

Abstract
Cyanovirin-N (CV-N) is a small, cyanobacterial lectin that neutralizes many enveloped viruses, including human immunodeficiency virus type I (HIV-1). This antiviral activity is attributed to two homologous carbohydrate binding sites that specifically bind high mannose glycosylation present on envelope glycoproteins such as HIV-1 gp120. We created obligate CV-N oligomers to determine whether increasing the number of binding sites has an effect on viral neutralization. A tandem repeat of two CV-N molecules (CVN2) increased HIV-1 neutralization activity by up to 18-fold compared to wild-type CV-N. In addition, the CVN2 variants showed extensive cross-clade reactivity and were often more potent than broadly neutralizing anti-HIV antibodies. The improvement in activity and broad cross-strain HIV neutralization exhibited by these molecules holds promise for the future therapeutic utility of these and other engineered CV-N variants.
Keywords: crystal structure, domain-swapped dimer, protein engineering
Cyanovirin-N (CV-N), a cyanobacterial lectin, is uniquely positioned to become a therapeutic and prophylactic for diseases caused by enveloped viruses. CV-N is a small, two-domain protein that neutralizes HIV by specifically binding to high mannose glycans on the envelope glycoprotein gp120, thereby preventing interaction of the virus with a host cell (1, 2). In addition to its potent activity against HIV, CV-N is also active against a number of other enveloped viruses including influenza (3, 4), Ebola (5, 6), hepatitis C (7), and herpesvirus 6 (2).
The two domains of CV-N are homologous in both their sequence [32% sequence identity and 58% sequence similarity (8)] and their three-dimensional structure (9, 10). Wild-type (WT) CV-N exists mainly as a monomer in solution and a domain-swapped dimer in crystals (Fig. S1). NMR structures of the monomer show that the protein is an ellipsoid with ten β-strands and four 310-helical turns, approximately 55 Å in length and 25 Å wide (Fig. S1A). Each CV-N monomer contains two symmetrically related structural domains (A and B), with each domain containing a carbohydrate binding site that specifically interacts with α(1-2) linked oligomannose moieties within Man-8 or Man-9 glycans (9, 11–13). Domain A contains the N and C termini and includes residues 1–39 and 90–101, and domain B contains residues 40–89. Although purified as a monomer, a trapped, metastable domain-swapped dimer can be formed during folding and crystallization, and WT CV-N crystallizes exclusively as a domain-swapped dimer (10, 14, 15). In the domain-swapped dimer structure, domain A interacts with B′ to form a “monomer-like unit,” whereas domain A′ and B interact to form the second “monomer-like unit” (Fig. S1 B and C). The domain swapping does not result in additional intramolecular interactions, but instead results from the extension of residues 50 to 53 across the interface. Carbohydrate binding is similar in the domain-swapped crystal structures and monomeric NMR structures, although binding in the B and B′ domains is not seen in crystal structures due to potential steric constraints or crystal packing artifacts (15) (Fig. S1 A and B).
The two binding sites in monomeric CV-N exhibit distinct affinities for carbohydrate in solution: the binding site in domain B, located distal from the N and C termini, has an equilibrium dissociation constant (KD) of approximately 140 nM for Manα1 → 2Man disaccharide, which is about 10-fold higher affinity than the binding site in domain A, located near the termini, which binds to Manα1 → 2Man disaccharide with a KD of about 1.5 μM (9). Numerous studies have shown, however, that both sites are necessary for viral neutralization and that destruction of either site renders the CV-N variant inactive (16, 17). However, a recent study showed that in the context of a CV-N dimer that was covalently crosslinked using disulfide bonds, two out of the four possible binding sites are sufficient to maintain neutralization activity, indicating that it is the number and not the identity of sites that is important for neutralization (18, 19). These results point toward a key role for avidity in the viral neutralization activity of CV-N.
A number of groups have attempted to study the oligomerization of CV-N to determine whether the domain swapping is a crystallization artifact or a biologically relevant state. However, because the domain-swapped dimer of WT CV-N is not stable at physiological temperatures, a significant amount of purified dimer may revert to monomer during the course of a viral neutralization assay (14). Therefore, mutations have been used to stabilize either the monomer (14) or the domain-swapped dimer (14, 20, 21). The effect of dimerization remains unclear, as some groups have concluded that the dimeric state is more active than monomeric WT CV-N (21), whereas others find that monomeric and dimeric variants have similar antiviral activities (20).
In this study, we show that by linking two CV-N molecules together in a head-to-tail fashion, we can stabilize the domain-swapped dimeric form of the protein in solution. These linked dimers show enhanced HIV neutralization compared to WT CV-N against 33 strains from 3 clades. In addition, we show that although two carbohydrate binding sites are sufficient for activity as previously reported (18, 19), variants with more binding sites (three or four) have increased neutralization activity.
Results
Design and Construction of CV-N Oligomers.
To directly assay the effects of multimerization on the activity of CV-N, we generated CV-N dimers (CVN2s) containing tandem repeats of CV-N in which the C terminus of one copy of CV-N was linked to the N terminus of the next copy through a flexible polypeptide linker. Because WT CV-N has the ability to domain swap, we hypothesized that the oligomeric molecules would adopt either a monomeric-like linked structure in which the two CV-N repeats are folded as monomers and connected through the linker (Fig. 1 A and B) or a domain-swapped linked structure that takes a form similar to the domain-swapped dimeric crystal structures of WT CV-N but contains a direct linkage between the two CV-N repeats (Fig. 1 C and D). We generated different versions of CVN2 proteins using 10 different linker lengths ranging from 0 to 20 amino acids (Table S1). We also constructed trimers (CVN3) with linkers containing 0, 5, or 10 amino acids and a tetramer (CVN4) in which two CVN2L0 molecules were linked through a 20 amino acid linker.
Fig. 1.

Two models of CVN2 proteins. (A, B) The linked monomer dimeric model for CVN2s. (C, D) The linked domain-swapped dimeric model for CVN2s. The CV-N repeats are shown in red and blue, and the flexible polypeptide linker is modeled in green. The N and C termini are labeled N and C. Each of the four carbohydrate binding domains are labeled A, B, A′, or B′ in each model. Models (A) and (C) are based on solved WT CV-N structures (10, 44) and (B) and (D) are block representations of the models in (A) and (C), respectively.
CV-N Oligomers Show Enhanced HIV Neutralization.
CV-N and CV-N oligomers were assayed for their ability to neutralize HIV in an in vitro luminescence-based neutralization assay (Fig. 2A, Table S2) (22). Initial characterizations of the CV-N proteins were conducted by assaying activity against the clade B HIV strain SC422661.8. WT CV-N showed half-maximal neutralization at concentrations (IC50s) between 1.0 and 9.4 nM (0.012 to 0.12 μg/mL) over 30 independent trials, with an average of 4.4 ± 2.6 nM (0.054 ± 0.032 μg/mL), consistent with published values (20, 23–25).
Fig. 2.

CV-N oligomers exhibit enhanced HIV neutralization. (A) Typical neutralization data and curve fits for WT CV-N and two variants run in triplicate on the same 96-well plate. (B) The IC50s of HIV neutralization for WT CV-N and CVN2 dimers containing varying linker lengths. All linked dimers show significant enhancements in their HIV neutralization compared to WT CV-N, and dimers containing 0 to 10 amino acid linkers are more potent than CVN2L20. N≥4; CVN2L20: N = 1. (C) IC50s of HIV neutralization for CVN2s and CVN3s of the same linker length. N≥3; CVN3L5 and CVN3L10: N = 1. (A–C) Error bars = SD.
Most dimeric variants (CVN2s) exhibited more potent HIV neutralization than WT CV-N, with enhancements of approximately 3- to 6-fold when correcting for the increased molecular weight of the dimers (Fig. 2B). The single exception was CVN2L20, which displayed an IC50 similar to that seen for WT CV-N. In this case, the long linker may allow a different domain-swapping state relative to variants with shorter linkers. Additionally, CVN2L20 was only assayed on a single occasion (in triplicate) and therefore the true behavior of the molecule may not be accurately represented. Although there were significant increases in potency for most of the dimers, the addition of the third CV-N repeat to make the CVN3 proteins did not improve HIV neutralization compared to the dimeric variants; in fact the trimers showed decreased potency compared to the lower molecular weight dimeric molecules (Fig. 2C). A tetrameric CV-N protein (CVN4) showed similar activity to the CVN3 molecules (data not shown).
CV-N Dimers Exhibit Broad Cross-Clade Reactivity and Are Comparable or More Potent than Anti-HIV Antibodies.
We then assayed WT CV-N and two of our more potent dimers (CVN2L0 and CVN2L10) for their ability to neutralize other strains of HIV across different clades. The proteins were tested against a total of 33 strains from three clades. WT CV-N and the dimeric mutants effectively neutralized all 33 HIV pseudoviruses (IC50s less than 300 nM) (Table S3). Consistent with earlier results, the dimeric variants were more potent than WT CV-N against all strains tested (Fig. 3A). In 32 out of 33 cases, CVN2L0 neutralized virus with greater potency than CVN2L10, whereas CVN2L10 was the most potent against one clade C strain.
Fig. 3.

CV-N dimers neutralize HIV broadly and potency is similar to broadly neutralizing anti-HIV antibodies. (A) The designed dimers show enhanced neutralization activity relative to WT CV-N across all 33 strains tested from 3 clades. CVN2L0 is more potent than CVN2L10 in 32 of 33 cases. (B) When the IC50s of CVN2L0 neutralization against a panel of HIV-1 strains were compared to the IC50s of seven broadly neutralizing antibodies (Table S3), we saw that most strains were as sensitive to CVN2L0 as they were to the broadly neutralizing antibodies. Because 2-fold differences in potency are generally not significant, similar potency (≥) is defined as a potency for CVN2L0 that is within 2-fold of the potency of the antibody or higher.
We were also interested in the overall potency of the CV-N proteins compared to known broadly neutralizing anti-HIV antibodies (NAbs): 4E10 (26), 2G12 (26, 27), 2F5 (26, 28), b12 (29), PG9 (30), PG16 (30), and VRC01 (31). IC50 values for these NAbs were taken from the literature (22, 32, 33) or determined for this study (PG9, PG16, and VRC01) and converted to molarity for comparisons with the smaller CV-N proteins. When compared to each of the seven broadly neutralizing anti-HIV antibodies, CVN2L0 showed similar or greater potency against most of the strains tested (Fig. 3B). Because 2-fold differences in IC50 values are generally not significant, we consider IC50s within 2-fold of the IC50 of the antibody to be similar. CVN2L0 has similar or greater potency against 100% of the viruses compared to 4E10, 2G12, and 2F5. Our molecule also fairs very well when compared to the newly discovered broadly neutralizing antibodies PG9 (72%), PG16 (66%), and VRC01 (77%).
CV-N Dimers Are Domain-Swapped in Crystal Structures.
To elucidate a mechanism for the enhanced neutralization activity of the dimeric proteins, we solved crystal structures of CVN2L0 and CVN2L10 (SI Text, Table S4, and Fig. S2). Both structures were very similar to each other and were intramolecularly domain-swapped with no major deviations relative to WT CV-N domain-swapped structures. Small differences between the CVN2 structures and previously published WT CV-N structures were noted near the termini, as expected from the linkage, and also in the region of the domain swap. These differences, however, are minor and are unlikely to be responsible for the observed differences in antiviral activity.
Binding Site Mutants of CVN2L0 Show That It Is Domain-Swapped in Solution.
Although crystallographic studies definitively showed that the CVN2 molecules are domain-swapped in crystals, it was still possible that the dimeric proteins were folded as two linked monomers (Fig. 1A) rather than as a domain-swapped dimer (Fig. 1B) in solution. To address this issue, we generated variants of CVN2L0 that contained previously described CV-N carbohydrate binding site knockouts (16, 34) (Fig. 4A). Each of the binding sites in a domain-swapped dimer (A, B, A′, and B′) are formed from residues in both CV-N repeats, so we created variants in which a binding site in either the A or B domain was knocked out completely in the context of the linked monomer dimeric model (CVN2L0ΔAmm, CVN2L0ΔBmm) or the domain-swapped dimeric model (CVN2L0ΔA, CVN2L0ΔB) (Table S5). In each case, if the model that the mutations were based on is correct, the mutations would form a single full binding site knockout (black squares in Fig. 4A). However, if the model is incorrect, the mutations would form two half-site knockouts (black triangles in Fig. 4A). We hypothesized that CVN2L0 mutants with a complete binding site knockout in solution would show decreased ability to neutralize HIV, whereas mutants with two half-site knockouts would be less severely affected (34). Control variants with only a single half-site knockout showed only modest decreases in potency, verifying that half-site knockouts could be distinguished from complete binding site knockouts (Table S5, Fig. S3).
Fig. 4.

Anti-HIV activity of CVN2L0 correlates with number of functional binding sites. (A) Schematic representation of variants to determine whether CVN2L0 is in a linked monomer dimeric structure (mm) or domain-swapped dimeric structure. The two CV-N repeats are represented in red and blue, as in Fig. 1 B and D. Black triangles represent partial carbohydrate binding site knockouts and black squares represent complete binding site knockouts. (B) HIV neutralization results for mutants in A. Mutants with full binding site deletions in the context of the domain-swapped dimer model have more significant increases in their HIV neutralization IC50s compared to mutants with full binding site deletions in the context of the monomer model. (C) Schematic representations of multiple binding site mutants. All variants contain one or more complete binding site knockouts according to the CVN2L0 domain-swapped dimer model. Black squares represent binding sites that have been knocked out, and squares containing red and blue triangles represent WT (functional) binding sites. (D) HIV neutralization results for mutants in C. The number of functional binding sites in CVN2L0 is proportional to its ability to neutralize HIV. Mutants with two functional binding sites are less active than those with three sites. Additionally, the deletion of a B binding site has a greater effect on activity than the deletion of an A binding site.
Variants with binding site knockouts made in the context of the domain-swapped dimeric model were significantly less active against HIV than those designed based on the monomeric model (Fig. 4B), indicating that the linked dimers are domain-swapped in solution as well as in crystals.
The Number of Functional Binding Sites in CVN2L0 Is Directly Proportional to Its Anti-HIV Activity.
To determine whether CVN2L0 has enhanced neutralization activity relative to WT CV-N due to its increased number of binding sites, we created a series of binding site knockout mutants in which 1, 2, or 3 of the sites were fully knocked out (Fig. 4C). Variants with only a single binding site knockout showed approximately 20- to 35-fold decreases in potency relative to CVN2L0, whereas those with two binding site knockouts exhibited decreases of 80- to almost 2,000-fold (Fig. 4D). Variants with three binding sites knocked out were unable to neutralize HIV at the concentrations tested (data not shown). These data are consistent with previously published accounts, which showed that at least two functional binding sites are required for activity (16, 19), and are also consistent with the hypothesis that avidity is an important factor for viral neutralization by CV-N. Interestingly, CVN2L0 variants that contain one functional A and one functional B binding site did not neutralize with the same potency as WT CV-N. CVN2L0ΔA∥B and CVN2L0ΔA×B in which the active A and B binding sites are on the same pseudomonomer or opposite pseudomonomer (Fig. 4C), respectively, were 15- to 75-fold less potent than monomeric WT CV-N, which also contains only a single A and single B binding site. This observation indicates that factors in addition to avidity, including possible steric occlusion of binding sites and/or the relative orientation of binding sites, contribute to the potency of an oligomeric CV-N molecule.
Discussion
By covalently linking two or more copies of CV-N together, we generated CV-N oligomers with significantly enhanced HIV neutralization activity compared to WT CV-N. CVN2L0 not only exhibited broadly neutralizing activity across three clades of HIV-1, but was also able to neutralize many HIV strains with potency similar to that of seven well studied broadly neutralizing antibodies (4E10, 2G12, 2F5, b12, PG9, PG16, and VRC01). In addition, our crystallographic and mutagenesis studies revealed that the CVN2s form domain-swapped dimers in solution as well as in crystal form. By increasing the local concentration of CV-N through linking two molecules together, we have stabilized the domain-swapped dimeric form of the protein, allowing it to be stable under physiological conditions.
Previous studies were divided about whether the domain-swapped dimer of WT CV-N is more active than the monomeric form (20, 21). However, because the CV-N domain-swapped dimer is metastable under physiological temperatures and significant amounts can revert to monomer over the course of an assay (14), the domain-swapped form has been difficult to evaluate with current HIV neutralization assays. In contrast, our variants are covalently linked at their termini and are thereby effectively forced into the domain-swapped dimeric form by the effective increase in local concentration. Because there were no major structural differences between the linked dimers and WT CV-N, our results suggest that the dimeric species of WT CV-N would also be a more potent neutralization agent if it were stable during the assay. Therefore, other methods in which the dimer is stabilized may also result in increased neutralization activity.
In addition to the potential benefit of domain swapping, the simple increase in avidity in the CVN2s significantly improves the neutralization activity. WT CV-N itself has a high affinity for gp120 (1, 23), but by doubling the number of carbohydrate binding sites in the CVN2 variants, the increase in avidity may prevent possible dissociation and escape of the virus. As shown by our knockout studies, CVN2L0 variants with more functional binding sites were significantly more potent at neutralizing HIV, indicating that higher avidity translates to greater potency. Interestingly, we also found that deletion of a binding site in the B domain had a greater effect on the ability to neutralize HIV than deletion of a binding site in the A domain. CVN2L0ΔBB, which contains only two functional A sites, is approximately 15-fold worse at neutralizing HIV than CVN2L0ΔAA, which contains only two functional B sites. This finding is consistent with earlier studies that showed that binding site B has an approximately 10-fold lower KD for Manα1-2Man than binding site A (9) and may indicate that the overall activity of CV-N could be improved by improving the affinity of site A.
An alternate mechanism for increased neutralization could result from the fact that the binding sites in CVN2s can potentially sample distances farther apart than the binding sites in monomeric WT CV-N. The wider spacing could allow CVN2s to crosslink glycosylation sites within a single gp120, across multiple gp120 subunits on an envelope spike or, less likely, across multiple spikes. This crosslinking would prevent a larger number of gp120 subunits from binding to CD4, the primary receptor for HIV, than would be blocked by WT CV-N, thus decreasing the IC50. An interesting note is that in one conformation of the domain-swapped structure of WT CV-N (Fig. S1B), every pair of carbohydrate binding sites is approximately 30 to 35 Å apart (Fig. 5A). The neutralizing antibody 2G12, which is also domain-swapped and binds carbohydrates on gp120, has carbohydrate binding sites that are also approximately 30 to 35 Å apart (35) (Fig. 5B). Perhaps by stabilizing the domain-swapped structure of CV-N, the carbohydrate binding sites of the CVN2 variants are optimally positioned to interact with gp120.
Fig. 5.

Similarity in carbohydrate binding site spacing in CV-N and the 2G12 anti-HIV (Fab)2. (A) Each of the four carbohydrate binding sites in one WT CV-N crystal structure (15) (P41212 space group) is approximately 30 to 35 Å from the other sites (structure is viewed from the bottom with respect to Fig. 1). Carbohydrates (shown as sticks with black carbons and red oxygens) were only resolved in the A binding sites in the crystal structure. (B) Ribbon diagram of the domain-swapped (Fab)2 from IgG 2G12, a broadly neutralizing antibody specific for carbohydrates on gp120 (35). The domain swapping creates a rigid (Fab)2 dimer in which the carbohydrate binding sites at the antigen combining sites are spaced approximately 30 to 35 Å apart. Carbohydrates are shown as sticks with black carbons and red oxygens and antibody domains are labeled.
While addition of a second CV-N molecule increases the potency of HIV neutralization significantly, the addition of a third or fourth CV-N repeat (CVN3, CVN4) does not increase it further. Although the mechanism for enhanced activity is not fully understood, perhaps nondomain-swapped CV-N repeats do not have a significant impact on the activity; for example, one of the three repeats in trimeric CVN3 molecules may have little effect on activity. Alternatively, due to the close proximity of the N and C termini in the WT structure and their proximity to the lower affinity carbohydrate binding site (site A), the additional CV-N molecule(s) may sterically occlude access to some of the carbohydrate binding sites in the molecule, rendering those sites nonfunctional and therefore inhibiting any additional effect. Longer linkers, including structured linkers, may be necessary to prevent steric occlusion of the binding sites.
WT CV-N and the CVN2 molecules show excellent cross-clade and cross-strain reactivity. This property is promising for the development of these or other variants for therapeutic use, as they can potentially be used throughout the world. Because of the increase in potency relative to WT CV-N, CVN2L0 could be more effective in any prophylactic treatment protocol that WT CV-N is currently being investigated for, including gels, suppositories, and in vivo Lactobacillus delivery (36, 37). In addition to the increase in potency of CVN2L0, the lack of a proteolytically sensitive linker between the CV-N repeats suggests that this variant will probably have similar stability in vivo as WT CV-N. CVN2L0 shows similar potency to many of the broadly neutralizing antibodies that have recently been reported but is easier to express than intact antibodies and therefore could be used for a range of therapeutics that are intractable for antibodies. CV-N variants could also theoretically be used in combination therapy with anti-gp120 antibodies to direct gp120 evolution toward decreased glycosylation. Glycosylation itself has been shown to be important in the folding and function of viral glycoproteins (38), and in the case of HIV, deglycosylation of gp120 diminishes its binding to CD4, making the virus less infective (39, 40). Alternatively, deglycosylation of gp120 could reveal protein epitopes that can be recognized by the adaptive immune system, allowing the immune system to fight off infection more effectively.
Materials and Methods
Construct Generation.
The gene for WT CV-N was constructed using a recursive PCR method with 40-mer synthesized oligos (41), then subcloned into the NdeI and BamHI sites of pET11a. The protein contained an N-terminal 6-histidine purification tag followed by a Factor Xa protease cleavage site. CVN2L5 and CVN2L10 were constructed using PCR-based cloning to insert a tandem repeat of the WT CV-N gene and sequence encoding the flexible polypeptide linker into the WT plasmid. The CVN3L5 gene was created by inserting an Escherichia coli-optimized WT CV-N DNA sequence between the two existing copies of the WT gene in CVN2L5. Other dimeric and trimeric genes of varying linker lengths were constructed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) to insert or delete codons corresponding to linker amino acids. All constructs were verified through DNA sequencing and restriction analysis to ensure the correct sequence and number of CV-N repeats.
Binding site knockout mutant constructs were generated in the background of a CVN2L0 template gene containing two distinct DNA sequences for each CV-N repeat. Mutations were made using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene).
Protein Expression and Purification.
The expression of WT CV-N and all oligomeric variants was induced with IPTG in BL21(DE3) E. coli cells in LB including ampicillin. The harvested cells were lysed using an EmulsiFlex-C5 (Avestin, Inc.), and the insoluble fraction was resuspended in buffer containing 6 M GnHCl and 10 mM imidazole and centrifuged to remove debris. The solubilized CV-N was then purified under denaturing conditions using a Ni-NTA gravity column (Qiagen) and refolded by dialyzing the Ni-NTA eluate against native buffer overnight at room temperature (42). Following refolding, proteins were additionally purified on a Superdex-75 column and eluted in 25 mM sodium phosphate pH 7.4, 150 mM NaCl. The N-terminal 6-histidine purification tag was not removed prior to functional or structural assays. Pure protein was concentrated or stored as eluted at 4 °C.
Amino acid analysis was performed on WT CV-N, CVN2L5, CVN2L10, CVN3L5, and CVN3L10 to determine extinction coefficients at 280 nm (Texas A&M University). These experimentally determined extinction coefficients (WT: 10,471 M-1 cm-1; CVN2s: 20,800 M-1 cm-1; CVN3s: 32,000 M-1 cm-1) were used to calculate the protein concentration.
HIV Neutralization Assays.
HIV-1 pseuodovirus particles from pseudotyped primary virus strains were prepared as described (22, 43). The SC422661.8 strain (clade B) was used for all assays unless otherwise noted. HIV neutralization assays were performed either in-house (Fig. 2, Table S2) or by the Collaboration for AIDS Vaccine Discovery (CAVD) core neutralization facility (Fig. 3, Table S3) as previously described (22). Briefly, 250 infectious viral units of virus per well were incubated with threefold dilutions of CV-N or a CV-N variant in triplicate (our assays) or duplicate (CAVD assays) for 1 h at 37 °C after which approximately 10,000 Tzm-Bl cells were added to each well and incubated for 48 h. The cells were then lysed using Bright Glo Luciferase Assay Buffer (Promega), and luciferase expression was assayed using a Victor3 Multilabel Counter (PerkinElmer).
To determine the IC50 of neutralization, the luminescence was first averaged across the replicates, then the percent neutralization (%Neutralization) was calculated using Eq 1, where RLU is the average relative luminescence for a given concentration, CC is the average luminescence from the cell control wells, and VC is the average luminescence from the viral control wells. The percent of virus neutralized was then plotted as a function of neutralizing protein concentration in Kaleidagraph (Synergy Software) and fitted to Eq. 2, where IC50 is the concentration of CV-N at which half of the virus is neutralized, C is the concentration of CV-N, and m is a Hill coefficient. IC50s are reported as the average of a minimum of four independent trials and the error reported is the standard deviation of the IC50s from those trials. CVN2L20, CVN3L5, CVN3L10, and the binding site mutants were tested on only one occasion and therefore a standard deviation is not reported.
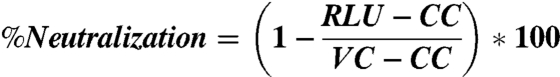 |
[1] |
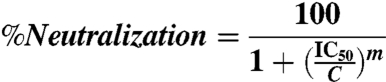 |
[2] |
Crystallization, Crystallographic Data Collection, and Refinement.
See SI Text.
Supplementary Material
Acknowledgments.
We thank Leonard Thomas, Pavle Nikolovski, and the Molecular Observatory at Caltech, which is supported by the Gordon and Betty Moore Foundation, for assistance in setting up crystal trays, collecting and processing diffraction data, and refining crystal structures. We also thank the Collaboration for AIDS Vaccine Discovery Neutralizing Antibody Core Laboratories for performing HIV neutralization assays and Marie Ary for critical review of the manuscript. This work was funded by the National Security Science and Engineering Faculty Fellowship program and the Defense Advanced Research Projects Agency Protein Design Processes program (to S.L.M.) and by the Bill and Melinda Gates Foundation Grant 38660 through the Grand Challenges in Global Health Initiative (to P.J.B.). X-ray data for CVN2L10 were collected at the Stanford Synchrotron Radiation Lightsource (Beam Line 12-2). Operations at Stanford Synchotron Radiation Lightsource are supported by the Department of Energy and the National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3S3Y, 3S3Z).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1108777108/-/DCSupplemental.
References
- 1.Boyd MR, et al. Discovery of cyanovirin-N, a novel human immunodeficiency virus-inactivating protein that binds viral surface envelope glycoprotein gp120: potential applications to microbicide development. Antimicrob Agents Chemother. 1997;41:1521–1530. doi: 10.1128/aac.41.7.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dey B, et al. Multiple antiviral activities of cyanovirin-N: blocking of human immunodeficiency virus type 1 gp120 interaction with CD4 and coreceptor and inhibition of diverse enveloped viruses. J Virol. 2000;74:4562–4569. doi: 10.1128/jvi.74.10.4562-4569.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Keefe BR, et al. Potent anti-influenza activity of cyanovirin-N and interactions with viral hemagglutinin. Antimicrob Agents Chemother. 2003;47:2518–2525. doi: 10.1128/AAC.47.8.2518-2525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smee DF, et al. Influenza A (H1N1) virus resistance to cyanovirin-N arises naturally during adaptation to mice and by passage in cell culture in the presence of the inhibitor. Antivir Chem Chemoth. 2007;18:317–327. doi: 10.1177/095632020701800604. [DOI] [PubMed] [Google Scholar]
- 5.Barrientos LG, Gronenborn AM. The highly specific carbohydrate-binding protein cyanovirin-N: structure, anti-HIV/Ebola activity and possibilities for therapy. Mini-Rev Med Chem. 2005;5:21–31. doi: 10.2174/1389557053402783. [DOI] [PubMed] [Google Scholar]
- 6.Barrientos LG, et al. Cyanovirin-N binds to the viral surface glycoprotein, GP1,2 and inhibits infectivity of Ebola virus. Antiviral Res. 2003;58:47–56. doi: 10.1016/s0166-3542(02)00183-3. [DOI] [PubMed] [Google Scholar]
- 7.Helle F, et al. Cyanovirin-N inhibits hepatitis C virus entry by binding to envelope protein glycans. J Biol Chem. 2006;281:25177–25183. doi: 10.1074/jbc.M602431200. [DOI] [PubMed] [Google Scholar]
- 8.Gustafson KR, et al. Isolation, primary sequence determination, and disulfide bond structure of cyanovirin-N, an anti-HIV (human immunodeficiency virus) protein from the cyanobacterium Nostoc ellipsosporum. Biochem Biophys Res Commun. 1997;238:223–228. doi: 10.1006/bbrc.1997.7203. [DOI] [PubMed] [Google Scholar]
- 9.Bewley CA, Otero-Quintero S. The potent anti-HIV protein cyanovirin-N contains two novel carbohydrate binding sites that selectively bind to Man(8) D1D3 and Man(9) with nanomolar affinity: implications for binding to the HIV envelope protein gp120. J Am Chem Soc. 2001;123:3892–3902. doi: 10.1021/ja004040e. [DOI] [PubMed] [Google Scholar]
- 10.Yang F, et al. Crystal structure of cyanovirin-N, a potent HIV-inactivating protein, shows unexpected domain swapping. J Mol Biol. 1999;288:403–412. doi: 10.1006/jmbi.1999.2693. [DOI] [PubMed] [Google Scholar]
- 11.Bolmstedt AJ, O’Keefe BR, Shenoy SR, McMahon JB, Boyd MR. Cyanovirin-N defines a new class of antiviral agent targeting N-linked, high-mannose glycans in an oligosaccharide-specific manner. Mol Pharmacol. 2001;59:949–954. doi: 10.1124/mol.59.5.949. [DOI] [PubMed] [Google Scholar]
- 12.Shenoy SR, et al. Multisite and multivalent binding between cyanovirin-N and branched oligomannosides: calorimetric and NMR characterization. Chem Biol. 2002;9:1109–1118. doi: 10.1016/s1074-5521(02)00237-5. [DOI] [PubMed] [Google Scholar]
- 13.Shenoy SR, O’Keefe BR, Bolmstedt AJ, Cartner LK, Boyd MR. Selective interactions of the human immunodeficiency virus-inactivating protein cyanovirin-N with high-mannose oligosaccharides on gp120 and other glycoproteins. J Pharmacol Exp Ther. 2001;297:704–710. [PubMed] [Google Scholar]
- 14.Barrientos LG, et al. The domain-swapped dimer of cyanovirin-N is in a metastable folded state: reconciliation of X-ray and NMR structures. Structure. 2002;10:673–686. doi: 10.1016/s0969-2126(02)00758-x. [DOI] [PubMed] [Google Scholar]
- 15.Botos I, et al. Structures of the complexes of a potent anti-HIV protein cyanovirin-N and high mannose oligosaccharides. J Biol Chem. 2002;277:34336–34342. doi: 10.1074/jbc.M205909200. [DOI] [PubMed] [Google Scholar]
- 16.Barrientos LG, Matei E, Lasala F, Delgado R, Gronenborn AM. Dissecting carbohydrate-Cyanovirin-N binding by structure-guided mutagenesis: functional implications for viral entry inhibition. Protein Eng Des Sel. 2006;19:525–535. doi: 10.1093/protein/gzl040. [DOI] [PubMed] [Google Scholar]
- 17.Fromme R, et al. A monovalent mutant of cyanovirin-N provides insight into the role of multiple interactions with gp120 for antiviral activity. Biochemistry. 2007;46:9199–9207. doi: 10.1021/bi700666m. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, et al. Multivalent interactions with gp120 are required for the anti-HIV activity of Cyanovirin. Biopolymers. 2009;92:194–200. doi: 10.1002/bip.21173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matei E, et al. Anti-HIV activity of defective cyanovirin-N mutants is restored by dimerization. J Biol Chem. 2010;285:13057–13065. doi: 10.1074/jbc.M109.094938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrientos LG, Lasala F, Delgado R, Sanchez A, Gronenborn AM. Flipping the switch from monomeric to dimeric CV-N has little effect on antiviral activity. Structure. 2004;12:1799–1807. doi: 10.1016/j.str.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 21.Kelley BS, Chang LC, Bewley CA. Engineering an obligate domain-swapped dimer of cyanovirin-N with enhanced anti-HIV activity. J Am Chem Soc. 2002;124:3210–3211. doi: 10.1021/ja025537m. [DOI] [PubMed] [Google Scholar]
- 22.Li M, et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79:10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colleluori DM, et al. Expression, purification, and characterization of recombinant cyanovirin-N for vaginal anti-HIV microbicide development. Protein Expres Purif. 2005;39:229–236. doi: 10.1016/j.pep.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 24.Mori T, et al. Functional homologs of cyanovirin-N amenable to mass production in prokaryotic and eukaryotic hosts. Protein Expres Purif. 2002;26:42–49. doi: 10.1016/s1046-5928(02)00513-2. [DOI] [PubMed] [Google Scholar]
- 25.Mori T, et al. Analysis of sequence requirements for biological activity of cyanovirin-N, a potent HIV (human immunodeficiency virus)-inactivating protein. Biochem Biophys Res Commun. 1997;238:218–222. doi: 10.1006/bbrc.1997.7202. [DOI] [PubMed] [Google Scholar]
- 26.Buchacher A, et al. Generation of human monoclonal antibodies against HIV-1 proteins; electrofusion and Epstein-Barr virus transformation for peripheral blood lymphocyte immortalization. AIDS Res Hum Retroviruses. 1994;10:359–369. doi: 10.1089/aid.1994.10.359. [DOI] [PubMed] [Google Scholar]
- 27.Trkola A, et al. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J Virol. 1996;70:1100–1108. doi: 10.1128/jvi.70.2.1100-1108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Purtscher M, et al. A broadly neutralizing human monoclonal antibody against gp41 of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 1994;10:1651–1658. doi: 10.1089/aid.1994.10.1651. [DOI] [PubMed] [Google Scholar]
- 29.Burton DR, et al. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc Natl Acad Sci USA. 1991;88:10134–10137. doi: 10.1073/pnas.88.22.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walker LM, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu X, et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li M, et al. Genetic and neutralization properties of subtype C human immunodeficiency virus type 1 molecular env clones from acute and early heterosexually acquired infections in Southern Africa. J Virol. 2006;80:11776–11790. doi: 10.1128/JVI.01730-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.West AP, Jr, et al. Evaluation of CD4-CD4i antibody architectures yields potent, broadly cross-reactive anti-human immunodeficiency virus reagents. J Virol. 2010;84:261–269. doi: 10.1128/JVI.01528-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang LC, Bewley CA. Potent inhibition of HIV-1 fusion by cyanovirin-N requires only a single high affinity carbohydrate binding site: characterization of low affinity carbohydrate binding site knockout mutants. J Mol Biol. 2002;318:1–8. doi: 10.1016/S0022-2836(02)00045-1. [DOI] [PubMed] [Google Scholar]
- 35.Calarese DA, et al. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science. 2003;300:2065–2071. doi: 10.1126/science.1083182. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, et al. Engineered vaginal lactobacillus strain for mucosal delivery of the human immunodeficiency virus inhibitor cyanovirin-N. Antimicrob Agents Chemother. 2006;50:3250–3259. doi: 10.1128/AAC.00493-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ndesendo VM, et al. A review of current intravaginal drug delivery approaches employed for the prophylaxis of HIV/AIDS and prevention of sexually transmitted infections. AAPS PharmSciTech. 2008;9:505–520. doi: 10.1208/s12249-008-9073-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vigerust DJ, Shepherd VL. Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 2007;15:211–218. doi: 10.1016/j.tim.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fenouillet E, Gluckman JC, Bahraoui E. Role of N-linked glycans of envelope glycoproteins in infectivity of human immunodeficiency virus type 1. J Virol. 1990;64:2841–2848. doi: 10.1128/jvi.64.6.2841-2848.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montefiori DC, Robinson WE, Jr, Mitchell WM. Role of protein N-glycosylation in pathogenesis of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1988;85:9248–9252. doi: 10.1073/pnas.85.23.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene. 1995;164:49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]
- 42.Barrientos LG, et al. Design and initial characterization of a circular permuted variant of the potent HIV-inactivating protein cyanovirin-N. Proteins. 2002;46:153–160. doi: 10.1002/prot.10024. [DOI] [PubMed] [Google Scholar]
- 43.Wei XP, et al. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 44.Bewley CA. Solution structure of a cyanovirin-N:Manα1-2Manα complex: structural basis for high-affinity carbohydrate-mediated binding to gp120. Structure. 2001;9:931–940. doi: 10.1016/s0969-2126(01)00653-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.