10.1098/rsbl.2010.0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sia SK, Whitesides GM. Microfluidic devices fabricated in poly(dimethylsiloxane) for biological studies. Electrophoresis. 2003;24:3563–3576. doi: 10.1002/elps.200305584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.